

Abstract
Extracellular DNA (eDNA) is an important component of the biofilm matrix produced by many bacteria. In general, the release of eDNA is associated with the activity of muralytic enzymes leading to obvious cell lysis. In the Gram-positive oral commensal Streptococcus gordonii, eDNA release is dependent on pyruvate oxidase generated hydrogen peroxide (H2O2). Addition of H2O2 to cells grown under conditions non-permissive for H2O2 production causes eDNA release. Furthermore, eDNA release is maximal under aerobic growth conditions known to induce pyruvate oxidase gene expression and H2O2 production. Obvious cell lysis, however, does not occur. Two enzymes have been recently associated with eDNA release in S. gordonii. The autolysin AtlS and the competence regulated murein hydrolase LytF. In the present report, we investigated the role of both proteins in the H2O2 dependent eDNA release process. Single and double mutants in the respective genes for LytF and AtlS released less eDNA under normal growth conditions, but the AtlS mutant was still inducible for eDNA release by external H2O2. Moreover, we showed that the AtlS mutation interfered with the ability of S. gordonii to produce eDNA release inducing amounts of H2O2. Our data support a role of LytF in the H2O2 eDNA dependent release of S. gordonii as part of the competence stress pathway responding to oxidative stress.
Introduction
The biofilm developmental process requires the release of extracellular polymeric substances (EPS) by the biofilm forming community [1], [2]. The EPS is commonly composed of protein, polysaccharides, lipids and extracellular DNA (eDNA) [3], [4]. The presence of eDNA during the developmental process is important [5], since treatment of developing or preformed biofilms with DNA degrading enzymes disrupts the biofilm structure and stability [6], [7], [8]. In general, eDNA found in the biofilm EPS seems to be of microbial chromosomal origin [9], [10], [11]. Investigations on the integrity of the eDNA revealed largely intact DNA still carrying genomic information, which is further supported by the observation that eDNA is also a source for horizontal gene transfer [12], [13]. A recent detailed oligonucleotide array based study using a non-domesticated Bacillus subtilis strain showed that the biofilm recovered eDNA includes the whole genome without specific gene preferences [14]. Earlier studies with other bacterial species showed that randomly selected genes on different chromosomal locations are present in eDNA [10] suggesting that chromosomal DNA is released to serve as eDNA during biofilm development.
Different release/production mechanisms for eDNA of chromosomal origin seem to exist. For example, the bacteriolytic dependency of eDNA release was demonstrated for several species and is ultimately linked to bacterial cell death [11], [15], [16]. The regulatory relationship between eDNA release and cell death has been studied in detail in Gram-positive Staphylococcus aureus [17], [18], [19]. The Staphylococcal Cid/Lrg system encodes proteins analogous to the bacteriophage-encoded holins and antiholins. Initial studies suggest that the LrgA and CidA proteins function in similar mechanisms as the holins/antiholins in S. aureus, ultimately activating murein hydrolases leading to bacterial cell lysis [17], [18], [19]. Homologs of Cid/Lrg can be found in several species, including cariogenic Streptococcus mutans. Interestingly, the Cid/Lrg system in S. mutans is involved in several other cellular processes including competence development and oxidative stress tolerance, suggesting a connection between general stress and lysis dependent eDNA release in oral streptococci [20], [21]. In the opportunistic pathogen Pseudomonas aeruginosa, phage inductions in biofilms are implicated in the release of DNA as a result of phage mediated cell lysis [11], [22]. Other examples of lysis dependent eDNA release are the autolysin AtlE dependent eDNA release in Staphylococcus epidermidis [23] and the gelatinase GelE and serine protease SprE dependent eDNA release in Enterococcus faecalis [16].
An alternative to the cell-lysis dependent eDNA release mechanisms has been suggested in two recent studies in E. faecalis [24] and B. subtilis [14] where a lysis-independent eDNA release mechanism has been proposed. In E. faecalis well-defined structures of eDNA were observed supporting early biofilm development. However, no intracellular components indicative of cell lysis could be detected in cell free supernatants during the early biofilm developmental stage. Furthermore, cells implicated in the release of eDNA had an active membrane potential excluding a connection with bacterial cell death [24]. B. subtilis on the other hand has a mechanism to release eDNA in the late exponential phase. The authors of this study confirmed lysis-independence genetically by constructing several mutant strains with genes involved in bacterial lysis showing they are not reduced in the eDNA release. The release process appeared to be regulated by the B. subtilis early competence genes [14].
Streptococcus gordonii belongs to the group of early oral biofilm formers and it’s presence is critical for subsequent biofilm development since it provides attachment sites for other species [25]. S. gordonii as well as several other oral streptococci are known for their ability to produce competitive amounts of hydrogen peroxide (H2O2) during aerobic growth [26]. H2O2 production inhibits growth of competing species, but also induces the release of eDNA [10], [27], [28]. Furthermore, H2O2 has been demonstrated as the sole agent responsible for triggering the release process. The addition of H2O2 to S. gordonii grown under non-H2O2-producing conditions during static growth induces eDNA release, but no detectable autolysis [12]. Involvement of bacteriolytic enzymes in the eDNA release of S. gordonii, however, has been shown by two recent studies [13], [29]. Deletion of the autolysin AtlS causes a major decrease in eDNA release [29]. In addition, inactivation of the competence dependent murein hydrolase LytF reduced the gene transfer in a co-culture of competent S. gordonii with the LytF mutant about 100 fold. However, direct eDNA concentrations were not determined [13].
The present report presents a further characterization of the role of LytF and AtlS in the H2O2 dependent eDNA release of S. gordonii.
Materials and Methods
Bacterial Species and Culture Conditions
All S. gordonii strains used in this study listed in Tab. 1 were routinely grown aerobically (5% CO2) at 37°C in BHI (Brain Heart Infusion; Difco, Sparks, MD) unless otherwise stated. For antibiotic selection, cultures were supplemented with the following antibiotics: erythromycin at 5 µg ml−1 and kanamycin at 300 µg ml−1.
Table 1. Strains and oligonucleotides used in this study.
| Strain | Relevant characteristics | Reference |
| DL1 | Wild-type S. gordonii | [29] |
| DL1 AtlS | atlS; Kanr | [29] |
| DL1 AtlS-1 | DL1 AtlS; kan resistance cassette replacedwith erm resistance cassette; Ermr | This study |
| DL1 LytF | Transformation of chrom. DNA from strain SGH24[13] carrying a lytF deletion, Kanr | This study |
| DL1 AtlS/LytF | DL1 AtlS-1 transformed with chrom. DNA fromstrain SGH24 [13] carrying a lytF deletion, Kanr, Ermr | This study |
| Oligonucleotides | Sequence | Purpose |
| gyrA RT-F | CCAAACCTTTTGGTCAATGG | Real time RT-PCR |
| gyrA RT-R | CCCAGGCAAAACTTCCATAA | Real time RT-PCR |
| 16S rRNA Sg-F | AAGGAACGCGAAGAACCTTA | Real Time PCR |
| 16S rRNA Sg-R | GTCTCGCTAGAGTGCCCAAC | Real Time PCR |
| spxB RT-F | GGATGCTTTGGCTGAAGAC | Real time RT-PCR |
| spxB RT-R | GGACCACCTGAACCTACTG | Real time RT-PCR |
| AtlS up-F | GAAATCCTGCGCAATAAAGC | AtlS K.O. |
| AtlS up-R | ATCAAACAAATTTTGGGCCCGGTGAA | AtlS K.O. |
| AtlS down-F | ATTCTATGAGTCGCTGCCGACTCCAA | AtlS K.O. |
| AtlS down –R | CATGCAGACATTATAGCA | AtlS K.O. |
| ermAM-F | CCGGGCCAAAAATTTGTTTGAT | AtlS K.O. |
| ermAM-F | CCGGGCCAAAAATTTGTTTGAT | AtlS K.O. |
| lytF RT-F | TTAATGGCCAAGACGACCTC | Real time RT-PCR |
| lytF RT-R | TACTTTCCCGCCAGATTGTC | Real time RT-PCR |
| Sg-ldh-up-F | GAAGGAGATGTTTAGAGAATGAC | ren-reporter |
| Sg-ldh-up-R-ren | TCTCTAAACATCTCCTTCTTAGTTTTTAGATGCTGCTTGGAATT | ren-reporter |
| Sg-ren-F | GAAGGAGATGTTTAGAGAATGACTTCGAAAGTTTATGATCCAG | ren-reporter |
| Sg-ren-R | CAAACAAATTTTGGGCCCGGTTATTGTTCATTTTTGAGAACTCGC | ren-reporter |
| Sg-ren-erm-F | GCGAGTTCTCAAAAATGAACAATAACCGGGCCCAAAATTTGTTTG | ren-reporter |
| Sg-ren-erm-R | CTCCTTTAATAAGGAGATGTTTTTATAAAGTCGGCAGCGACTCATAG | ren-reporter |
| Sg-ldh-down-F | CTATGAGTCGCTGCCGACTTTATAAAAACATCTCCTTATTAAAGGAG | ren-reporter |
| Sg-ldh-down-R | CCAAAAGATGTCTTGTCAGTTGG | ren-reporter |
Growth Kinetics
The growth of static wild type and mutant strains was monitored using a Bioscreen C analyzer version 2.4 (Oy Growth Curves AB Ltd., Finland), which measures the turbidity in multiple cultures in parallel for static cultures. Growth kinetics were monitored at 37°C in BHI in 20 min intervals. To measure growth under aerobic conditions with maximal H2O2 production, cells were grown in 15 ml screw cap tubes with a starting volume of 10 ml. The tubes were placed on a rocking table, promoting horizontal movement (Barnstead Thermolyne Vari-Mix), at 20 rpm and incubated at 37°C. Bacterial cell density was determined with a Genesys 20 spectrophotometer (Thermo Spectronic) at A600 nm.
DNA Manipulations
Standard recombinant DNA manipulations were used [30]. PCR was performed with a G-Storm GS1 thermocycler (GeneTechnologies; Essex, UK) according to the manufacturer’s protocol. Phusion® DNA polymerase was obtained from New England Biolab. Oligonucleotides (Tab. 1) were designed using sequence data obtained from the Los Alamos National Laboratory Oral Pathogens Sequence Database (http://www.oralgen.lanl.gov) and synthesized by Integrated DNA Technologies (Coralville, IA).
Construction of Mutant Strains
The lytF mutant strain was constructed by transforming chromosomal DNA from strain SGH24 [13] carrying a lytF deletion using a transformation protocol reported earlier [31]. To construct a double AtlS/LytF K.O. mutant, the AtlS mutant was chosen to replace the kan antibiotic cassette with an ermAM cassette for compatibility with the lytF K.O. Replacement of the antibiotic cassette was done via double-crossover homologous recombination using a overlap PCR strategy. To generate the overlap PCR constructs, two fragments corresponding to around 500 bp of the upstream and downstream sequences of atlS were amplified by PCR, using Phusion® DNA polymerase with the oligonucleotides AtlS up-F/AtlS up-R and AtlS down-F/AtlS down-R. Each of the oligonucleotides listed in Tab.1 as up-R and down-F incorporated 25 bases complementary to the erythromycin resistance cassette, ermAM [32]. The erythromycin resistance cassette ermAM was amplified by PCR using the primers ermAM F and ermAM R as described before [31]. All three PCR amplicons were purified with the QIAGEN PCR purification kit and mixed in a 1∶1:1 ratio. The mixture served as a template for a second round PCR with the appropriate up F and down R primers. The resulting PCR amplicons were transformed into DL1 AtlS to generate the deletion mutant DL1 AtlS-1. To create the double mutant, the LytF mutation was transformed into DL1 AtlS-1 as described above.
Construction of Renilla Bioluminescent Reporter-strain and Renilla Assay
The renilla reporter strain was constructed via a four-piece overlapping PCR ligation strategy similar to the strategy described above. The renilla gene was set under the control of the ldh (lactate dehydrogenase) promoter from S. gordonii. The renilla gene was inserted downstream of the ldh stop codon to leave the ldh gene intact. Briefly, about 1000 bp of the 5′ ldh open reading frame including the ribosome binding site (rbs) of the ldh promoter were amplified with oligonucleotides Sg-ldh-up-F/Sg-ldh-up-R-ren, the renilla gene was amplified from plasmid pRL-TK (gift from Dr. Ralf Janknecht, University of Oklahoma Health Sciences Center) with oligonucleotides Sg-ren-F/Sg-ren-R. The Sg-ren-F primer introduced the ldh rbs on the 5′ end of the renilla gene. The ermAM gene cassette for selection of PCR product integration into the chromosome was amplified using oligonucleotides Sg-ren-erm-F/Sg-ren-erm-R and the ldh downstream fragment (about 1000 bp) was amplified with oligonucleotides Sg-ldh-down-F/Sg-ldh-down-R. Oligonucleotides were constructed with overlapping sequences as shown in Tab. 1. All four PCR amplicons were purified with the QIAGEN PCR purification kit and mixed in a 1∶1:1∶1 ratio. The mixture served as a template for a second round PCR with the appropriate up F and down R primers. The resulting PCR amplicons were transformed into DL1. Successful transformation was confirmed by testing several transformants for renilla reporter gene activity. The LytF and AtlS mutation were generated by transformation of chromosomal DNA from DL1-AtlS and SGH24, respectively. To assay for renilla activity, 100 µl of an exponentially growing culture was mixed with 0.5 µl of ViviRen™ Live Cell Substrate (Promega) from a 3.7 µg/1 µl stock solution. Bioluminescence was determined with a Modulus Luminometer (Turner BioSystems).
RNA Isolation, cDNA Synthesis, and Real-time RT PCR
RNA was isolated using a Qiagen RNeasy kit, and cDNA was synthesized using qScript™ cDNA synthesis kit (Quanta Biosciences) according to the manufacturer’s protocol. Real-time RT PCR was performed to determine specific cDNA copies with the comparative threshold cycle (CT) method using a MyiQ single-color real-time PCR detection system (Bio-Rad) and PerfeCta™ SYBR® Green SuperMix for iQ™ (Quanta Biosciences). Relative changes in cDNA copies representing differential gene expression were calculated using the ΔCT method described previously (62). The 16S rRNA gene was used as the housekeeping reference gene using the 16S rRNA oligonucleotides described in Tab. 1.
Determination of H2O2 Concentration
The concentration of H2O2 in liquid cultures was determined using a modification of the protocol described by Gilliland (12). Cell-free culture supernatants (40 µl) were mixed with 160 µl of freshly prepared 0.1 M sodium acetate (pH 4.5) containing 0.1 µg horseradish peroxidase (Thermo Scientific) and 10 µl of 1 mg/ml o-dianisidine (Alfa Aesar) in methanol. The reaction mixture was incubated at room temperature for 10 min and protected from light before A415 nm was determined using a microplate reader (model 680; Bio-Rad). The concentration was calculated from a standard curve prepared in the same medium or buffer using a serial dilution of a commercial 30% H2O2 solution in MilliQ water. The concentration of the initial dilution was determined spectrophotometrically (ε240 = 43.6/M·cm) using a SmartSpec Plus UV-visible spectrophotometer (Bio-Rad) before each new experiment. The detectable range was 0.1 to 4.0 mM H2O2 in BHI.
Observation of eDNA Release
The amount of eDNA in liquid cultures was measured directly by quantitative real-time PCR. A 2-µl aliquot of cell-free culture supernatant was mixed with 8 µl molecular-grade water (G-Biosciences), 12.5 µl PerfeCta™ SYBR® Green SuperMix for iQ™ (Quanta Biosciences), 1.25 µl of primer 16S rRNA Sg-F, and 1.25 µl primer 16S rRNA Sg-R from a 10 mM stock solution. The PCR was performed in a MyiQ single-color real-time PCR detection system (Bio-Rad) and included one cycle of 95°C for 3 min, followed by 40 cycles of 95°C for 15 s and 55°C for 1 min. The DNA concentration was calculated based on average threshold cycle values against a 10-fold dilution series of purified DL1 genomic DNA in the same medium. The detectable range was 0.001 to 100 µg/ml DNA. The concentration of the standard was adjusted using a NanoDrop-1000 spectrophotometer (Thermo Scientific).
Statistical Analysis
Statistical significance was calculated using a two-sided Student’s t-test and Quickcalcs online calculator (http://www.graphpad.com/quickcalcs). P values less than 0.05 were considered statistically significant.
Results
Influence of Aeration on Growth of Wild Type, AtlS, LytF and AtlS/LytF Mutants
Previous experimental results linked the production of H2O2 to the release of eDNA in S. gordonii [12]. H2O2 is produced by the pyruvate oxidase (SpxB) under aerobic growth conditions and has a self-inhibitory effect on the producing species [12], [28]. To determine whether the introduction of the respective AtlS, LytF and AtlS/LytF mutations into S. gordonii causes any growth defects, growth was monitored under aerobic and static conditions, which has been shown to abolish H2O2 production [12]. During static growth, both the wild type and the LytF mutant showed nearly identical growth patterns (Fig. 1A). The AtlS and AtlS/LytF mutant strains showed a slightly reduced growth rate, but all four strains reached the same final bacterial density. In contrast, when cells were grown aerobically with maximal H2O2 production, the wild type and the LytF mutant strains reached stationary phase earlier than the AtlS and AtlS/LytF mutants, while the growth rate was identical (Fig. 1B). This observation is reminiscent of results obtained with aerobically grown streptococcal cells in the presence of catalase, which allows for increased cell density by avoiding the growth inhibitory effect of H2O2 [33]. The observed higher cell densities of the AtlS and AtlS/LytF mutants suggest an impaired H2O2 production. To exclude, however, that bacterial aggregation and therefore increased precipitation of bacterial aggregates is a result of the here observed growth phenotype cells were examined microscopically. Aerobically grown cells showed tangled and elongated streptococcal chains for the AtlS and AtlS/LytF mutants, while the wild type and LytF mutant grew in short chains (Fig. 2). This excludes a possible effect of cell aggregation on the bacterial density measurements. In conclusion, the observed growth phenotypes suggest that either the AtlS mutation confers some kind of resistance to the produced H2O2 or a lower H2O2 production in the AtlS mutants during aerobic growth allowing for higher final cell densities.
Figure 1. Growth curves of S. gordonii wild type, AtlS, LytF and AtlS/LytF mutants.

A. Cells were grown as static cultures in ambient air at 37°C. Absorption was measured automatically using Bioscreen C every 20 min. B. Cells were grown aerobically as shaking cultures on a platform rocker at 37°C for maximal H2O2 production.
Figure 2. Elongated chain formation in the AtlS and AtlS/LytF mutant.

Cells were grown to mid-logarithmic phase and phase contrast images taken at 400 fold magnification. The images are adjusted for contrast and brightness. Images were taken using an Olympus BX51 microscope, Olympus DP72 digital camera and cellSens 1.3 software. Shown is a representative of 2 independent experiments with similar outcome.
Differential H2O2 Production by the Wild Type, AtlS, LytF and AtlS/LytF Mutants
The observed difference in cell density of aerobically grown cultures could be the result of changes in H2O2 production of the respective mutant strains. Supporting a difference in H2O2 production, growth under aerobic conditions also leads to a clear difference in colony size between wild type, AtlS, LytF and AtlS/LytF mutants (Fig. 3). This was not observed during incubation in an anaerobic growth chamber (data not shown). The production of H2O2 was therefore monitored and no H2O2 production was observed during static growth as reported before [12]. During aerobic growth, both the wild type and the LytF mutant showed peak production of H2O2 during exponential growth, reaching about 1.4 mM when entering the stationary phase (Fig. 4), consistent with earlier reports [12]. In contrast, the AtlS and AtlS/LytF mutants only produced up to 0.6 mM when stationary phase was reached, about 40% of the wild type production capacity (Fig. 4). The net H2O2 production of the AtlS mutant was determined with cells grown aerobically to mid-exponential phase, showing about a 25% reduction when compared to the wild type (data not shown). These results suggest that the introduction of the AtlS mutation had a general effect on the H2O2 production capacity of strain DL1.
Figure 3. Oxygen dependent growth phenotype.

Cells were grown under aerobic conditions on a TH plate overnight. The image is adjusted for contrast and brightness. Shown is a representative of 2 independent experiments with similar outcome.
Figure 4. H2O2 production.

H2O2 concentration was determined during growth under aerobic conditions. Error bars represent standard deviations from the mean (n = 3).
Reduced Production of H2O2 is a Result of Decreased spxB Expression
H2O2 in S. gordonii originates mostly from the enzymatic activity of the pyruvate oxidase, SpxB (gene spxB) [34], [35]. SpxB catalyzes the conversion of pyruvate to acetyl phosphate, which subsequently is converted to acetate by acetate kinase [34]. The expression of the spxB gene was determined from cells grown aerobically to mid-exponential phase. In agreement with the observed lower H2O2 production shown in Fig. 4, cells carrying the AtlS mutation showed a statistically significant 2.5 to 4 fold lower spxB expression, respectively (Fig. 5).
Figure 5. Expression of spxB under aerobic conditions.

For comparative real-time RT PCR analysis, wild type, AtlS, and AtlS/LytF mutant cells were grown in BHI until mid-logarithmic phase. The expression level for wild type spxB was arbitrarily assigned a value of 1. The gyrA gene was used as the housekeeping reference gene. Error bars represent standard deviations from the mean (n = 3). Asterisks indicate statistically significant differences (p = 0.05) in spxB expression in comparison to the wild type.
Release of eDNA
AtlS and LytF have been implicated in the release of eDNA in S. gordonii [13], [29]. However, the eDNA release of the lytF mutant was not determined directly. To learn how the AtlS, LytF and AtlS/LytF mutation interferes with the ability of S. gordonii to release eDNA, cells were first grown under aerobic conditions to mid-exponential phase and the eDNA concentration measured in the supernatant. All mutant strains had a significant reduction in eDNA release, ranging from a 26-fold reduction for the AtlS mutant, 10-fold reduction for the LytF mutant and 14-fold reduction for the AtlS/LytF double mutant strain (Fig. 6A).
Figure 6. Release of eDNA during aerobic growth and as a result of H2O2 treatment.
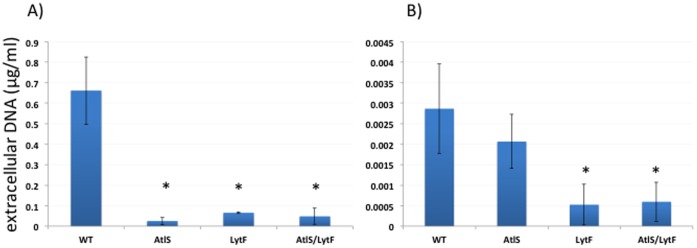
A. Wild type, AtlS, LytF and AtlS/Lytf mutant were grown until early stationary phase under aerobic conditions and the eDNA in the supernatant determined using Real-Time PCR. B. Cells were grown as static cultures under non-producing conditions and 2 mM H2O2 added after cells reached an A600 of 0.3. After further incubation of 5.5 hours, the eDNA concentration was determined in the supernatant using Real-Time PCR. Error bars represent standard deviations from the mean (n = 3). Asterisks indicate statistically significant differences (p = 0.05) in the eDNA concentration in comparison to the wild type.
Since the release of eDNA in S. gordonii can be induced by H2O2 [12], it was important to determine if the eDNA release could still be induced in the mutant strains. Thus, cells were grown under static, non-producing conditions and 2 mM H2O2 was added to the cultures during mid-exponential growth. The cells were further incubated for 5.5 hours and the concentration of eDNA determined in the supernatant. As previously reported, the amount of eDNA released is lower when non-producing cells are challenged with H2O2 as compared to aerobically grown cells [12]. The AtlS mutant showed a reduced amount of released eDNA compared to the wild type, albeit not statistically significant (Fig. 6B). In contrast, the LytF and AtlS/Lytf mutant strains showed a statistically significant 5-fold reduction in the detectable eDNA (Fig. 6B), indicating that the LytF mutation interfered with the H2O2 induced eDNA release.
Contribution of AtlS and LytF to S. gordonii Bacteriolysis
To determine the contribution of AtlS and LytF to cell lysis, a renilla luciferase reporter protein strain was constructed [36]. This renilla luciferase (36 kDa) catalyzes the emission of visible light in the presence of oxygen and the substrate coelenterazine, and does not require any other co-factors [37]. In addition, the enzyme is highly stable over hours in supernatants [38]. The measurement of reporter protein activities in supernatants has been used before to determine bacterial lysis in connection with eDNA release [14], [15], [39]. The cells were grown as shaken cultures for maximal eDNA release and to avoid any oxygen limitation for the renilla enzyme. No difference in renilla activity was determined when cell suspensions were measured (Fig. 7A). The expression of the renilla reporter-fusion is therefore not influenced by the introduced mutations allowing for direct comparison of all strains. Next, the filter-sterilized supernatant was measured to determine the activity of the released renilla protein and normalized to the renillla activity of the respective cell suspension to determine the percentage of released renilla activity (Fig. 7B). The percentage of extracellular renilla activity was 0.52% ±0.2 for the wild type, 0.22% ±0.1 for the AtlS mutant and 0.49% ±0.2 for the LytF mutant. The difference between wild type and AtlS mutant is statistically significant.
Figure 7. Detection of renilla reporter protein activity.
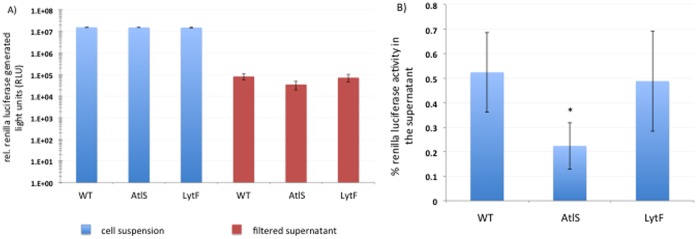
A) Relative light units (RLUs) were measured with 100 µl cell suspension or filter-sterilized supernatants. B) Percentage of renilla luciferase activity in the supernatant normalized to the respective cellular renilla activity. Error bars represent standard deviations from the mean (n = 3). Asterisk indicates statistically significant difference in normalized renilla luciferase activity (p = 0.05).
Discussion
The present report aims to better understand the mechanism of eDNA release in the oral commensal S. gordonii. We initially reported the observation of growth phase dependent eDNA release in S. gordonii and Streptococcus sanguinis [10]. The eDNA release was linked to the oxygen dependent metabolic enzyme pyruvate oxidase (SpxB). A deletion of the spxB gene diminished the release of eDNA in both organisms. Further examination revealed that the metabolic by-product of SpxB activity, H2O2, is responsible for the observed eDNA release based on several lines of evidence: i) addition of the H2O2 degrading catalase or peroxidase decreased the released eDNA over 100 fold ([10] and unpublished result), ii) cells grown under anaerobic conditions producing no H2O2 failed to generate eDNA [12], [29], and iii) addition of physiological amounts of H2O2 to either anaerobically grown cells or to the spxB mutant triggered the release of eDNA [10], [12]. Interestingly, we did not observe an eDNA release associated cell density decline as reported for other species. Incubation for more than 90 hours under maximal H2O2 producing conditions did not result in changes to the final cell density determined spectrophotometrically. An autolysis assay known to induce bacteriolytic activity in other species failed to induce lysis in S. gordonii and S. sanguinis regardless if H2O2 was present or not [10]. Furthermore, no difference of the intracellular nucleoside triphosphate ATP was measured in the supernatant of the wild type vs. the SpxB mutant, further confirming that no general lysis is associated with eDNA release in both oral streptococci [10]. We concluded that both streptococcal species used in our studies did not lyse substantially under conditions known to cause lysis of other firmicutes, for example E. faecalis and S. aureus [10], [16], [40].
Our data, however, is in contrast to two recent observations implicating bacteriolytic activity to the release of eDNA in S. gordonii [13], [29]. First, the autolysin AtlS was identified to be essential for autolysis in S. gordonii. A knock-out mutant did not release eDNA at all [29]. Second, the muralytic LytF protein seemed to be involved in the competence dependent release of eDNA [13]. LytF is a functional analog to the well-characterized Streptococcus pneumoniae murein hydrolase CbpD [41]. Both AtlS and LytF have proven murein-hydrolyzing activity in in vitro zymorgraphic assays [13], [29]. LytF however, is only active during competence development and its muralytic activity seems to be limited to a sub-fraction of cells [13]. The here used DL1 wild type and AtlS mutant were identical to the one used in [29]; however, the LytF mutation, originally in another S. gordonii strain, NCTC 7865, was transferred to the DL1 strain. The original AtlS study used 1/4-strength BHI medium supplemented with 10 mM sucrose [29]. Since sucrose causes carbon catabolite dependent repression of spxB expression [42], our study used full strength BHI without any added carbohydrates.
Quantitation of the released eDNA in the wild type, AtlS, LytF and AtlS/LytF mutant strains showed an obvious reduction in the produced eDNA in the mutants, ranging from 10 to 26 fold less eDNA in the supernatant compared to the wild type. This initially supported an involvement of AtlS and LytF in the eDNA release process. Further investigation, however, of the H2O2 production pattern showed that the AtlS mutation affected the concentration of H2O2 during the exponential growth phase leading to a twofold lower end concentration of H2O2 in the supernatant. The reduction was the result of a decreased spxB expression in the mutants carrying the AtlS mutation. This has a profound effect on the eDNA release, since our earlier data showed that a threshold H2O2 concentration is required for eDNA release [12]. Detectable amounts of eDNA were only released when the H2O2 concentration reached amounts higher then 0.6 mM, with the maximal release of eDNA at concentrations around 1 to 2 mM [12]. We suspected that the AtlS mutant did not produce sufficient amounts of H2O2 to trigger the eDNA release process. To confirm this, we added H2O2 to exponentially growing AtlS mutant cells under non eDNA releasing conditions and confirmed that the AtlS mutant was still inducible for eDNA production comparable to wild type amounts. Our new results argue against an involvement of AtlS in the H2O2 dependent eDNA release process, however, the effect of AtlS on H2O2 production and spxB expression requires further investigation to understand the causal relationship between these observed phenotypes.
The eDNA release was not associated with a detectable cell lysis as reported in this study and earlier [10], suggesting that only a subpopulation of cells lyse and releases eDNA or that the actual release process is not caused by complete bacterial lysis, leaving the cell envelope mostly intact. The study by Berg et al. demonstrated that most of the S. gordonii cells are not affected by the muralytic activity of LytF and that only a fraction of the cell population is lysed. To determine the contribution of AtlS and LytF to cell lysis, we measured the release of a reporter protein into the medium during aerobic growth. This growth condition promoted the highest amount of eDNA release [10], [12], [29]. Interestingly, the activity measured in the supernatant was comparable between the wild type, the AtlS mutant and the LytF mutant and about 1000 fold over growth medium background. After calculating the percentage of renilla activity in the supernatant a two-fold significant reduction of activity for the AtlS mutant was detectable with 0.52% ±0.2 for the wild type and 0.22% ±0.1 for the AtlS mutant. The LytF mutant was comparable to the wild type with 0.49% ±0.2, respectively. For comparison, the CbpD mutant encoding the functional LytF analog of S. pneumoniae had a 80-fold reduction in a β-galactosidase release assay, when compared to the wild type [43]. The S. aureus cidA mutant encoding the murein hydrolase regulator involved in DNA release had a 10-fold difference in the β-galactosidase release assay, when compared to the wild type [15]. The low extracellular activity of the renilla enzyme of the wild type, AtlS and LytF mutant suggests that complete cell lysis is not a major factor in the eDNA release process.
Our data, however, suggests that LytF is the responsible enzyme for the here-observed eDNA release, since the LytF mutant strain is no longer inducible for eDNA release. An involvement of LytF also makes sense considering that its gene is part of the competence system. The S. gordonii competence system is induced under H2O2 producing conditions [12]. Competence in general is considered a major stress response allowing for the uptake of environmental DNA for repair and recombination [44]. S. gordonii appears to release eDNA as part of competence development under stress situations. Further research is required to understand to what extend LytF lyses cells and releases eDNA, but not cellular content such as enzymes and ATP as shown before.
Acknowledgments
We would like to thank Prof. Robert R. Burne and Dr. Yaling Liu (College of Dentistry, University of Florida, Gainesville, Florida) for providing the AtlS mutant and the respective wild type DL1, and Prof. Leiv Sigve Håvarstein (Norwegian University of Life Sciences, Ås, Norway) for providing chromosomal DNA of strain SGH24. The authors thank Dr. J. Ferretti (Department of Microbiology & Immunology, University of Oklahoma HSC) for helpful comments.
Funding Statement
Funding came from R00 DE018400 & R03 DE022601 from National Institutes of Health/National Institute of Dental and Craniofacial Research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Flemming HC, Wingender J (2010) The biofilm matrix. Nat Rev Microbiol 8: 623–633. [DOI] [PubMed] [Google Scholar]
- 2. Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS (2002) Extracellular DNA required for bacterial biofilm formation. Science 295: 1487. [DOI] [PubMed] [Google Scholar]
- 3. Flemming HC, Neu TR, Wozniak DJ (2007) The EPS matrix: the “house of biofilm cells”. J Bacteriol 189: 7945–7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mann EE, Wozniak DJ (2012) Pseudomonas biofilm matrix composition and niche biology. FEMS Microbiol Rev 36: 893–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Das T, Sharma PK, Busscher HJ, van der Mei HC, Krom BP (2010) Role of extracellular DNA in initial bacterial adhesion and surface aggregation. Appl Environ Microbiol 76: 3405–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dominiak DM, Nielsen JL, Nielsen PH (2011) Extracellular DNA is abundant and important for microcolony strength in mixed microbial biofilms. Environ Microbiol 13: 710–721. [DOI] [PubMed] [Google Scholar]
- 7. Kaplan JB, Jabbouri S, Sadovskaya I (2011) Extracellular DNA-dependent biofilm formation by Staphylococcus epidermidis RP62A in response to subminimal inhibitory concentrations of antibiotics. Res Microbiol 162: 535–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Klein MI, DeBaz L, Agidi S, Lee H, Xie G, et al. (2010) Dynamics of Streptococcus mutans transcriptome in response to starch and sucrose during biofilm development. PLoS One 5: e13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harmsen M, Lappann M, Knochel S, Molin S (2010) Role of extracellular DNA during biofilm formation by Listeria monocytogenes . Appl Environ Microbiol 76: 2271–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kreth J, Vu H, Zhang Y, Herzberg MC (2009) Characterization of hydrogen peroxide-induced DNA release by Streptococcus sanguinis and Streptococcus gordonii . J Bacteriol 191: 6281–6291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Allesen-Holm M, Barken KB, Yang L, Klausen M, Webb JS, et al. (2006) A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Mol Microbiol 59: 1114–1128. [DOI] [PubMed] [Google Scholar]
- 12. Itzek A, Zheng L, Chen Z, Merritt J, Kreth J (2011) Hydrogen peroxide-dependent DNA release and transfer of antibiotic resistance genes in Streptococcus gordonii . J Bacteriol 193: 6912–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berg KH, Ohnstad HS, Havarstein LS (2012) LytF, a novel competence-regulated murein hydrolase in the genus Streptococcus. J Bacteriol 194: 627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zafra O, Lamprecht-Grandio M, de Figueras CG, Gonzalez-Pastor JE (2012) Extracellular DNA Release by Undomesticated Bacillus subtilis Is Regulated by Early Competence. PLoS One 7: e48716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rice KC, Mann EE, Endres JL, Weiss EC, Cassat JE, et al. (2007) The cidA murein hydrolase regulator contributes to DNA release and biofilm development in Staphylococcus aureus. . Proc Natl Acad Sci U S A 104: 8113–8118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thomas VC, Thurlow LR, Boyle D, Hancock LE (2008) Regulation of autolysis-dependent extracellular DNA release by Enterococcus faecalis extracellular proteases influences biofilm development. J Bacteriol 190: 5690–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rice KC, Bayles KW (2008) Molecular control of bacterial death and lysis. Microbiol Mol Biol Rev 72: 85–109, table of contents. [DOI] [PMC free article] [PubMed]
- 18. Bayles KW (2007) The biological role of death and lysis in biofilm development. Nat Rev Microbiol 5: 721–726. [DOI] [PubMed] [Google Scholar]
- 19. Rice KC, Bayles KW (2003) Death’s toolbox: examining the molecular components of bacterial programmed cell death. Mol Microbiol 50: 729–738. [DOI] [PubMed] [Google Scholar]
- 20. Ahn SJ, Qu MD, Roberts E, Burne RA, Rice KC (2012) Identification of the Streptococcus mutans LytST two-component regulon reveals its contribution to oxidative stress tolerance. BMC Microbiol 12: 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ahn SJ, Rice KC, Oleas J, Bayles KW, Burne RA (2010) The Streptococcus mutans Cid and Lrg systems modulate virulence traits in response to multiple environmental signals. Microbiology 156: 3136–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Webb JS, Thompson LS, James S, Charlton T, Tolker-Nielsen T, et al. (2003) Cell death in Pseudomonas aeruginosa biofilm development. J Bacteriol 185: 4585–4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Qin Z, Ou Y, Yang L, Zhu Y, Tolker-Nielsen T, et al. (2007) Role of autolysin-mediated DNA release in biofilm formation of Staphylococcus epidermidis . Microbiology 153: 2083–2092. [DOI] [PubMed] [Google Scholar]
- 24. Barnes AM, Ballering KS, Leibman RS, Wells CL, Dunny GM (2012) Enterococcus faecalis produces abundant extracellular structures containing DNA in the absence of cell lysis during early biofilm formation. MBio 3: e00193–00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kolenbrander PE, Palmer RJ Jr, Rickard AH, Jakubovics NS, Chalmers NI, et al. (2006) Bacterial interactions and successions during plaque development. Periodontol 2000 42: 47–79. [DOI] [PubMed] [Google Scholar]
- 26. Zhu L, Kreth J (2012) The role of hydrogen peroxide in environmental adaptation of oral microbial communities. Oxid Med Cell Longev 2012: 717843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kreth J, Merritt J, Shi W, Qi F (2005) Competition and coexistence between Streptococcus mutans and Streptococcus sanguinis in the dental biofilm. J Bacteriol 187: 7193–7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kreth J, Zhang Y, Herzberg MC (2008) Streptococcal antagonism in oral biofilms: Streptococcus sanguinis and Streptococcus gordonii interference with Streptococcus mutans . J Bacteriol 190: 4632–4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu Y, Burne RA (2011) The major autolysin of Streptococcus gordonii is subject to complex regulation and modulates stress tolerance, biofilm formation, and extracellular-DNA release. J Bacteriol 193: 2826–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sambrook J, Fritsch EF, Maniatis T, editors (1989) Molecular Cloning: A Laboratory Manual, 2nd edn. NY: Cold Spring Harbor Laboratory.
- 31. Zheng L, Chen Z, Itzek A, Ashby M, Kreth J (2011) Catabolite control protein A controls hydrogen peroxide production and cell death in Streptococcus sanguinis . J Bacteriol 193: 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martin B, Alloing G, Mejean V, Claverys JP (1987) Constitutive expression of erythromycin resistance mediated by the ermAM determinant of plasmid pAM beta 1 results from deletion of 5′ leader peptide sequences. Plasmid 18: 250–253. [DOI] [PubMed] [Google Scholar]
- 33. Regev-Yochay G, Trzcinski K, Thompson CM, Lipsitch M, Malley R (2007) SpxB is a suicide gene of Streptococcus pneumoniae and confers a selective advantage in an in vivo competitive colonization model. J Bacteriol 189: 6532–6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carlsson J, Edlund MB, Lundmark SK (1987) Characteristics of a hydrogen peroxide-forming pyruvate oxidase from Streptococcus sanguis . Oral Microbiol Immunol 2: 15–20. [DOI] [PubMed] [Google Scholar]
- 35. Carlsson J, Edlund MB (1987) Pyruvate oxidase in Streptococcus sanguis under various growth conditions. Oral Microbiol Immunol 2: 10–14. [DOI] [PubMed] [Google Scholar]
- 36. Tannous BA, Teng J (2011) Secreted blood reporters: insights and applications. Biotechnol Adv 29: 997–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Srikantha T, Klapach A, Lorenz WW, Tsai LK, Laughlin LA, et al. (1996) The sea pansy Renilla reniformis luciferase serves as a sensitive bioluminescent reporter for differential gene expression in Candida albicans . J Bacteriol 178: 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu J, Escher A (1999) Improved assay sensitivity of an engineered secreted Renilla luciferase. Gene 237: 153–159. [DOI] [PubMed] [Google Scholar]
- 39. Steinmoen H, Knutsen E, Havarstein LS (2002) Induction of natural competence in Streptococcus pneumoniae triggers lysis and DNA release from a subfraction of the cell population. Proc Natl Acad Sci U S A 99: 7681–7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bose JL, Lehman MK, Fey PD, Bayles KW (2012) Contribution of the Staphylococcus aureus Atl AM and GL murein hydrolase activities in cell division, autolysis, and biofilm formation. PLoS One 7: e42244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eldholm V, Johnsborg O, Haugen K, Ohnstad HS, Havarstein LS (2009) Fratricide in Streptococcus pneumoniae: contributions and role of the cell wall hydrolases CbpD, LytA and LytC. Microbiology 155: 2223–2234. [DOI] [PubMed] [Google Scholar]
- 42. Zheng L, Itzek A, Chen Z, Kreth J (2011) Environmental influences on competitive hydrogen peroxide production in Streptococcus gordonii . Appl Environ Microbiol 77: 4318–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kausmally L, Johnsborg O, Lunde M, Knutsen E, Havarstein LS (2005) Choline-binding protein D (CbpD) in Streptococcus pneumoniae is essential for competence-induced cell lysis. J Bacteriol 187: 4338–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Claverys JP, Prudhomme M, Martin B (2006) Induction of competence regulons as a general response to stress in Gram-positive bacteria. Annu Rev Microbiol 60: 451–475. [DOI] [PubMed] [Google Scholar]