

Abstract
Cyanobacterial harmful blooms (CyanoHABs) that produce microcystins are appearing in an increasing number of freshwater ecosystems worldwide, damaging quality of water for use by human and aquatic life. Heterotrophic bacteria assemblages are thought to be important in transforming and detoxifying microcystins in natural environments. However, little is known about their taxonomic composition or pathways involved in the process. To address this knowledge gap, we compared the metagenomes of Lake Erie free-living bacterioplankton assemblages in laboratory microcosms amended with microcystins relative to unamended controls. A diverse array of bacterial phyla were responsive to elevated supply of microcystins, including Acidobacteria, Actinobacteria, Bacteroidetes, Planctomycetes, Proteobacteria of the alpha, beta, gamma, delta and epsilon subdivisions and Verrucomicrobia. At more detailed taxonomic levels, Methylophilales (mainly in genus Methylotenera) and Burkholderiales (mainly in genera Bordetella, Burkholderia, Cupriavidus, Polaromonas, Ralstonia, Polynucleobacter and Variovorax) of Betaproteobacteria were suggested to be more important in microcystin degradation than Sphingomonadales of Alphaproteobacteria. The latter taxa were previously thought to be major microcystin degraders. Homologs to known microcystin-degrading genes (mlr) were not overrepresented in microcystin-amended metagenomes, indicating that Lake Erie bacterioplankton might employ alternative genes and/or pathways in microcystin degradation. Genes for xenobiotic metabolism were overrepresented in microcystin-amended microcosms, suggesting they are important in bacterial degradation of microcystin, a phenomenon that has been identified previously only in eukaryotic systems.
Introduction
Freshwater lakes are ecologically important and a major source of drinking water; thus maintaining and improving water quality in lakes is critical. Cyanobacterial (blue-green algal) harmful blooms (CyanoHABs) threaten water quality and the frequency and extent of these blooms are increasing worldwide [1]. One important harmful effect of CyanoHABs is production of cyanotoxins, such as microcystins, which have strong hepatotoxicity that can severely damage mammalian liver cells. Microcystins (MCs) are produced by several bloom-forming cyanobacteria that are common in freshwater lakes, including Microcystis, Anabaena, Planktonthrix and Nostoc [2].
There are over 80 chemical variants of MCs, which all share a cyclic structure consisting of five constant non-protein amino acids and two variable protein amino acids [3]. Microcystin-LR (MC-LR) is the most abundant and well-studied form of MCs and contains leucine (Leu or L) and arginine (Arg or R) in the two variable positions (Figure 1). Due to its cyclic structure, MC-LR is chemically stable under the environmental range of pH, light radiation and temperature [4]. Heterotrophic bacterial assemblages are thought as major agents that regulate MC degradation in in lakes [5], [6], estuaries [7] and water treatment units [8].
Figure 1. MC-LR structure and potential degradation pathways in bacteria.

Steps of the known cleavage pathway are linked by solid arrows. Steps through xenobiotic metabolism are linked by dotted arrows. Overrepresented genes in the MC metagenomes are labeled with asterisks and bold fonts. Unknown genes are labeled with question marks. Triangle indicates the cleavage site of mlrA. Ala: alanine, Arg: arginine, Adda: 3-amino-9-methoxy-2, 6, 8-trimethyl-10-phenyldeca-4, 6-dienoic acid, Cys: cysteine, Glu: glutamic acid, GSH: glutathione, Leu: leucine, MeAsp: methylaspartic acid.
Most previous studies on MC-degrading bacteria are culture-based and many of them are conducted in artificial environments, such as water treatment units [8], [9]. These studies have suggested that MC-degrading assemblages are mainly consisted of a narrow group of alphaproteobacteria in order Sphingomonadales. However, indirect evidence from studies on bacteria associated with MC-producing CyanoHABs has suggested that much broader bacterial taxa may be involved in MC degradation [10], [11]. Direct studies on in situ taxonomic composition of MC-degrading assemblages are scarce.
To date, a single pathway has been identified in bacterial systems for MC-LR degradation. This cleavage pathway is encoded by a cluster of genes (mlrABCD) and has been identified in all MC-degrading Sphingomonas species and several other strains of Gammaproteobacteria and non-Sphingomonas Alphaproteobacteria [3], [9], [12]. However, the specificity and ubiquity of mlr among environmental MC-degrading bacteria remain unclear [13]. This study aims to identify taxa, genes and pathways involved in microbially mediated MC transformation, using a comparative metagenomic approach on free-living bacterial assemblages from Lake Erie. Our results suggest that diverse taxa of free-living bacterioplankton, especially members of Methylophilales and Burkholderiales, might be important in MC degradation and that they likely employ different pathways from the mlr-based cleavage.
Materials and Methods
Sample Collection and Nutrient Analysis
Surface water samples were collected in carboys from the Western Basin of Lake Erie (Latitude 41.7423, Longitude −83.4019; Station MB18) on Aug. 27th, 2010, where CyanoHABs were reported throughout the summer, including at the time of this sampling trip (NOAA, Harmful Algal Bloom Events Response; http://www.glerl.noaa.gov). Before use, the carboys were acid washed in the lab and rinsed with ambient lake water three times. Standard limnological data were collected using a YSI 6600 Water Quality Sonde and included temperature, dissolved oxygen concentration, pH and turbidity (Table S1). The Secchi depth was also measured at the time of sampling. Water samples for nutrient analyses were filtered through 0.2 µm-pore-size membrane filters (Pall Life Sciences, Port Washington, NY) and stored on ice or at 4°C. Concentrations of nutrients, including dissolved organic carbon, total dissolved nitrogen, soluble reactive phosphorus, nitrate/nitrite, ammonium, were measured using standard methods for water quality analyses [14] and reported in Table S1.
Microcosm Setup and Incubation
Lake water was filtered through 1.0 µm-pore-size membrane filters (Pall Life Sciences, Port Washington, NY) immediately after sampling to obtain free-living bacterioplankton proportion, and to exclude bacterivores and other large particles. Filtrate was collected in carboys and amended with a mixture of inorganic nitrogen and phosphorus compounds (5 µM NH4Cl, 5 µM NaNO3, and 1 µM NaH2PO4, final concentrations) and incubated in the dark at room temperature (22±1C°) with for 7 days. The water was agitated every 4–12 hours by shaking the carboys by hand. This pre-incubation was done to allow the bacteria to consume labile dissolved organic carbon compounds and to become growth limited by carbon availability.
At the end of the pre-incubation, microcosms were set up in six 20 L carboys. Two microcosms, designated as MC-1 and MC-2, were constructed of pre-incubated lake water and amended with MC-LR (∼15 µg L−1, final concentration, Axxora LLC, Farmingdale, NY). Two microcosms, designated as CT-1 and CT-2, served as controls and were constructed of pre-incubated lake water without further amendments. The remaining two microcosms were designated as FW-MC-1 and FW-MC-2; these received pre-incubated lake water that was in turn filtered by passage through 0.2 µm pore-size membrane filters to remove most of the bacterial cells and then amended with MC-LR (∼15 µg L−1, final concentration). The final volume of each microcosm was 18 L.
Microcosms were incubated in the dark at room temperature for a total of 48 hours and agitated every 4–12 hours by shaking the carboys by hand. Samples (10 ml) were taken in triplicates from each microcosm after 0 hour, 12 hours, 24 hours and 48 hours of incubation for subsequent MC-LR concentration measurement and flow cytometric analysis.
All plasticware was acid washed then rinsed with sample waters for three times before use. All glassware was ashed at 500°C for 5 hours then rinsed with sample waters for three times before use.
Microcystin Concentration Measurement
Samples collected as above were filtered through 0.2-µm-pore size filters. MC-LR concentration in filtrate was measured using the Microcystins/Nodularins (ADDA) ELISA Kits (Abraxis BioScience, Warminster, PA) following the manufacturer’s instruction. Technical duplicates were measured for each sample.
Flow Cytometric Analysis
Flow-cytometric analysis (FCM) was performed with a FACSAria (BD, Franklin Lakes, NJ) to measure the abundance, size and metabolic activity of bacterioplankton in the microcosms. Before FCM analysis, samples were preserved with 1% (final concentration) freshly made paraformaldehyde at room temperature for 2 hours. Preserved cells were stained with Sybr Green II (1∶5 000 dilution of the commercial stock; Molecular Probes Inc.) in the dark at room temperature for 20 min and mixed with an internal standard of beads that have a known density (1-µm-diameter Fluoresbrite YG Microspheres; Polysciences, Warrington, PA). FCM data acquisition was triggered by green fluorescence intensity of Sybr Green II staining (GFI). All FCM signals were collected on a logarithmic scale. Bacterial cell numbers were calculated based on ratios between the counts of bacterial cells and the internal bead standard.
FCM populations were defined based on GFI and side scatter (SSC) using a procedure described previously [15]. FCM population notation was based on the value of GFI from Sybr Green II staining, a proxy for intracellular nucleic acid content (largely RNA), which was taken as a surrogate indicator of cell activity [16]. Two FCM populations were gated for each sample, one was designated as “high intensity cells” (HI) and the other was designated as “low intensity cells” (LI). HI and LI populations thereby were corresponding to cells with higher and lower activity, respectively. Technical duplicates were analyzed for each sample.
Bacterial Growth Rate Estimation
Bacterial growth rate (µ) during the incubation experiment was calculated using a linear regression formula: µ = (lnNt –lnN0)/t, where t is the incubation time at the time of sampling, N0 and Nt are bacterial abundance at initial (0 hour) and at the time of sampling (t).
DNA Extraction
For PCR amplification, bacteria cells in 1 L water samples were collected from each microcosm and filtered onto 47 mm-diameter, 0.2 µm-pore-size membrane filters (Pall Life Sciences, Port Washington, NY). Filters were changed after approximately every 500 ml of water filtered.
For metagenomic analysis, bacteria cells in ∼17 L water samples were collected from each microcosm and filtered on to 142 mm-diameter, 0.2 µm-pore-size membrane filters (Pall Life Sciences). Filters were changed after approximately every 9 L of water filtered. DNA was extracted from the filters using a PowerMax Soil DNA Isolation Kit (Mobio Inc, Carlsbad, CA) and served as templates for PCR amplification and metagenomic sequencing.
16S rRNA Gene Amplification and T-RFLP Analysis
16S rRNA gene amplification and terminal restriction fragment polymorphism (T-RFLP) analysis were performed following a protocol described previously with minor modifications [15]. Briefly, PCR was carried out with Illustra PuRe Taq Ready-to-go PCR beads (GE Healthcare, Piscataway, NJ) using 0.4 µM of 6-carboxyfluorescein (FAM) labeled 8F (5′-FAM-AGAGT TTGAT CCTGG CTCAG-3′) and unlabeled 1492R (5′-TACGG YTACC TTGTT ACGAC TT-3′) primers. A touchdown PCR program was used with the annealing temperature sequentially decreasing from 62°C to 52°C by 1°C per cycle, followed by 15 cycles at 52°C. Each PCR cycle included denaturing (at 95°C for 50s), annealing (at 62 to 52°C for 50s), and extension (at 72°C for 50s) steps. An initial 3-min denaturation and final 7-min extension step were also included. For each sample, triplicate PCR amplifications were performed and resulting amplicons were pooled before being examined on ethidium bromide-stained 1% agarose gels.
FAM-labeled PCR amplicons were purified with the QIAquick gel extraction kits (QIAGEN, Valencia, CA) and then digested with the CfoI restriction enzyme (Roche Applied Science, Indianapolis, IN) at 37°C for 3 hours. Afterwards, the digestion products were purified using ethanol precipitation. The length and relative abundance of each terminal restriction fragment (T-RF) were determined using a 3730 DNA Analyzer (Applied Biosystems) at the Plant-Microbe Genomic Facility, Ohio State University, Columbus, OH.
T-RFLP profiles among bacterial FCM populations were quantitatively compared using a hierarchical cluster analysis using the Primer v5 program (Primer-E Ltd, Plymouth, United Kingdom). The relative peak area of each terminal restriction fragment (T-RF) from the output of T-RFLP data was used as a proxy for the relative abundance of bacterial taxa associated with that T-RF peak. The relative peak areas were square-root transformed before analysis. T-RFs with <2% relative peak areas were excluded from the analysis.
Metagenomic Sequencing and Sequence Annotation
Genomic DNA of metagenomes collected at the end of microcosm incubations, i.e., 48 hours after the MC-LR was added, was sequenced together by one full plate run of 454 multiplex pyrosequencing with titanium chemistry at the Georgia Genomics Facility, University of Georgia, Athens, GA. The metagenomic sequences were deposited in the CAMERA database under the project CAM_P_0000956.
Low quality reads (<200 bp or Phred quality scores <20) were removed from the metagenomic library. Identical reads that were generated as artifacts during pyrosequencing [17] were also removed using the CD-HIT-454 identifiers [18]. Remaining sequences were analyzed by BLASTn against the RDPII database to identify putative rRNA gene sequences (cutoff value of E <10−5). The taxonomic affiliations of each putative rRNA gene sequence was assigned based on the best hit of the BLASTn against the Greengenes database [19], using the E value <10−10 and identity >85%. The taxonomic annotation was further confirmed by consultation with RDP taxonomy classifier (>80% confidence).
Putative protein-coding sequences were identified from non-rRNA sequences by BLASTx against the NCBI RefSeq protein database (E ≤0.01, identity ≥40% and overlapping length ≥65 nt) [20]. The protein-encoding sequences were further categorized into Clusters of Orthologous Groups (COG) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways by BLASTx against the NCBI’s COG database and the KEGG databases (E ≤0.1, similarity ≥40% and overlapping length ≥65 nt). The taxonomic affiliations were obtained by BLASTx against the NCBI RefSeq database using the MEGAN program [21]. Sequences that did not meet any of the criteria for rRNA or functional genes were excluded from further analysis.
Functional Gene Identification
Putative mlrABCD, a cluster of genes that encode MC degradation in bacterial systems, were identified in the metagenomic libraries using tBLASTx with a bit score cutoff of 50. Putative glutathione S-transferase (GST) genes, key genes in xenobiotic metabolism, were identified using BLASTx with a bit score cutoff of 50. Accession numbers for reference gene sequences are provided in Table S2.
Shannon-Wiener Index
The taxonomic diversity of microbes was estimated at the order level using the formula H’ = −∑ (Pi * ln Pi), where Pi is the relative abundance of the sequences belonging to the ith microbial order, and R is the total number of unique orders.
Statistical Analyses
A Student’s t test for two samples of unequal variance was performed to compare total bacterial abundance, relative abundance of each FCM population and MC-LR loss between the MC and CT microcosms.
A t test with Bonferroni correction for two samples of unequal variance [22] was used to compare the relative abundance of bacterial taxa at two levels, e.g., between the within-treatment metagenome replicates (MC1 vs. MC2 and CT1 vs. CT2) and between the pooled metagenomes of different treatments (MCs vs. CTs). Significant differences between MC and CT microcosms were reported at P<0.05 with Bonferroni correction. Taxa with significant within-treatment differences were removed from the final list of responsive taxa.
The Xipe-TOTEC program, a statistical method that has been specifically developed to compare metagenomes [23], was used to identify overrepresented COGs and KEGGs in the MC metagenomes. Pair-wise comparisons were performed between within-treatment metagenome replicates and between the pooled metagenomes of different treatments, based on the occurrence of gene categories. In each comparison, a total of 20,000 re-samplings were made, with the sample size equal to the average number of sequences in the two metagenome sequence libraries being compared. Significant differences between the metagenome datasets were reported at the level of P<0.02, after removing those gene categories that had significant within-treatment differences.
The results of Xipe-TOTEC are affected by sample size, i.e., the number of sequences in randomly formed pools [24], and the copy number of target genes [20]. Therefore, changes in relative abundance of gene categories were also assessed by a statistical analysis that is free of these concerns, which is based on calculating the odds ratio (OR) and binominal distribution probabilities [25]. OR was calculated using the equation [(nmc/(Nmc-nmc)]/[nct/(Nct-nct)], where nmc and nct were the number of targeted gene sequences in the pooled MC and CT metagenomes, respectively; Nmc and Nct were the total number of sequences in the pooled MC and CT metagenomes, respectively. Binomial distribution of genes was assumed in each metagenomic sequence library. The binomial distribution probability (P) was calculated within Microsoft Excel, using the [nmc/(Nmc-nmc)] as the observed gene sequence frequency and [nct/(Nct-nct)] as the expected gene sequence frequency. Genes or gene groups were reported as significantly overrepresented in MC metagenomes when the corresponding OR >1 and P<0.02. Genes or gene groups that had significant within-treatment differences were removed from the final list of overrepresented gene categories.
Results
Response of Bacterial Assemblages to Microcystin
Added MC-LR was consumed rapidly in microcosms with a pre-established carbon-limited condition (Figure 2A). Within 12 hours of incubation, over 75% of MC-LR was lost and MC-LR became nearly undetectable after 24 hours in the MC microcosms. In contrast, MC-LR that was added to microcosms with filter-sterilized lake water (FW-MC) remained untransformed throughout the incubation (t test, P<0.05).
Figure 2. Variation of MC-LR concentration and total bacterial cell number during microcosm incubation.

(A) Average MC-LR concentrations and standard deviations in the MC and FW-MC microcosms. (B) Average bacterial abundance and standard deviations in MC and CT microcosms. Shaded areas indicate periods of pre-incubation with inorganic N and P for establishing carbon-limited conditions in microcosms.
Concomitant with MC-LR consumption, number of bacteria in the MC microcosms significantly increased. Bacterioplankton in the MC microcosms nearly doubled within 12 hours following addition of MC-LR (growth rate; µ = 1.2 day −1) and the cell density reached 2.2×106 cells ml−1 after 48 hours of incubation (∼3.4-fold increase from initial cell density). Meanwhile, cell density in the control microcosms (CTs) was unchanged at 6.1×105 cells ml−1 (t test, P<0.05; Figure 2B).
MC-LR addition also led to compositional differentiation between the MC and CT metagenomes (Figure 3). In the MC microcosms, the relative abundance of HIs increased from 14.3% of total cells (1.0×105 cells ml−1) at 0 hour to 45.9% (3.1×105 cells ml−1) at 48 hours of incubation. Meanwhile, no significant change was found for the relative abundance of HIs in the CT microcosms (t test, P<0.05; Figures 3B and 3C), indicating the observed increase in HI cells in the MC microcosms was due to growth of bacterioplankton on added MC-LR. At the end of the 48-hour incubation, LIs in the MC microcosms accounted for a smaller percentage (54.1%) than those in the CTs (85.7%), however, it contained nearly 3 times more cells than LIs in the CT microcosms (Figure 3C). This suggests that a considerable fraction of LI cells in the MC microcosms were also microcystin-responsive. Thus, rather than analyzing only the HI populations, the total bacterial communities (HI plus LI) in the MC and CT microcosms were examined to elucidate the taxa and genes involved in microcystin degradation.
Figure 3. Distribution of bacterial cells of high (HI) and low (LI) metabolic activities during microcosm incubation.

(A) Flow cytometric analysis of bacterial cell distribution in the MC and CT microcosms after 48 hours of incubation. (B) Average relative abundance of HI and LI cells in the MC and CT microcosms during the course of incubation. (C) Average numbers of HI and LI cells and standard deviations in the MC and CT microcosms during the course of incubation.
T-RFLP analysis, based on 16S rRNA genes, was performed to examine the potential shifts in total bacterial community structures during inorganic nutrient pre-incubation and MC-LR incubation experiment. Cluster analysis of T-RFLP data closely grouped duplicates for each sample to each other, indicating good within-treatment reproducibility (Figure 4). The original water (Ori) and pre-incubated water samples (Pre-incub) had highly similar T-RFLP data. These two samples were moderately similar with the samples from the CT microcosms at the end of the incubation experiment (CT-48 h), but were distant from samples of the MC microcosms (MC-48 h) (Figure 4). These findings indicated that pre-incubation had less effect on bacterial community structure than MC-LR amendments.
Figure 4. Clustering pattern of bacterioplankton 16S rRNA gene contents based on T-RFLP analysis.
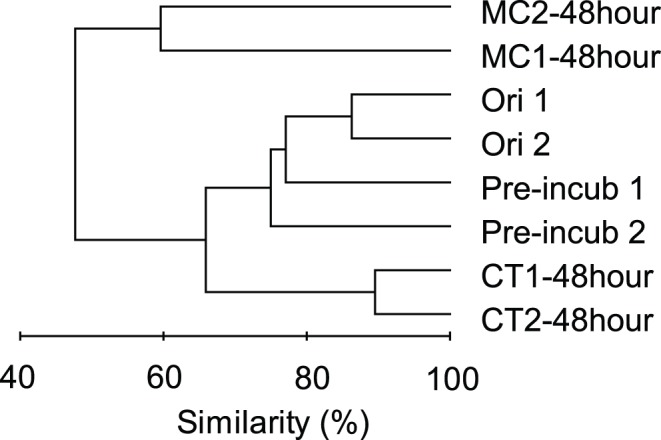
Cluster analysis of T-RFLP data for original water samples (Ori 1 and 2), samples at the end of inorganic nutrient pre-incubation (Pre-incub 1 and 2) and samples at the end of MC-LR incubation experiments in the microcystin amended (MC1–48 h and MC2–48 h) and control (CT1–48 h and CT2–48 h) microcosms.
General Structure of Metagenomes
A total of 815,435 metagenomic sequences with average length of 386 bp were recovered, after removing low quality and artificial reads (Table 1). More sequences were recovered for the MC libraries than the CTs, although starting amounts of genomic DNAs were similar (∼1 µg). About 0.4% and 0.2% of the sequences of the MC and CT metagenomes, respectively, were affiliated with16S rRNA genes, in accordance with their expected frequency in prokaryotes (Mou et al., 2008). Most of the non-16S rRNA gene sequences in the MC (∼70%) and CT (∼50%) metagenomes were identified as putative protein-coding sequences.
Table 1. Sequence annotation statistics for the MC and CT metagenomes.
| Parameter | MC-1 | MC-2 | CT-1 | CT-2 |
| Number of unique sequences | 251,154 | 201,543 | 164,026 | 198,712 |
| Average sequence length (bp) | 386 | 366 | 414 | 377 |
| Number (%) of total rRNA genes | 1000 (0.4%) | 771 (0.4%) | 381 (0.2%) | 359 (0.2%) |
| Number (%) of total predicted protein-coding genes | 182,25 (73%) | 140,86 (70%) | 83,621 (51%) | 91,778 (46%) |
| Number (%) of protein-coding genes categorized by COG groups | 121,80 (67%) | 92,719 (66%) | 53,754 (64%) | 61,742 (67%) |
| Number (%) of protein-coded genes categorized by KEGG pathways | 168,118 (92%) | 129,469 (92%) | 72,542 (87%) | 83,391 (91%) |
Out of 498,519 putative protein-coding gene sequences, 90% were further assigned into a total of 3,259 unique COG groups and 182 unique KEGG pathways (Table 1). These were further classified into 23 COG (A-V, Z) and 21 KEGG classes within the networks of metabolism, genetic information processing, environmental information processing and cellular processes. COG and KEGG assignments consistently revealed that functional categories, including cell motility, signal transduction, metabolisms of organic and inorganic molecules were significantly overrepresented in the MC relative to the CT metagenomes (Figure 5).
Figure 5. Significantly overrepresented gene categories in the MC metagenomes, relative to those in the CT metagenomes.

(A) General COG categories. (B) General KEGG categories. Calculations were based on relative abundance of each gene categories between the MC and CT metagenomes. Significance overrepresentation was reported when OR>1, P<0.02.
Microcystin Responsive COGs, KEGGs and Genes
Metagenomic contents of COG groups (COGs) and KEGG pathways (KEGGs) were compared between the MC and CT metagenomes using the Xipe-TOTEC (Rodriguez-Brito et al., 2006; P<0.02) and OR calculation analyses (Gill et al., 2006; OR >1, P<0.02). A total of 80 COGs and 14 KEGGs were found significantly overrepresented in the MC metagenomes relative to the CT metagenomes (Tables 2, 3,S3 and S5). These COGs and KEGGs were affiliated with metabolisms, mostly those for energy, amino acids, carbohydrates and lipids (28 COGs, 10 KEGGs), information processing (8 COGs, 2 KEGGs) and cellular processes, especially, bacterial motility and chemotaxis (33 COGs, 2 KEGGs; Figure 5). Several overrepresented COGs have been found in metagenomes associated with a microcystin-producing cyanobacterial bloom (Li et al., 2011), including COG0243 (anaerobic dehydrogenase), COG5013 (nitrate reductase alpha subunit), COG3696 (putative silver efflux pump), COG0583 (transcriptional regulator), COG0642 (signal transduction histidine kinase) and COG0664 (cAMP-binding proteins) (Table 2).
Table 2. Overrepresented COG groups in the MC metagenomes relative to the CT metagenomes, based on odds ratios (OR) calculated between the copy number of putative gene sequences in the MC and CT metagenomes.
| COGa | COG description | Class | Class description | MC | CT | ORMC/CT |
| Metabolism | ||||||
| 0243* | Anaerobic dehydrogenases, typically selenocysteine-containing | C | Energy production and conversion | 361 | 97 | 2.0 |
| 5013* | Nitrate reductase alpha subunit | C | Energy production and conversion | 110 | 6 | 9.9 |
| 1362 | Aspartyl aminopeptidase | E | Amino acid transport and metabolism | 94 | 1 | 50.6 |
| 3696* | Putative silver efflux pump | P | Inorganic ion transport and metabolism | 938 | 254 | 2.0 |
| Information storage and processing | ||||||
| 0583* | Transcriptional regulator | K | Transcription | 888 | 182 | 2.6 |
| Cellular processes and signaling | ||||||
| 1291 | Flagellar motor component | N | Cell motility | 128 | 10 | 6.9 |
| 0643 | Chemotaxis protein histidine kinase andrelated kinases | NT | Cell motility/Signal transduction mechanisms | 348 | 64 | 2.9 |
| 0840 | Methyl-accepting chemotaxis protein | NT | Cell motility/Signal transduction mechanisms | 442 | 43 | 5.5 |
| 2804 | Type II secretory pathway, ATPase PulE/Tfppilus assembly pathway, ATPase PilB | NU | Cell motility/Intracellular trafficking, secretion,and vesicular transport | 551 | 166 | 1.9 |
| 3419 | Tfp pilus assembly protein, tip-associatedadhesin PilY1 | NU | Cell motility/Intracellular trafficking, secretion,and vesicular transport | 113 | 11 | 5.5 |
| 5008 | Tfp pilus assembly protein, ATPase PilU | NU | Cell motility/Intracellular trafficking, secretion,and vesicular transport | 256 | 21 | 6.6 |
| 0625 | Glutathione S-transferase | O | Posttranslational modification, protein turnover, chaperones | 352 | 92 | 2.1 |
| 1391 | Glutamine synthetase adenylyltransferase | OT | Posttranslational modification, protein turnover, chaperones/Signal transduction mechanisms | 238 | 46 | 2.8 |
| 0642* | Signal transduction histidine kinase | T | Signal transduction mechanisms | 962 | 343 | 1.5 |
| 0664* | cAMP-binding proteins - catabolite geneactivator and regulatory subunit ofcAMP-dependent protein kinases | T | Signal transduction mechanisms | 367 | 95 | 2.1 |
| 0841 | Cation/multidrug efflux pump | V | Defense mechanisms | 1693 | 512 | 1.8 |
| 1566 | Multidrug resistance efflux pump | V | Defense mechanisms | 376 | 104 | 1.9 |
Only those COG groups that were discussed in the present study or have been reported previously (labeled with asterisks) are shown. A full list is provided in Table S3.
Table 3. Significantly enriched KEGG pathways in the MC metagenomes relative to the CT metagenomes, based on odds ratios (OR) calculated between the copy number of putative gene sequences in the MC and CT metagenomes.
| KEGG Pathway | General Processes | Functional Description | MC | CT | ORMC/CT |
| Cellular Processes | |||||
| 2030 | Cell Motility | Bacterial chemotaxis | 2257 | 343 | 3.7 |
| 2040 | Cell Motility | Flagella assembly | 2458 | 518 | 2.7 |
| Environmental Information Processing | |||||
| 2020 | Signal Transduction | Two-component system | 8185 | 2297 | 2.1 |
| 3070 | Membrane Transport | Bacterial secretion system | 3063 | 1264 | 1.4 |
| Metabolism | |||||
| 0480 | Metabolism of Other Amino Acids | Glutathione metabolism | 2254 | 954 | 1.3 |
| 0540 | Glycan Biosynthesis and Metabolism | Lipopolysaccharide biosynthesis | 1822 | 539 | 1.9 |
| 0550 | Glycan Biosynthesis and Metabolism | Peptidoglycan biosynthesis | 2815 | 1279 | 1.2 |
| 0564 | Lipid Metabolism | Glycerophospholipid metabolism | 1542 | 613 | 1.4 |
| 0680 | Energy Metabolism | Methane metabolism | 3805 | 1807 | 1.2 |
| 0780 | Metabolism of Cofactors and Vitamins | Biotin metabolism | 469 | 136 | 1.9 |
| 0860 | Metabolism of Cofactors and Vitamins | Porphyrin and chlorophyll metabolism | 3160 | 1122 | 1.6 |
| 0910 | Energy Metabolism | Nitrogen metabolism | 2900 | 184 | 1.3 |
| 0920 | Energy Metabolism | Sulfur metabolism | 1379 | 535 | 1.4 |
| 0980 | Xenobiotics Biodegradation | Xenobiotics metabolism by Cytochrom P450 | 342 | 124 | 1.5 |
Xenobiotic metabolism-related COGs and KEGGs were overrepresented in the MC metagenomes (Table 2). KEGG0980 and COG0625 are associated with cytochrome P450 oxidase and glutathione S-transferase (GST), respectively. These two enzymes have been found to catalyze the synthetic conversion of MC-LR into glutathione (GSH) and cysteine (Cys) conjugates in animal cells [26]. COG0841 and COG1566 are both affiliated with multidrug efflux pumps, which have been found to regulate the excretion of final degradation products of GSH and Cys conjugates from animal cells (Figure 1).
The known pathway of MC-LR cleavage in bacterial systems involves expression of a cluster of genes, e.g., mlrABCD [3], [9]. Putative mlr genes had similar relative abundance in the MC (0.22‰ of protein-coding sequences) and CT (0.19‰) metagenomes (OR >1, P<0.02). On the other hand, putative GST genes, which are involved in MC-LR degradation in animal cells but has yet unreported in bacteria [26], were overrepresented in the MC (0.54‰ of protein-coding sequences) than in the CT (0.24‰) metagenomes (OR >1, P<0.02).
Microcystin Responsive Bacterial Taxa
Most of the putative protein sequences in the MC (90%) and CT (80%) metagenomes received taxonomic assignments at least to the phylum level, and ∼64% of these sequences had COG assignment. Patterns of taxonomic affiliation of metagenomic sequences were conserved, regardless of whether all protein-coding sequences or just those subsets assigned to significantly overrepresented COG categories were considered (Figure S1). In addition, even though 16S rRNA gene sequences were a much smaller fraction of total sequences than the protein-coding gene sequences (Table S4), they revealed a similar bacterial taxonomic structure (Figure S1 and Table S6).
COG sequences were affiliated with 89 unique bacterial orders, but about 65% of them were from only 22 orders of the phyla of Acidobacteria, Actinobacteria, Bacteroidetes, Planctomycetes, Proteobacteria (in subdivision of alpha, beta, gamma and delta/epsilon) and Verrucomicrobia (Figure 6A). Archaeal sequences occurred in low abundance (0.08% COGs in the MCs; 0.4% in the CTs) and 95% of them were affiliated with Euryarchaeota. The richness of the MC and CT metagenomes was similar at the order level. However, the COG sequences of the MC metagenomes were taxonomically less diverse (Shannon-Wiener Index, H’ = 2.0) than the CT metagenomes (H’ = 3.3), because evenness of the MC metagenomes was lower.
Figure 6. Taxonomic distribution of COG sequences in the MC and CT metagenomes.

(A) At the order level. (B) At the family level. (C) At the genus level. Only major taxa are shown (collectively accounted for >4% of total metagenomic sequences). Asterisks are to label bacterial taxa with different relative abundance between the MC and CT metagenomes (OR >1, P<0.02).
Over half (53.2%) of COG sequences in the MC metagenomes were affiliated with Methylophilales (Betaproteobacteria), a taxon that was significantly less abundant in the CT metagenomes (9.6% of COG sequence; t test with Bonferroni correction, P<0.05). Burkholderiales (Betaproteobacteria; 18.1% of COG sequences) and Xanthomonadales (Gammaproteobacteria; 9.0%) were the second and third most abundant taxa affiliated with COG sequences in the MC metagenomes. Their relative abundances were similar to those in the CT metagenomes (15.9% and 2.2%, respectively) (Figure 6).
Although representing fewer sequences (Table S4), similar distribution patterns of bacterial taxa were observed at the family and genus levels (Figures 6B and 6C). Like their parent order Methylophilales, the family Methylophilaceae and genus Methylotenera were the most abundant members in the MC metagenomes and were significantly more abundant than those in the CT metagenomes (t test with Bonferroni correction, P<0.05). On the other hand, underrepresentation of Actinobacteria in the MC metagenomes at the order level was not observed at the family or species level (Figure 6). This may be partly due to the fact that only a limited number of environmental Actinobacteria species have been isolated and sequenced [27].
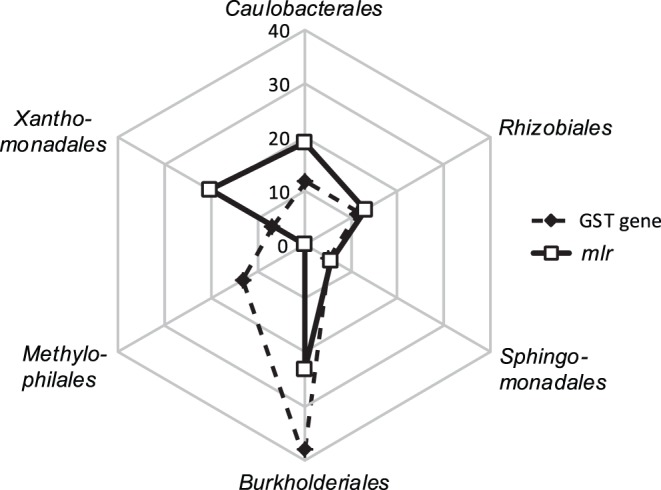
Putative genes of MC-LR cleavage pathway (mlr) and xenobiotic metabolisms (GST genes) were affiliated with 13 and 16 bacterial orders, respectively (Figure 7). About 80% of the putative mlr sequences were affiliated with only 5 orders, including Burkholderiales (in genera Burkholderia, Cupriavidus, and Variovorax), Caulobacterales (Phenylobacterium), Rhizobiales (Mesorhizobium, Methylobacterium and Rhodopseudomonas), Sphingomonadales (Sphingopyxis), and Xanthomonadales (Stenotrophomonas). These orders and genera also represented major taxa for putative GST gene sequences. Taxonomic affiliations of mlr and GST genes were statistically similar between the MC and CT metagenomes (OR >1, P<0.02). However, a significant difference was found for Methylophilales-affiliated sequences. They accounted for over 13% of putative GST gene sequences but were not identified among the putative mlr sequences (Figure 7).
Figure 7. Percent distribution of major bacterial orders that were affiliated with GST and mlr genes.
Discussion
Bacterially mediated microcystin degradation has been studied primarily on bacterial cultures or in artificial environments. Related studies in natural environments have generally assumed that bacteria associated with CyanoHABs are predominant microcystin degraders [25], [26], [27], [28]. Using microcosm incubations, our study provides empirical data to identify bacterial genes and taxa that are involved in microcystin degradation in nature.
Microcosms are widely used in ecological research because they can be readily replicated and examined under controlled laboratory conditions, permitting experimental manipulations as in this study. However, the reliability of conclusions drawn from microcosms can be compromised by artifacts of confinement (“bottle effects”), which are exacerbated as the ratio of bottle surface to microcosm volume increases [29]. For this reason we constructed microcosms as large as possible to be manipulated in the laboratory: 18 L microcosms in 20 L carboys. Because of the uniformly large size of our microcosms we assumed that “bottle effects” would be consistent among the treatments and have low impacts on our overall conclusion. Other manipulations, i.e., pre-filtration and pre-incubation, were found necessary to establish contrasting results of cell abundance, size and nucleic acid content distributions, and MC-LR degradation activities between the MC and CT microcosms. However, these processing steps also made the experimental systems less in situ-like. Nonetheless, our approach allowed culture-independent identification of MC-degrading bacterial taxa and genes without constrains from prior knowledge.
In this study, free-living bacterioplankton grew substantially at the expense of added MC-LR, indicating they were actively using microcystin as carbon and/or energy sources (Figure 2). Cell distribution observed from FACS analysis also indicated that MC-LR affected the bacterial taxa differently and stimulated the growth of a subset of bacterial taxa present in the samples (Figure 3). The average growth rate of bacterioplankton in MC microcosms was 0.94 day−1, which is comparable to rates that have been reported for Lake Erie bacteria [30], [31], [32]. The consumption rate of MC-LR (∼15 µg L−1 in 48 hours) in microcosms was similar to previous observations in pure cultures growing on MC-LR at similar concentrations [8], [33]. If MC-LR were supplied at higher concentrations (>25 µg L−1) and/or to un-manipulated ambient lake water, which contains diverse labile dissolved organic compounds, MC-LR degradation rate would likely be slower and with a lengthy initial lag phase [8], [33].
Base on the amount of MC-LR addition and the growth of bacterial cells, we estimated that the average carbon content per bacterial cell (Cc) in the MC microcosms was 6 fg/cell. The widely used average Cc for bacteria is 20 fg/cell [34], but many studies have proposed lower values (7–13 fg/cell) [35], [36], [37]. Studies have shown that Cc values of bacterial cells can range across three orders of magnitude, from1.5 fg/cell to 1.9 pg/cell [36]. Lower Cc values are typically associated with cells that are small in size [34], [36] and/or growing under nutrient limited conditions [38], similar as those in the MC microcosms. This calculation also indicated that bacterial growth in the MC microcosms can be largely explained by active incorporation of added MC-LR by bacterial cells.
Over twenty strains of MC-degrading bacterial isolates are currently available and they are affiliated with a narrow group of bacterial orders, including Actinomycetales, Bacillales, Sphingomonadales and Burkholderiales and Methylophilales [13], [39], [40]. Our culture-independent study suggests a highly heterogeneous composition of MC-responsive bacteria, including members from over 89 orders with in the phyla of Actinobacteria, Bacteroidetes, Firmicutes, Planctomycetes, Proteobacteria of the alpha, beta, gamma and delta/epsilon subdivisions, and Verrucomicrobia. Recent studies on CyanoHAB-associated bacteria have similarly indicated a high taxonomic diversity of MC degraders in a number of freshwater lakes [10], [41], [42].
Previous culturing studies and surveys of CyanoHAB-associated bacteria have suggested a dominant role of Sphingomonadales (mainly within the genus Sphingomonas) in MC degradation. However, a recent survey by 16S rRNA gene pyrotag sequencing has indicated a low relative abundance of Sphingomonadales (∼1% of total bacterial community) during a CyanoHAB event in Lake Erie [43]. Our metagenomic data also indicate that Sphingomonadales may be less important than Methylophilales and Burkholderiales in bacterioplankton-mediated MC degradation in Lake Erie (Figures 6 and S1; Table S6). The latter two Betaproteobacterial orders are common to freshwater environments [44], and each has cultured MC-degrading representatives [7], [40]. Moreover, although often at low abundance, Methylophilales have been frequently found to be associated with freshwater CyanoHABs [25], [26], [27], [28], [41]. Differences between our study and those of others most likely are due to variation between one site and another in physical, chemical and biotic conditions and the targeted fraction of the bacterioplankton (free-living bacterioplankton vs. total community). Notwithstanding those differences, our findings emphasize that MC-degrading bacteria and pathways likely are broader than earlier studies indicated.
Archaea have been identified as important in situ microbial taxa during a MC-producing CyanoHAB, but their high abundance has declined to undetectable levels after being incubated in MC-amended microcosms [10]. The authors have attributed this to high sensitivity of Archaeal cells to “bottle effects”. In our study, MC and CT metagenomes were subjected to the same container incubation conditions. Archaea-affiliated sequences occurred in low quantities in all microcosms, and their relative abundance was significantly lower in the MC (0.08% of total protein-coding sequences) than in the CT microcosms (0.5%) (t test with Bonferroni correction, P<0.05). This suggests an insignificant role of archaea in microcystin degradation.
Actinobacteria have several MC-degrading species and are common taxa associated with MC-producing CyanoHABs [10], [26]. They have been found more important during CyanoHABs in lakes with water temperature below 20°C than in warmer lakes. In our study, water temperatures at time of sampling and during incubation were at 22°C or above. Significantly fewer Actinobacteria were found in MC metagenomes, indicating that this taxon was insignificant in MC degradation in the samples examined.
The genes (mlrABCD), intermediates and products of an enzymatic pathway for bacterial MC degradation have been identified based on works on Sphingomonadales strains [12], [45]. mlrA genes are considered as the most important within the mlr cluster because they encode the ring-cleavage step that leads to opening of the microcystin ring structure (Figure 1). Probes/primers of mlrA have been developed and used to study in situ activity of MC-degrading bacteria in various environments [46], [47], [48]. However, PCR amplification of mlrA genes from microcystin-degrading Actinobacteria isolates has failed [13]. Two factors may be contributing to this failure: first, existing primers may be inefficient for broad identification of mlrA in non-Sphingomonas taxa [40] and second, microcystin degradation genes and/or pathways may vary among bacterial taxa. Our results support the latter hypothesis. In this study, putative mlrA genes were identified based on full-length amino acid sequence homology, which should have largely bypassed the bias of primer specificity that is inherent in the PCR method. In accordance, recovered mlrA genes in our metagenomes were broadly affiliated with Proteobacteria (in subdivisions of alpha, beta, gamma and delta) and Bacteroidetes. In addition, none of the mlrA or other mlr sequences was affiliated with Methylophilales, even though Methylophilales represented the most abundant taxon in the MC metagenomes. These suggest that bacteria, especially members of Methylophilales, may employ an alternative microcystin degradation pathway.
Our results suggest that this alternative pathway may involve xenobiotic metabolism (Figure 1). Xenobiotic metabolism-related genes and gene categories, e.g., GST gene and COG0625, COG0841, COG1566 and KEGG0980, were significantly overrepresented in the MC metagenomes. Moreover, Methylophilales were affiliated with a large proportion of xenobiotic metabolism related sequences, but none of the putative mlr sequences. Xenobiotic metabolism is widely distributed among living organisms of all three life domains and refers to intracellular processes that neutralize and eliminate toxic effects of foreign compounds by altering their chemical structures. A long list of substrates has been identified for xenobiotic metabolizing systems in bacteria, including halogenated compounds [49], drugs [50] and numerous environmental pollutants [51]. Our metagenomic study suggests adding MC-LR to this list. Although it is novel for bacterial systems, GST-mediated xenobiotic metabolisms are known for their critical role in MC detoxification by various aquatic eukaryotes, including higher plants, invertebrate and vertebrate animals [26]. The wide distribution of GST genes and related xenobitc metabolism have been largely attributed to horizontal gene transfer, and the parallel and independent evolution of these essential genes among different phylogenic groups [52].
It is noted that our results did not rule out the involvement of mlr gene-based pathway in MC-LR degradation, but suggested that alternative pathway, such as xenobiotic metabolism, may also be important in the process. Further studies, especially those that identify degradation intermediates and measure gene expression, are required to confirm the occurrence of MC degradation by xenobiotic metabolism in bacteria and to examine its importance relative to mlr gene-based cleavage pathway.
Supporting Information
Relative abundance of major bacterial taxa at order levels in the MC and CT metagenomes. Taxonomic affiliations are based on (A) total protein sequences, (B) total protein sequences with COG group assignment, (C) COG sequences that were overrepresented in the MC metagenomes, relative to the CT metagenomes and (D) putative 16S rRNA genes.
(EPS)
Basic physiochemical parameters of surface water samples at the time of collection.
(DOC)
NCBI database accession numbers for reference sequences used to identify homologs to 16S rRNA and functional genes.
(DOC)
Full list of overrepresented COG groups in the MC metagenomes relative to the CT metagenomes.
(DOC)
Number and percent of metagenomic sequences with taxonomic assignment at different resolution levels.
(DOC)
List of underrepresented COG groups in the MC metagenomes relative to the CT metagenomes.
(DOC)
Relative abundance (% of total sequences) of bacterial taxa revealed by 16S rRNA gene sequences in each metagenomes.
(DOC)
Acknowledgments
We thank T Bridgman for his assistance in collecting water samples and providing hydrological data, E Mintz for assistance in statistical analysis, R Newton for assistance in 16S rRNA gene sequence annotation using the ARB tree analysis and L Leff for comments on the manuscript.
Funding Statement
This study was supported by Lake Erie Protection Fund SG374-09 and KSU Farris Family Innovation Research Fellowship to XM. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Carmichael W (2008) A world overview: One hundred twenty seven years of research on toxic cyanobacteria, where do we go from here? Adv Exp Med Biol 619: 105–25. [DOI] [PubMed] [Google Scholar]
- 2. Rinta-Kanto JM, Wilhelm SW (2006) Diversity of microcystin-producing cyanobacteria in spatially isolated regions of Lake Erie. Appl Environ Microbiol 72: 5083–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bourne DG, Jones GJ, Blakeley RL, Jones A, Negri AP, et al. (1996) Enzymatic pathway for the bacterial degradation of the cyanobacterial cyclic peptide toxin microcystin-LR. Appl Environ Microbiol 62: 4086–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manage PM, Edwards C, Lawton LA (2010) Bacterial degradation of microcystin. In Hamamura N, Suzuki S, Mendo S, Barroso CM, Iwata H, Tanabe, S. (eds) Interdisciplinary Studies on Environmental Chemistry-Biological Responses to Contaminants. TERRAPUB: Tokyo, 97–104.
- 5. Christoffersen K, Lyck S, Winding A (2002) Microbial activity and bacterial community structure during degradation of microcystins. Aqua Microb Ecol 27: 125–136. [Google Scholar]
- 6. Chen W, Song LR, Peng L, Wan N, Zhang XM, et al. (2008) Reduction in microcystin concentrations in large and shallow lakes: Water and sediment-interface contributions. Water Res 42: 763–773. [DOI] [PubMed] [Google Scholar]
- 7. Lemes GAF, Kersanach R, Pinto LD, Dellagostin OA, et al. (2008) Biodegradation of microcystins by aquatic Burkholderia sp. from a South Brazilian coastal lagoon. Ecotoxicol Environ Saf 69: 358–365. [DOI] [PubMed] [Google Scholar]
- 8. Ho L, Hoefel D, Saint CP, Newcombe G (2007) Isolation and identification of a novel microcystin-degrading bacterium from a biological sand filter. Water Res 41: 4685–4695. [DOI] [PubMed] [Google Scholar]
- 9. Ho L, Hoefel D, Palazot S, Sawade E. Newcombe G, et al. (2010) Investigations into the biodegradation of microcystin-LR in wastewaters. J Hazard Mat 180: 628–633. [DOI] [PubMed] [Google Scholar]
- 10. Dziallas C, Grossart HP (2011) Temperature and biotic factors influence bacterial communities associated with the cyanobacterium Microcystis sp. Environ Microbiol 13: 1632–1641. [DOI] [PubMed] [Google Scholar]
- 11. Cheng C, Zaichao Z, Aizhong D, Jiayan W, Jingfa X, et al. (2011) Bar-Coded Pyrosequencing reveals the bacterial community during Microcystis water bloom in Guanting Reservoir, Beijing. Procedia Eng 18: 341–346. [Google Scholar]
- 12. Bourne DG, Riddles P, Jones GJ, Smith W, Blakeley RL (2001) Characterisation of a gene cluster involved in bacterial degradation of the cyanobacterial toxin microcystin-LR. Environ Toxicol 16: 523–534. [PubMed] [Google Scholar]
- 13. Manage PM, Edwards C, Singh BK, Lawton LA (2009) Isolation and identification of novel microcystin degrading bacteria. Appl Environ Microbiol 75: 6924–6928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.APHA (1999) Standard methods for the examination of water and wastewater. American Public Health Association: Washington, DC.
- 15. Mou X, Moran MA, Stepanauskas R, Gonzalez JM, Hodson RE (2005) Flow-cytometric cell sorting and subsequent molecular analyses for culture-independent identification of bacterioplankton involved in dimethylsulfoniopropionate transformations. Appl Environ Microbiol 71: 1405–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lebaron P, Parthuisot N, Catala P (1998) Comparison of blue nucleic acid dyes for flow cytometric enumeration of bacteria in aquatic systems. Appl Environ Microbiol 64: 1725–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gomez-Alvarez V, Teal TK, Schmidt TM (2009) Systematic artifacts in metagenomes from complex microbial communities. ISME J 3: 1314–1317. [DOI] [PubMed] [Google Scholar]
- 18. Niu B, Fu F, Sun S, Li W (2010) Artificial and natural duplicates in pyrosequencing reads of metagenomic data. BMC Bioinformatics 11: 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, et al. (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mou XZ, Sun S, Edwards RA, Hodson RE, Moran MA (2008) Bacterial carbon processing by generalist species in the coastal ocean. Nature 451: 708–711. [DOI] [PubMed] [Google Scholar]
- 21. Huson DH, Auch AF, Qi J, Schuster SC (2007) MEGAN analysis of metagenomic data. Genome Res 17: 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rice WR (1989) Analyzing tables of statistical tests. Evolution 43: 223–225. [DOI] [PubMed] [Google Scholar]
- 23. Rodriguez-Brito B, Rohwer F, Edwards R (2006) An application of statistics to comparative metagenomics. BMC Bioinformatics 7: 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Parks DH, Beiko RG (2010) Identifying biologically relevant differences between metagenomic communities. Bioinformatics 26: 715–721. [DOI] [PubMed] [Google Scholar]
- 25. Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ (2006) Metagenomic analysis of the human distal gut microbiome. Science 312: 1355–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Campos A, Vasconcelos V (2010) Molecular mechanisms of microcystin toxicity in animal cells. Int J Mol Sci 11: 268–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Philosof A, Sabehi G, Béjà O (2009) Comparative analyses of actinobacterial genomic fragments from Lake Kinneret. Environ Microbiol 11: 3189–3200. [DOI] [PubMed] [Google Scholar]
- 28. Eiler A, Bertilsson S (2004) Composition of freshwater bacterial communities associated with cyanobacterial blooms in four Swedish lakes. Environ Microbiol 6: 1228–1243. [DOI] [PubMed] [Google Scholar]
- 29.Elliott ET, Hunt HW, Walter DE, Moore JC (1986) Microcosms, mesocosms and ecosystems: linking the laboratory to the field. In: Megušar F and Gantar M (eds.) Perspectives in Microbial Ecology. Slovene Society for Microbiology, Lubljana: 472–480.
- 30. Hwang SJ, Heath RT (1999) Zooplankton bacterivory at coastal and offshore sites of Lake Erie. J Plank Res 21: 699–719. [Google Scholar]
- 31. Wilhelm SW, Smith REH (2000) Bacterial carbon production in Lake Erie is influenced by viruses and solar radiation. Can J Fish Aquat Sci 57: 317–326. [Google Scholar]
- 32. Lavrentyev PJ, McCarthy MJ, Klarer DM, Jochem F, Gardner WS (2004) Estuarine microbial food web patterns in a Lake Erie coastal wetland. Microb Ecol 48: 567–577. [DOI] [PubMed] [Google Scholar]
- 33. Eleuterio L, Batista JR (2009) Biodegradation studies and sequencing of microcystin-LR degrading bacteria isolated from a drinking water biofilter and a fresh water lake. Toxicon 55: 1434–1442. [DOI] [PubMed] [Google Scholar]
- 34. Lee S, Fuhrman JA (1987) Relationships between biovolume and biomass of naturally derived marine bacterioplankton. Appl Environ Microbiol 53: 1298–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Christian JR, Karl DM (1994) Microbial community structure at the U.S.-Joint Global Ocean Flux Study Station ALOHA: Inverse methods for estimating biochemical indicator ratios. J Geophys Res 99: 14269–14276. [Google Scholar]
- 36. Fagerbakke KM, Heldal M, Norland S (1996) Content of carbon, nitrogen, oxygen, sulfur and phosphorus in native aquatic and cultured bacteria. Aquat Microb Ecol 10: 15–27. [Google Scholar]
- 37. Carlson CA, Bates NR, Ducklow, Hansell DA (1999) Estimation of bacterial respiration and growth efficiency in the Ross Sea, Antarctica. Aquat Microb Ecol 19: 229–244. [Google Scholar]
- 38. Troussellier M, Bouvy M, Courties C, Dupuy C (1997) Variation of carbon content among bacterial species under starvation condition. Aquat Microb Ecol 13: 113–119. [Google Scholar]
- 39. Ho L, Gaudieux AL, Fanok S, Newcombe G, Humpage AR (2007) Bacterial degradation of microcystin toxins in drinking water eliminates their toxicity. Toxicon 50: 438–441. [DOI] [PubMed] [Google Scholar]
- 40. Hu LB, Yang JD, Zhou W, Yin YF, Chen J, et al. (2009) Isolation of a Methylobacillus sp. that degrades microcystin toxins associated with cyanobacteria. New Biotechnol 26: 205–211. [DOI] [PubMed] [Google Scholar]
- 41. Li N, Zhang L, Li F, Wang Y, Zhu Y, et al. (2011) Metagenome of microorganisms associated with the toxic cyanobacteria Microcystis aeruginosa analyzed using the 454 sequenicng platform. Chin J Oceanol Limnol 29: 505–513. [Google Scholar]
- 42. Wilhelm SW, Farnsely SE, LeCleir GR, Layton AC, Satchwell MF, et al. (2011) The relationship between nutrients, cyanobacterial toxins and the microbial community in Taihu (Lake Lai), China. Harm Algae 10: 207–215. [Google Scholar]
- 43.Mou X, Jacob J, Lu X, Robbins R, Sun S, et al.. (2013) Diversity and distribution of free-living and particle-associated bacterioplankton in Sandusky Bay and adjacent waters of Lake Erie Western Basin. J Great Lakes Res (In revision).
- 44. Newton RJ, Jones SE, Eiler A, McMahon KD, Bertilsson S (2011) A guide to the natural history of freshwater lake bacteria. Microbiol Mol Biol Rev 75: 14–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shimizu K, Maseda H, Okano K, Kurashima T, Kawauchi Y, et al. (2012) Enzymatic pathway for biodegrading microcystin LR in Sphingopyxis sp. C-1. J Biosci Bioeng 114: 630–634. [DOI] [PubMed] [Google Scholar]
- 46. Saito T, Okano K, Park H, Itayama T, Inamori Y, et al. (2003) Detection and sequencing of the microcystin LR-degrading gene, mlrA, from new bacteria isolated from Japanese lakes. FEMS Microbiol Lett 229: 271–276. [DOI] [PubMed] [Google Scholar]
- 47. Hoefel D, Adriansen CMM, Bouyssou MAC, Saint CP, Newcombe G, et al. (2009) Development of an mlrA gene-directed Taqman PCR assay for quantitative assessment of microcystin-degrading bacteria within water treatment plant sand filter biofilms. Appl Environ Microbiol 75: 5167–5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jimbo Y, Okano K, Shimizu K, Maseda H, Fujimoto N, et al. (2010) Quantification of microcystin-degrading bacteria in a biofilm from a practical biological treatment facility by real-time PCR. J Water Environ Technol 8: 193–201. [Google Scholar]
- 49. Janssen DB, Dinkla IJ, Poelarends GJ, Terpstra P (2005) Bacterial degradation of xenobiotic compounds: evolution and distribution of novel enzyme activities. Environ microbiol 7: 1868–82. [DOI] [PubMed] [Google Scholar]
- 50. Maurice CF, Haiser HJ, Turnbaugh PJ (2013) Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 152: 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Aksu Z (2005) Application of biosorption for the removal of organic pollutants: a review. Proc Biochem 40: 997–1026. [Google Scholar]
- 52. Vuilleumier S (1997) Bacterial glutathione S-transferases: what are they good for? J Bacteriol 179: 1431–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Relative abundance of major bacterial taxa at order levels in the MC and CT metagenomes. Taxonomic affiliations are based on (A) total protein sequences, (B) total protein sequences with COG group assignment, (C) COG sequences that were overrepresented in the MC metagenomes, relative to the CT metagenomes and (D) putative 16S rRNA genes.
(EPS)
Basic physiochemical parameters of surface water samples at the time of collection.
(DOC)
NCBI database accession numbers for reference sequences used to identify homologs to 16S rRNA and functional genes.
(DOC)
Full list of overrepresented COG groups in the MC metagenomes relative to the CT metagenomes.
(DOC)
Number and percent of metagenomic sequences with taxonomic assignment at different resolution levels.
(DOC)
List of underrepresented COG groups in the MC metagenomes relative to the CT metagenomes.
(DOC)
Relative abundance (% of total sequences) of bacterial taxa revealed by 16S rRNA gene sequences in each metagenomes.
(DOC)