

Abstract
αB-crystallin is a small heat shock protein that exhibits chaperone activity and can protect multiple cell types against oxidative stress damage. Altered levels and specific mutations of αB-crystallin are associated with multiple degenerative diseases. We previously found that αB-crystallin translocates to lens and retinal cell mitochondria upon oxidative stress exposure where it provides protection against oxidative stress damage. To date, the role of the chaperone function of αB-crystallin in mitochondrial translocation and protection has not been established. Here, we sought to determine the relationship between the chaperone activity of αB-crystallin and its ability to translocate to and protect retinal cell mitochondria against oxidative stress damage. Our data provide evidence that three forms of αB-crystallin exhibiting different chaperone activity levels including wild-type, R120G (decreased chaperone activity) and M68A (increased chaperone activity) provide comparable levels of mitochondrial translocation and protection to retinal cells exposed to oxidative stress. The results provide evidence that mitochondrial translocation and protection by αB-crystallin is independent of its chaperone activity and that other functions of αB-crystallin may also be independent of its chaperone activity.
Keywords: αB-crystallin, small heat shock protein, chaperone, mitochondria, oxidative stress, RPE, retinal cells
1. Introduction
αB-crystallin is a small heat shock protein that can prevent aggregation of proteins (Horwitz, 1992; Jakob et al., 1993). It is comprised of individual 20 kDa peptide subunits that multimerize to form higher molecular weight complexes (Horwitz, 2003). It is mainly found in the eye lens, retina, heart and neural tissues (Bhat and Nagineni, 1989; Dubin et al., 1989). Altered expression and/or accumulation of αB-crystallin is associated with the etiology of many diseases including mammary metaplastic carcinoma (Chan et al., 2011), hepatocellular carcinoma (Tang et al., 2009), and renal cell carcinoma (Shi et al., 2004), age related macular degeneration (Nakata et al., 2005; Alge et al., 2002; De et al, , 2007), age-related cataract (Chen et al., 1997; Truscott et al., 1998), Parkinson’s disease (Braak et al, 2001), Alexander disease (Iwaki et al., 1989), Lewy body disease (Dabir et al., 2004), Alzheimer disease (Renkawek et al., 1994), Creutzfeldt–Jakob disease (Renkawek et al., 1992), multiple sclerosis (Ousman et al., 2007), congestive heart failure (Dohke et al., 2006) and Huntington’s disease (Iwaki et al., 1992; Zabel et al., 2002; Dabir et al., 2004).
αB-crystallin protects multiple cell types against a variety of environmental stresses. It can defend cells against UV-light (Liu et al., 2004), peroxide (Yaung et al., 2007; McGreal et al., 2012) and ischemic reperfusion (Martin et al., 1997; Jin et al., 2008) damage. It preserves the viability of lens (McGreal et al., 2012), retina (Yaung et al., 2007; McGreal et al., 2012), cardiac myocytes (Martin et al., 1997), kidney (Arai and Atomi, 1997), and glial cells (Kegal et al., 1996) exposed to oxidative stress treatments. The precise mechanisms that orchestrate its protective function have yet to be fully elucidated, however, αB-crystallin has been demonstrated to modulate the function of key apoptotic control proteins (Andley et al., 2000; Kamradt et al., 2002; Kamradt et al., 2005; Mao et al., 2004; Liu et al., 2004) and to be important for cytoskeletal assembly (Ghosh et al., 2007; Houck and Clark, 2010).
Recently αB-crystallin has been shown to protect lens and retinal pigmented epithelial cells (RPE) against oxidative damage through its ability to translocate to and preserve the function of the mitochondria under oxidative conditions (McGreal et al., 2012). Based on these findings, we hypothesized that the chaperone activity of αB-crystallin would be required for mitochondrial translocation, mitochondrial protection or both. To test this hypothesis we evaluated the in vitro and ex vivo chaperone activities of two mutant forms of αB-crystallin and evaluated the potential of these mutants to translocate to and preserve the function of mitochondria in RPE cells. RPE cells were chosen for these studies over lens cells since they have barely detectable levels of αA-crystallin allowing us to specifically examine the function of αB-crystallin. The αB-crystallin R120G mutant that causes Desmin related myopathy and cataract has previously been characterized and found to have decreased chaperone function (Bova et al, 1999). Oxidation of methionine residues in α-crystallin has been shown to abolish chaperone function (Brennan et al., 2009) while substitution of methionine 68 in αB-crystallin has differential effects on chaperone function depending on the hydrophobicity of the substituted residue (Schroff et al., 2001). Here, we substituted methionine at position 68 with alanine and assessed the chaperone function of this mutant for the first time. We demonstrate that, as previously reported, R120G αB-crystallin exhibits diminished chaperone activity relative to wild-type (wt) αB-crystallin and that the novel M68A αB-crystallin mutant exhibits increased chaperone activity relative to wt αB-crystallin. These mutant forms of αB-crystallin exhibited similar ex vivo chaperone activities; however, they translocate to the mitochondria with equal efficiencies and provide equal levels of mitochondrial protection under oxidative conditions. These results provide evidence that mitochondrial translocation and protection by αB-crystallin under oxidative conditions is not dependent on chaperone activity levels and that other functions of αB-crystallin could be chaperone-independent.
2. Methods
2.1 Site directed mutagenesis
Human αB-crystallin cDNA in a pET-20b(+) expression vector was utilized to generate the R120G and M68A missense mutations. GeneEditor™ in vitro Site-Directed Mutagenesis System (Promega, Madison, WI) was used according to the manufacturer’s instructions using the following primers: αB R120G: 5’-GAGTTCCACGGGAAATACCGG-3’ and αB M68A: 5’-ACTGGACTCTCAGAGGCGCGCCTGGAGAAGGAC-3’. Sequences were confirmed by automated sequencing.
2.2 Cloning, expression, and purification of wt αB-crystallin, αB-crystallin R120G and αB-crystallin M68A
pET-20b(+) expression vectors containing wt αB-crystallin, αB-crystallin R120G and αB-crystallin M68A cDNA were used to produce recombinant protein. Sequences were confirmed by automated sequencing, the vectors were transformed into BL21 (DE3) competent E. coli (Invitrogen, Carlsbad, CA), and protein expression was induced using Isopropyl-β-D-thio-galactoside (IPTG)(Sigma-Aldrich, St Louis, MO). The induced proteins were purified by gel filtration chromatography using a Sephacryl G300 packed column (GE lifesciences, Piscataway, NJ), fractions were collected, and concentrated using an Amicon® Ultra-15 centrifugal filter device with a nominal molecular weight cutoff of 10 kDa. Protein purity was confirmed by SDS-PAGE and concentrations of protein measured using a Bradford protein assay with BSA as a standard.
2.3 In vitro chaperone function of wt αB-crystallin, αB-crystallin R120G and αB-crystallin M68A
Chaperone activity was assayed by monitoring the aggregation of lysozyme (Sigma-Aldrich) in the presence or absence of wt αB-crystallin, αB-crystallin R120G or αB-crystallin M68A. The ability of each αB-crystallin form to prevent DTT-induced (20 mM) aggregation of lysozyme at 37 °C was monitored by measuring light scattering at 360 nm as a function of time in a Shimadzu UV 1700 spectrophotometer (Columbia, MD) equipped with a temperature regulated cell holder. Lysozyme is destabilized by reduction of its disulfide bonds using DTT (Abgar et al., 2000). The αB-crystallin lysozyme ratio was 1:1 (w/w) for all experiments.
2.4 Cell culture
Human retinal pigmented epithelial cells (ARPE-19) (ATCC, Manassas, Va., USA) were cultured in DMEM (Invitrogen, Carlsbad, CA) supplemented with 15% FBS (Invitrogen), gentamicin (50 units/ml; Invitrogen), penicillin-streptomycin antibiotic mix (50 units/ml; Invitrogen), and fungizone (5 μl/ml; Invitrogen) at 37 °C in the presence of 5% CO2. Lentiviral over-expressing cell lines were maintained in complete medium as described above containing blasticidin antibiotic selection (ARPE-19, 3 μg/ml).
2.5 Production of over-expressing ARPE-19 cell lines
Wt αB-crystallin, αB-crystallin R120G and αB-crystallin M68A overexpressing ARPE-19 cell lines were developed using the ViraPower Lentiviral Expression System (Invitrogen), utilizing the pLenti6/V5-D-Topo plasmid according to the manufacturer’s instructions. Primers were designed to amplify full-length wt αB-crystallin, αB-crystallin R120G and αB-crystallin M68A transcripts. The resulting cDNA inserts were cloned into the expression vector and used to transfect HEK293-FT kidney cells to generate the viral construct. Virus particles were harvested and used to infect ARPE-19 cells. To generate the control cell line (N-LV), ARPE-19 cells were infected with the control virus LVP-Null (GenTarget Inc, San Diego). Over-expressing cells were selected for using blasticidin (Invitrogen), and over-expression was confirmed by RT-PCR and western blot analysis.
2.6 Transfection of mutant huntingtin protein in RPE cells and analysis of ex vivo protein aggregation
Control N-LV, wt αB-crystallin, αB-crystallin R120G and αB-crystallin M68A over-expressing RPE cells were plated onto coverslips in 12 well plates at a density of 200,000 cells per well and incubated overnight in complete media. Cells were washed and media replaced with serum free, phenol free DMEM before being transfected with a mutant huntingtin protein (htt-103QeGFP) vector using Lipofectamine 2000 (Invitrogen) following manufacturer’s instructions. 48 h after transfection cells were washed and fixed with 3.7% paraformaldehyde in PBS, mounted using ProLong Gold Antifade Reagent (Invitrogen), and visualized using the Zeiss LSM700 confocal microscope (Zeiss, Peabody, MA) for visible signs of aggregation of the GFP labeled protein.
2.7 Monitoring of translocation of αB-crystallin R120G and αB-crystallin M68A to the mitochondria of RPE cells
To analyze mitochondrial translocation under oxidative stress conditions, RPE cells were incubated for 2 h in serum free DMEM followed by treatment with either 0 μM, 100 μM or 200 μM H2O2 for 1 h. After treatment, cells were immediately harvested and mitochondrial and cytosolic fractions were isolated and analyzed by SDS-PAGE and western blotting using an antibody for αB-crystallin.
2.8 Isolation of cytosolic and mitochondrial proteins
Mitochondria were isolated using the Mitosciences (Eugene, Or) mitochondrial isolation kit for cultured cells according to the manufacturer’s protocol. RPE cells were detached from the culture dish by trypsinization and centrifuged at 1,000 g for 3 min. The resulting pellet was frozen at −80 °C to weaken the cell membrane. Cells were re-suspended with Reagent A followed by homogenization for 30 strokes. The homogenate was centrifuged at 1,000 g for 10 min. The supernatant was removed and the pellet was resuspended in Reagent B, homogenized, and centrifuged at 1,000 g for 10 min. The combined supernatants were further centrifuged at 12,000 g for 15 min. The resulting supernatant was collected as the cytosolic fraction and the pellet was removed and resuspended in Reagent C as the mitochondrial fraction. Protein concentrations were determined by Bradford protein assay with BSA as a standard.
2.9 SDS-PAGE and western blotting
Unless specified, all electrophoresis reagents and apparatus were purchased from Bio-Rad (Richmond, CA). Protein samples were mixed with 2X sample buffer at a 1:1 v/v ratio and heated at 100 °C for 5 min. The samples were separated by electrophoresis on 15% sodium dodecyl sulfate (SDS) gels at room temperature for 1.5 h at 120 V. Proteins were transferred onto nitrocellulose membranes (Amersham-Pharmacia, Piscataway, NJ) using a Mini Trans-Blot electrophoresis transfer cell apparatus. The membrane was equilibrated in Tris-buffered saline (TBS), pH 7.4, 0.05% Tween-20 for 15 min then blocked in TBS (pH 7.4), 5% Carnation nonfat milk, and 0.05% Tween-20, for 1 h. The membrane was incubated over night with the primary anti-αB-crystallin (Stressgen) at a concentration of 1:20,000, followed by incubation for 1 h with the anti-rabbit (αB-crystallin) secondary antibody (GE Healthcare) at a concentration of 1:5,000. The blot was visualized using ECL western blotting reagents (Amersham-Pharmacia) as specified by the manufacturer.
2.10 Analysis of mitochondrial membrane potential
Control N-LV, wt αB-crystallin, αB-crystallin R120G and αB-crystallin M68A overexpressing RPE cells were plated at a density of 200,000 cells/dish on 35 mm glass bottomed dishes. The cells were incubated in serum free media for 2 h before treatment with H2O2 at either 0 μM or 150 μM for 24 h. For mitochondrial membrane potential analysis, the cells were stained with 5 mM JC-1 (Invitrogen) for 20 min. Mitochondrial membrane potential (MMP) changes were examined using the Zeiss LSM700 confocal microscope (Zeiss, Thornwood, NY) by observing a red to green color shift which resulted in an increase in yellow emission in the merged images, indicating decreased MMP. Mean intensities, in relative light units, of the red and green channels were calculated using the Zen 2009 software (Zeiss) and the red/green ratio calculated by dividing the red intensity by the green intensity. Differences among red/green ratios for each cell line were determined using Student’s t test assuming equal variance. p < 0.05 was considered statistically significant.
3. Results
3.1 R120G has decreased and M68A increased levels of chaperone activity
To survey the chaperone activities of wt αB-crystallin, αB-crystallin R120G (Bova et al, 1999) and αB-crystallin M68A, we assayed the ability of these αB-crystallin forms to prevent lysozyme aggregation in the presence of DTT (Horwitz, 2003). M68A is a previously uncharacterized αB-crystallin mutant made here for the present study. Lysozyme was incubated with wt αB-crystallin, αB-crystallin R120G, or αB-crystallin M68A at a 1:1 ratio (w/w) prior to treatment with DTT (20 mM). Wt αB-crystallin protected lysozyme against DTT-induced aggregation (Fig. 1). αB-crystallin R120G showed reduced chaperone activity relative to wt αB-crystallin as previously demonstrated (Bova et al, 1999) (Fig. 1). Interestingly, the chaperone activity of αB-crystallin M68A was greater than wt αB-crystallin or R120G αB-crystallin (Fig. 1).
Fig. 1. αB-crystallin R120G has decreased and αB-crystallin M68A increased chaperone activity levels relative to wt αB-crystallin.
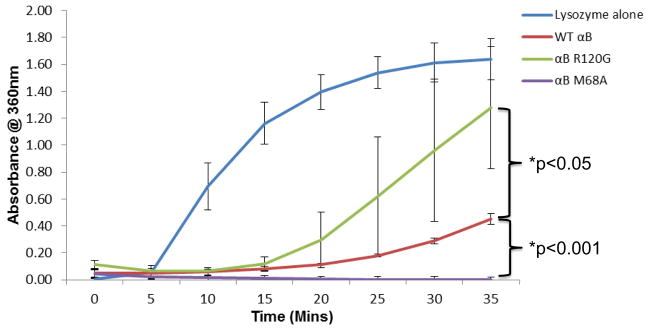
Chaperone activities of purified recombinant wt αB-crystallin, αB-crystallin R120G, and αB-crystallin M68A using lysozyme as a target substrate. Lysozyme was incubated in the presence or absence of wt αB-crystallin or αB-crystallin mutants R120G and M68A in the presence of absence of 20 mM DTT and protein aggregation monitored by measuring absorbance (turbidity) at 360nm. Graph shows mean values of n=3 ± SD. p values were calculated using two sample Student t-test assuming equal variance. Significant differences between wt and mutants are shown wt - R120G, p<0.05, wt - M68A, p<0.001.
3.2 The ex vivo chaperone activities of R120G and M68A αB-crystallins in RPE cells are similar to those detected in vitro
To establish the relative ex vivo chaperone levels of the R120G and M68A mutant forms of αB-crystallin, RPE cells stably overexpressing each mutant form of αB-crystallin were transiently transfected with a mutant huntingtin-GFP fusion protein (htt-103QeGFP) that spontaneously aggregates in RPE cells. Aggregation of the mutant huntingtin protein in each cell line was visualized by confocal microscopy. As shown in Fig. 2a both cell lines expressed significant amounts of endogenous (20 kDa) αB-crystallin, αB-crystallin R120G or αB-crystallin M68A as evidenced by the detection of an αB-crystallin-positive 23 kDa (his-tagged mutant αB-crystallins) band by western analysis. Images showing visible fluorescent htt-103QeGFP foci (aggregates) in the R120G and M68A overexpressing RPE cells are shown in Fig. 2B. Cells over-expressing αB-crystallin R120G transfected with htt-103QeGFP showed increased numbers of mutant huntingtin protein aggregates relative to the cells stably overexpressing αB-crystallin M68A, which had fewer aggregates per cell, consistent with their respective decreased and increased in vitro chaperone activities (Fig. 1). A histogram representing the numbers of htt-103QeGFP transfected cells containing htt-103QeGFP foci for each mutant cell line is shown in Fig 2C, where it can be seen that the majority of stably overexpressing R120G RPE cells transfected with htt-103QeGFP contain foci while the majority of M68A stably overexpressing RPE cells do not.
Fig. 2. αB-crystallin mutants have similar chaperone activity levels in RPE cells ex vivo as detected in vitro.

(A) SDS-PAGE and immunoblot of protein extracts from stably over-expressing αB-crystallin R120G RPE and αB-crystallin M68A RPE, probed with an αB-crystallin-specific antibody. (B) RPE cells stably overexpressing αB-crystallin R120G and αB-crystallin M68A were transiently transfected with mutant huntingtin protein htt-103QeGFP. 48 hours post transfection cells were fixed and imaged by confocal microscopy. Representative images for each cell line are shown, aggregates of the GFP-tagged htt-103Qe are circled, white circles indicate intracellular aggregates, red circles indicate extracellular aggregates that were not counted. (C) A semi-quantitative depiction of the number of aggregates per cell is shown in Fig 2C. Images and bar graph are representative of two independent experiments.
3.3 αB-crystallin R120G and αB-crystallin M68A translocate to the mitochondria of RPE cells upon oxidative stress exposure
It has previously been demonstrated that cobalt chloride treatment of RPE cells results in translocation of αB-crystallin to the mitochondria (Yaung et al., 2008). Mitochondrial translocation of αB-crystallin has also been detected upon hydrogen peroxide (H2O2) treatment of lens and RPE cells (McGreal et al., 2012). To determine the relative abilities of the R120G and M68A αB-crystallin mutants to translocate to the mitochondria of RPE cells upon oxidative stress treatment, stably over-expressing αB-crystallin R120G and αB-crystallin M68A cells were treated for 1 h with H2O2 and mitochondrial and cytosolic cellular fractions were isolated. SDS-PAGE and western blot analysis revealed that native αB-crystallin (20 kDa), αB-crystallin R120G (23 kDa) and αB-crystallin M68A (23 kDa) exhibited H2O2-dependent increases in mitochondrial localization relative to cytosolic localization upon H2O2 treatment (Fig. 3A and B). The mutant R120G and M68A αB-crystallin proteins migrate at 23 kDa due to the addition of a his-tag that adds about 3kD to the molecular weight of the resulting fusion protein. Although quantitative comparisons are not possible in this assay system, no differences in translocation efficiency are apparent between the chaperone active αB-crystallin M68A mutant and the chaperone deficient αB-crystallin R120G mutant form of αB-crystallin. The translocation of endogenous αB-crystallin (20 kDa) is also indicated and as control, colloidal blue stained gels are shown to demonstrate equal protein loading.
Fig. 3. αB-crystallin M68A and αB-crystallin R120G translocate to the mitochondria of RPE cells upon oxidative stress exposure.

SDS-PAGE and immunoblot of mitochondrial and cytosolic fractions from RPE cells overexpressing (A) αB-crystallin R120G and (B) αB-crystallin M68A treated with 0 μM, 100 μM, or 200 μM H2O2 for 1 h. Immunoblots were probed using an αB-crystallin-specific antibody and 10 μg of protein was loaded. Colloidal blue staining is shown as a control for equal protein loading. The mutant R120G and M68A αB-crystallin proteins migrate at 23 kDa due to the addition of a his-tag.
3.4 αB-crystallin R120G and αB-crystallin M68A preserve mitochondrial membrane potential in RPE cells upon oxidative stress treatment
It has previously been demonstrated that αB-crystallin protects lens and retinal cell mitochondrial function upon oxidative stress treatment (McGreal et al., 2012). We therefore, sought to determine whether the R120G and M68A mutant forms of αB-crystallin would provide comparable levels of mitochondrial protection to RPE cells exposed to oxidative stress relative to RPE cells over-expressing wt αB-crystallin. As a control to account for the protective effect of endogenously expressed αB-crystallin, vector alone transfected cells (N-LV) were also examined. To examine mitochondrial membrane potential (MMP), cells were stained with JC-1 (Marchetti et al., 2006; McGreal et al., 2012). In the mitochondria JC-1 exists as an aggregate and emits red fluorescence, however, when it is released into the cytosol due to loss of MMP, it exists as a monomer that emits green fluorescence. Therefore, loss of MMP results in increased green fluorescence in the cytosol, and decreased red fluorescence in the mitochondria, known as a red to green shift. In control N-LV cells expressing only endogenous αB-crystallin, increased green fluorescence was detected when the cells were treated with 150 μM H2O2 (Fig. 4A), demonstrating decreased MMP and therefore decreased mitochondrial function upon oxidative stress treatment. This can be visualized by the increase in yellow in the merged image of the red and green channels. Overexpression of wt αB-crystallin preserved MMP upon treatment with 150 μM H2O2 above that detected for endogenous αB-crystallin (N-LV) alone indicated by decreased green fluorescence (Fig. 4B) relative to the N-LV cells. Interestingly, cells overexpressing only αB-crystallin R120G (Fig. 4C) or αB-crystallin M68A (Fig. 4D) were also protected against oxidative stress induced loss of MMP when cells were treated with 150 μM H2O2, despite their opposite chaperone activity levels. The data are summarized graphically as a function of the red to green fluorescence shift in Fig. 4E by measuring the overall green intensities of three separate fields of cells.
Fig. 4. Over-expression of wt αB-crystallin, αB-crystallin R120G and αB-crystallin M68A mutant proteins protects RPE mitochondria against oxidative stress damage.

Confocal microscopic images of RPE cells overexpressing control N-LV (A), wt αB-crystallin (B), αB-crystallin R120G (C), and αB-crystallin M68A (D) treated with 0 μM or 150 μM H2O2 for 24 h in serum free media. Cells were stained with JC-1 for 20 min to detect changes in mitochondrial membrane potential (MMP) as indicated by red mitochondrial staining (increased potential) or green cytosolic staining (decreased potential). This is reflected in the merged image as increased yellow fluorescence indicating decreased MMP. (B) Representative bar graph indicating the red/green ratio calculated from the mean intensities of three different fields for each experiment. Error bars represent standard deviation and p-values were calculated using the students t-test (n=3). Differences between intensity of the control N-LV and the αB-crystallin overexpressing cell lines were determined. p < 0.05 was considered statistically significant.
4. Discussion
In previous studies we demonstrated that preservation of mitochondrial function is required for lens and retinal cell viability upon oxidative stress insult (Marchetti et al., 2006, McGreal et al., 2012). Recently, we demonstrated that the small heat shock protein αB-crystallin translocated to the mitochondria of lens and retinal cells under oxidative stress conditions (McGreal et al., 2012) and was important for the maintenance and preservation of mitochondrial membrane potential in these cells under oxidative stress conditions (McGreal et al., 2012). We also demonstrated that oxidation of M68A resulted in loss of αB-crystallin chaperone activity and that the mitochondrial enzyme MsrA could repair and restore the chaperone activity of αB-crystallin (Brennan et al., 2009b) further implicating αB-crystallin as an important regulator of mitochondrial function and suggesting that M68 was important for modulating the chaperone activity level of αB-crystallin. Previous studies showed that replacing M68 in αB-crystallin with amino acids of varying hydrophobicity resulted in marked changes in αB-crystallin chaperone activity levels (Shroff et al., 2001).
The chaperone activity of αB-crystallin has been shown to be essential for its ability to prevent apoptosis in lens cells (Andley et al., 2000), and mutations in αB-crystallin that diminish its chaperone activity result in lens cataract formation (Bova et al., 1999). In addition, αA-crystallin has been shown to prevent apoptosis in HeLa and Chinese hamster ovary cells through its chaperone activity (Pasupuleti et al., 2010). Based on these studies, we hypothesized that the chaperone activity levels of αB-crystallin could modulate its translocation to the mitochondria upon oxidative stress treatment and/or its ability to preserve mitochondrial function under oxidative stress conditions. To test this hypothesis, we measured the chaperone activity levels of two mutant forms of αB-crystallin, R120G and M68A, in vitro and in RPE cells ex vivo and correlated their respective chaperone activity levels with their abilities to translocate to RPE mitochondria under oxidative stress conditions and preserve mitochondrial membrane potential in these cells upon oxidative stress treatment.
Surprisingly, we found no relationship between the chaperone activity levels of αB-crystallin and its ability to translocate to the mitochondria under oxidative conditions. We demonstrate that αB-crystallin R120G exhibits diminished chaperone activity relative to wt αB-crystallin in vitro and ex vivo and that M68A exhibits increased chaperone activity relative to wt αB-crystallin in vitro and ex vivo (Fig. 1 and 2). These results are consistent with previous studies demonstrating decreased chaperone activity for αB-crystallin R120G (Bova et al., 1999) and they confirm the importance of M68 for modulating the chaperone activity of αB-crystallin. Although we cannot rule out the possibility that different chaperone levels might be detected for these mutant αB-crystallin forms on different target proteins and/or in different cell types or ex vivo chaperone assays, our data demonstrate that M68A has increased chaperone activity relative to R120G in vitro and ex vivo under our assay conditions.
We next compared the chaperone activities of the mutant forms of αB-crystallin with their abilities to translocate to the mitochondria of RPE cells under oxidative stress conditions by western analysis of mitochondrial and cytosolic cellular subfractions isolated from H2O2-treated RPE cells that separately overexpress each form of αB-crystallin. Our results demonstrated that each form of αB-crystallin was capable of mitochondrial translocation upon oxidative stress conditions and no differences could be detected in the translocation ability of either R120G, M68 or wt αB-crystallin. These data suggest that the mitochondrial translocation ability of αB-crystallin is independent of chaperone activity levels (Fig. 3).
Lastly, we monitored the relative abilities of RPE cell mitochondria to resist oxidative stress-induced loss of mitochondrial membrane potential when stably overexpressing wt, R120G and M68A αB-crystallins relative to the protection afforded by endogenous αB-crystallin (N-LV) alone (Fig. 4). Equally increased levels of RPE mitochondrial protection against oxidative stress were detected for all three αB-crystallin overexpressing cell lines suggesting that mitochondrial protection by αB-crystallin is independent of chaperone activity level (Fig. 4). We cannot rule that hetero-aggregate formation occurs at some level between endogenous w+-type αB-crystallin and the overexpressed mutatnt forms of αB-crystallin examined. Since αB-crystallin is a ubiquitously protein this possibility would extend to all but αB-crystallin deleted cells that were not available for the present study. We are confident that our results are indeed specific for the mutant αB-crystallin form examined since mitochondrial changes were detected upon over-expression of the mutated αB-crystallin forms to higher levels than those detected for endogenous αB-crystallin alone. Therefore, whether homo-aggregates of mutant αB-crystallin or some percentage of hetero-aggregates of mutant αB-crystallin containing some level of endogenous αB-crystallin mediate altered mitochondrial function, we are still measuring the effect of the αB-crystallin mutation on this function. Taken together the present data suggest that mitochondrial protection by αB-crystallin under oxidative stress conditions is independent of chaperone activity level since both R120G, which exhibits decreased chaperone function, and M68A, which exhibits increased chaperone function, both provide comparable levels of mitochondrial translocation and protection of mitochondrial function in RPE cells exposed to oxidative stress. Interestingly, a slightly higher level of mitochondrial protection was exhibited by R120G than M68A demonstrating a possible inverse relationship between chaperone activity and mitochondrial protection in the RPE cells (Fig. 4).
The possible chaperone independent ability of αB-crystallin to preserve mitochondrial function suggests that some in vivo functions of αB-crystallin may be separate from its chaperone functions. These functions could be important for the multitude of cellular roles that αB-crystallin is known to modulate. These include αB-crystallin’s well-established roles cytoskeletal assembly (Ghosh et al., 2007; Houck and Clark, 2010), membrane binding (Simon et al., 2007; Maddala and Rao, 2005), anti-apoptotic function (Andley et al., 2000; Kamradt et al., 2002; Kamradt et al., 2005; Mao et al., 2004; Liu et al., 2004), cell cycle control (Bai et al., 2004) and stress protection (Liu et al., 2004; Yaung et al., 2007; McGreal et al., 2012). This potential chaperone-independent function could also play a role in many diseases associated with αB-crystallin including age-related macular degeneration (Wang et al., 2008; Lin et al., 2011; Liang and Godley 2003), cataract (Huang et al., 2006), Parkinson disease (Schapira, 2011; Correia et al., 2012), Alexander disease, (Cáceres-Marzal et al., 2006), Alzheimer disease (Zhu et al. 2006; Blass, 2000; Sheng et al., 2012; Correia et al., 2012), Huntington’s disease (Horton et al., 1995; Costa and Scorrano, 2012; Correia et al., 2012), and multiple sclerosis (Mao and Reddy, 2010).
Highlights.
αB-crystallin is a chaperone protein that protects lens and retinal cells against oxidative stress.
αB-crystallin protects lens and retinal cell mitochondria against oxidative stress damage.
The chaperone activity of αB-crystallin is not required for mitochondrial protection
Many functions of αB-crystallin may be independent of its chaperone function.
Acknowledgments
We wish to thank Mason Posner for αB-crystallin pET20-b(+) vector. This work was funded by grant EY13022 to MK.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abgar S, Yevlampieva N, Aerts T, Vanhoudt J, Clauwaert J. Chaperone-like activity of bovine lens alpha-crystallin in the presence of dithiothreitol-destabilized proteins: characterization of the formed complexes. Biochem Biophys Res Comm. 2000;276:619–625. doi: 10.1006/bbrc.2000.3518. [DOI] [PubMed] [Google Scholar]
- Alge C, Priglinger S, Neubauer A, Kampik A, Zillig M, Bloemendal H, Welge-Lussen U. Retinal pigment epithelium is protected against apoptosis by αB-crystallin. Invest Ophthalmol Vis Sci. 2002;43:3575–3582. [PubMed] [Google Scholar]
- Andley UP, Song Z, Wawrousek EF, Fleming TP, Bassnett S. Differential protective activity of alphaA- and alphaB-crystallin in lens epithelial cells. J Biol Chem. 2000;275:36823–36831. doi: 10.1074/jbc.M004233200. [DOI] [PubMed] [Google Scholar]
- Arai H, Atomi Y. Chaperone activity of alpha B-crystallin suppresses tubulin aggregation through complex formation. Cell Struct Funct. 1997;22 (5):539–44. doi: 10.1247/csf.22.539. [DOI] [PubMed] [Google Scholar]
- Bai F, Xi J, Higashikubo R, Andley UP. Cell kinetic status of mouse lens epithelial cells lacking alphaA- and alphaB-crystallin. Mol Cell Biochem. 2004;265 (1–2):115–22. doi: 10.1023/b:mcbi.0000044365.48900.82. [DOI] [PubMed] [Google Scholar]
- Bhat SP, Nagineni CN. alpha B subunit of lens-specific protein alpha-crystallin is present in other ocular and non-ocular tissues. Biochem Biophys Res Commun. 1989;158 (1):319–25. doi: 10.1016/s0006-291x(89)80215-3. [DOI] [PubMed] [Google Scholar]
- Blass JP. The mitochondrial spiral. An adequate cause of dementia in the Alzheimer’s syndrome. Ann NY Acad Sci. 2000;924:170–183. doi: 10.1111/j.1749-6632.2000.tb05576.x. [DOI] [PubMed] [Google Scholar]
- Bova MP, Yaron O, Huang Q, Ding L, Haley DA, Stewart PL, Horwitz J. Mutation R120G in alphaB-crystallin, which is linked to a desmin-related myopathy, results in an irregular structure and defective chaperone-like function. Proc Nat Acad Sci USA. 1999;96:6137–6142. doi: 10.1073/pnas.96.11.6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Tredici K, Sandmann-Keil D, Schultz C. Nerve cells expressing heatshock proteins in Parkinson’s disease. Acta Neuropathol. 2001;102:449–454. doi: 10.1007/s004010100395. [DOI] [PubMed] [Google Scholar]
- Brennan LA, Lee W, Cowell T, Giblin F, Kantorow M. Deletion of mouse MsrA results in HBO-induced cataract: MsrA repairs mitochondrial cytochrome c. Mol Vis. 2009a;15:985–999. [PMC free article] [PubMed] [Google Scholar]
- Brennan LA, Lee W, Giblin FJ, David LL, Kantorow M. Methionine sulfoxide reductase A (MsrA) restores alpha-crystallin chaperone activity lost upon methionine oxidation. Biochim Biophys Acta. 2009b;1790:1665–1672. doi: 10.1016/j.bbagen.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cáceres-Marzal C, Vaquerizo J, Galán E, Fernández S. Early mitochondrial dysfunction in an infant with Alexander disease. Pediatr Neurol. 2006;35:293–296. doi: 10.1016/j.pediatrneurol.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Chan S, Lui P, Tan P, Yamaguchi R, Moriya T, Yu A, Shao M, Hliang T, Wong S, Tse G. Increased αB-crystallin expression in mammary metaplastic carcinomas. Histopathology. 2011;59:247–255. doi: 10.1111/j.1365-2559.2011.03882.x. [DOI] [PubMed] [Google Scholar]
- Chen Y, Reid G, Simpson R, Truscott R. Molecular evidence for the involvement of α-crystallin in the colouration/crosslinking of crystallins in age-related nuclear cataract. Exp Eye Res. 1997;65:835–840. doi: 10.1006/exer.1997.0393. [DOI] [PubMed] [Google Scholar]
- Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA. Mitochondrial importance in Alzheimer’s, Huntington’s and Parkinson’s diseases. Adv Exp Med Biol. 2012;724:205–21. doi: 10.1007/978-1-4614-0653-2_16. [DOI] [PubMed] [Google Scholar]
- Costa V, Scorrano L. Shaping the role of mitochondria in the pathogenesis of Huntington’s disease. EMBO J. 2012;31:1853–1864. doi: 10.1038/emboj.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabir D, Trojanowski J, Richter-Landsberg C, Lee V, Forman M. Expression of the small heat-shock protein αB-crystallin in tauopathies with glial pathology. Am J Pathol. 2004;164:155–166. doi: 10.1016/s0002-9440(10)63106-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S, Rabin D, Salero, Lederman EP, Temple S, Stern J. Human retinal pigment epithelium cell changes and expression of αB-crystallin: a biomarker for retinalpigment epithelium cell change in age-related macular degeneration. Arch Ophthalmol. 2007;125:641–5. doi: 10.1001/archopht.125.5.641. [DOI] [PubMed] [Google Scholar]
- Dohke T, Wada A, Isono T, Fujii M, Yamamoto T, Tsutamoto T, Horie M. Proteomic Analysis Reveals Significant Alternations of Cardiac Small Heat Shock Protein Expression in Congestive Heart Failure. J Card Fail. 2006;12:77–84. doi: 10.1016/j.cardfail.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Dubin RA, Wawrousek EF, Piatigorsky J. Expression of the murine alpha B-crystallin gene is not restricted to the lens. Mol Cell Biol. 1989;9 (3):1083–91. doi: 10.1128/mcb.9.3.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh JG, Houck SA, Clark JI. Interactive sequences in the stress protein and molecular chaperone human αB crystallin recognize and modulate the assembly of filaments. Int J Biochem. 2007;39:1804–1815. doi: 10.1016/j.biocel.2007.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton TM, Graham BH, Corral-Debrinski M, Shoffner JM, Kaufman AE, Beal MF, Wallace DC. Marked increase in mitochondrial DNA deletion levels in the cerebral cortex of Huntington’s disease patients. Neurology. 1995;45:1879–1883. doi: 10.1212/wnl.45.10.1879. [DOI] [PubMed] [Google Scholar]
- Horwitz J. Alpha-Crystallin can function as a molecular chaperone, Proc. Natl Acad Sci USA. 1992;89:10449–10453. doi: 10.1073/pnas.89.21.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz J. Alpha-crystallin. Exp Eye Res. 2003;76:145–153. doi: 10.1016/s0014-4835(02)00278-6. [DOI] [PubMed] [Google Scholar]
- Houck SA, Clark JI. Dynamic Subunit Exchange and the Regulation of Microtubule Assembly by the Stress Response Protein Human alphaB Crystallin. PloS One. 2010;5:e11795. doi: 10.1371/journal.pone.0011795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Tang D, Yappert MC, Borchman D. Oxidation-induced changes in human lens epithelial cells 2. Mitochondria and the generation of reactive oxygen species. Free Radical Biol Med. 2006;41:926–396. doi: 10.1016/j.freeradbiomed.2006.05.023. [DOI] [PubMed] [Google Scholar]
- Iwaki T, Kume-Iwaki A, Liem R, Goldman J. αB-crystallin is expressed and accumulates in Alexander’s. Cell. 1989;57:71–78. doi: 10.1016/0092-8674(89)90173-6. [DOI] [PubMed] [Google Scholar]
- Iwaki T, Wisniewski T, Iwaki A, Corbin E, Tomokane N, Tateishi J, Goldman J. Accumulation of αB-crystallin in central nervous system glia and neurons in pathologic conditions. Am J Pathol. 1992;140:345–356. [PMC free article] [PubMed] [Google Scholar]
- Jakob U, Gaestel M, Engel K, Buchner J. Small heat shock proteins are molecular chaperones. J Biol Chem. 1993;268:1517–1520. [PubMed] [Google Scholar]
- Jin JK, Whittaker R, Glassy MS, Barlow SB, Gottlieb RA, Glembotski CC. Localization of phosphorylated alphaB-crystallin to heart mitochondria during ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2008;294 (1):H337–44. doi: 10.1152/ajpheart.00881.2007. [DOI] [PubMed] [Google Scholar]
- Kamradt MC, Chen F, Sam S, Cryns VL. The small heat shock protein alpha B-crystallin negatively regulates apoptosis during myogenic differentiation by inhibiting caspase-3 activation. J Biol Chem. 2002;277:38731–38736. doi: 10.1074/jbc.M201770200. [DOI] [PubMed] [Google Scholar]
- Kamradt MC, Lu M, Werner ME, Kwan T, Chen F, Strohecker A, Oshita S, Wilkinson JC, Yu C, Oliver PG, Duckett CS, Buchsbaum DJ, LoBuglio AF, Jordan VC, Cryns VL. The small heat shock protein alpha B-crystallin is a novel inhibitor of TRAIL-induced apoptosis that suppresses the activation of caspase-3. J Biol Chem. 2005;280:11059–11066. doi: 10.1074/jbc.M413382200. [DOI] [PubMed] [Google Scholar]
- Kegel K, Iwaki A, Iwaki T, Goldman JE. Alpha B crystallin protects glial cells from hypertonic stress. Am J Physiol. 1996;270:903–909. doi: 10.1152/ajpcell.1996.270.3.C903. [DOI] [PubMed] [Google Scholar]
- Liang FQ, Godley BF. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp Eye Res. 2003;76:397–403. doi: 10.1016/s0014-4835(03)00023-x. [DOI] [PubMed] [Google Scholar]
- Lin H, Xu H, Liang FQ, Liang H, Gupta P, Havey AN, Boulton ME, Godley B. Mitochondrial DNA damage and repair in RPE associated with aging and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52:3521–3529. doi: 10.1167/iovs.10-6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Schlosser R, Ma W, Dong Z, Feng H, Liu L, Huang X, Liu Y, Li D. Human αA- and αB-crystallins prevent UVA-induced apoptosis through regulation of PKCα, RAF/MEK/ERK and AKT signaling pathways. Exp Eye Res. 2004;79:393–403. doi: 10.1016/j.exer.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Maddala R, Rao VP. alpha-Crystallin localizes to the leading edges of migrating lens epithelial cells. Exp Cell Res. 2005;306 (1):203–15. doi: 10.1016/j.yexcr.2005.01.026. [DOI] [PubMed] [Google Scholar]
- Mao Y, Xiang H, Li D. Human alphaA- and alphaB-crystallins bind to Bax and Bcl-XS to sequester their translocation during staurosporine-induced apoptosis. Cell Death Differ. 2004;11:512–526. doi: 10.1038/sj.cdd.4401384. [DOI] [PubMed] [Google Scholar]
- Mao P, Reddy PH. Is multiple sclerosis a mitochondrial disease? Biochim Biophys Acta. 2010;1802:66–79. doi: 10.1016/j.bbadis.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti MA, Lee W, Cowell TL, Wells TM, Weissbach H, Kantorow M. Silencing of the methionine sulfoxide reductase A gene results in loss of mitochondrial membrane potential and increased ROS production in human lens cells. Exp Eye Res. 2006;83:1281–1286. doi: 10.1016/j.exer.2006.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JL, Mestril R, Hilal-Dandan R, Brunton LL, Dillmann WH. Small Heat Shock Proteins and Protection Against Ischemic Injury in Cardiac Myocytes. Circulation. 1997;96:4343–4348. doi: 10.1161/01.cir.96.12.4343. [DOI] [PubMed] [Google Scholar]
- McGreal RS, Kantorow WL, Chauss DC, Wei J, Brennan LA, Kantorow M. αB-Crystallin/sHSP Protects Cytochrome c and Mitochondrial Function Against Oxidative Stress in Lens and Retinal Cells. Biochim Biophys Acta. 2012;1820:921–930. doi: 10.1016/j.bbagen.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata K, Crabb JW, Hollyfield JG. Crystallin distribution in Bruch’s membrane–choroid complex from AMD and age-matched donor eyes, Exp. Eye Res. 2005;80:821–826. doi: 10.1016/j.exer.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Ousman S, Tomooka B, van Noort J, Wawrousek E, O’Connor K, Hafler D, Sobel R, Robinson W, Steinman L. Protective and therapeutic role for αB-crystallin in autoimmune demyelination. Nature. 2007;448:474–479. doi: 10.1038/nature05935. [DOI] [PubMed] [Google Scholar]
- Pasupuleti N, Matsuyama S, Voss O, Doseff A, Song K, Danielpour D, Nagaraj RH. The anti-apoptotic function of human aA-crystallin is directly related to its chaperone activity. Cell Death Dis. 2010;1:e31. doi: 10.1038/cddis.2010.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renkawek K, de Jong W, Merck K, Frenken C, van Workum F, Bosman G. αB-crystallin is present in reactive glia in Creutzfeldt–Jakob disease. Acta Neuropathol. 1992;83:324–327. doi: 10.1007/BF00296796. [DOI] [PubMed] [Google Scholar]
- Renkawek K, Voorter C, Bosman G, van Workum F, de Jong W. Expression of αB-crystallin in Alzheimer’s disease. Acta Neuropathol. 1994;87:155–160. doi: 10.1007/BF00296185. [DOI] [PubMed] [Google Scholar]
- Schapira AHV. Mitochondrial pathology in Parkinson’s disease, Mt. Sinai J Med. 2011;78:872–881. doi: 10.1002/msj.20303. [DOI] [PubMed] [Google Scholar]
- Sheng B, Wang X, Su B, Lee H, Casadesus G, Perry G, Zhu X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem. 2012;120:419–429. doi: 10.1111/j.1471-4159.2011.07581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T, Dong F, Liou L, Duan Z, Novick A, DiDonato J. Differential Protein Profiling in Renal-Cell Carcinoma. Mol Carcinoge. 2004;40:47–61. doi: 10.1002/mc.20015. [DOI] [PubMed] [Google Scholar]
- Shroff NP, Bera S, Cherian-Shaw M, Abraham EC. Substituted hydrophobic and hydrophilic residues at methionine-68 influence the chaperone-like function of alphaB-crystallin. Mol Cell Biochem. 2001;220:127–133. doi: 10.1023/a:1010834107809. [DOI] [PubMed] [Google Scholar]
- Simon S, Fontaine JM, Martin JL, Sun X, Hoppe AD, Welsh MJ, Benndorf R, Vicart P. Myopathy-associated alphaB-crystallin mutants: abnormal phosphorylation, intracellular location, and interactions with other small heat shock proteins. J Biol Chem. 2007;282 (47):34276–87. doi: 10.1074/jbc.M703267200. [DOI] [PubMed] [Google Scholar]
- Tang Q, Liu Y, Zhub X, Li Y, Zhu J, Zhang J, Feng Z, Guan X. Expression and prognostic significance of the αB-crystallin gene in human hepatocellular carcinoma. Hum Pathol. 2009;40:300–305. doi: 10.1016/j.humpath.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Truscott R, Chen Y, Shaw D. Evidence for the participation of αB-crystallin in human age-related nuclear cataract. Int J Biol Macromol. 1998;22:321–330. doi: 10.1016/s0141-8130(98)00030-0. [DOI] [PubMed] [Google Scholar]
- Wang AL, Lukas TJ, Yuan M, Neufeld AH. Increased mitochondrial DNA damage and down-regulation of DNA repair enzymes in aged rodent retinal pigment epithelium and choroid. Mol Vis. 2008;14:644–651. [PMC free article] [PubMed] [Google Scholar]
- Yaung J, Jin M, Barron E, Spee C, Wawrousek EF, Kannan R, Hinton DR. alpha-Crystallin distribution in retinal pigment epithelium and effect of gene knockouts on sensitivity to oxidative stress. Mol Vis. 2007;13:566–577. [PMC free article] [PubMed] [Google Scholar]
- Yaung J, Kannanb R, Wawrousek E, Speec C, Sreekumarc P, Hinton D. Exacerbation of retinal degeneration in the absence of alpha crystallins in an in vivo model of chemically induced hypoxia. Exp Eye Res. 2008;86:355–365. doi: 10.1016/j.exer.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel C, Chamrad DC, Priller J, Woodman B, Meyer HE, Bates GP, Klose J. Alterations in the mouse and human proteome caused by Huntington’s disease. Mol Cell Proteomics. 2002;1 (5):366–75. doi: 10.1074/mcp.m200016-mcp200. [DOI] [PubMed] [Google Scholar]
- Zhu X, Perry G, Moreira PI, Aliev G, Cash AD, Hirai K, Smith MA. Mitochondrial abnormalities and oxidative imbalance in Alzheimer disease. J Alzheimer’s Dis. 2006;9:147–153. doi: 10.3233/jad-2006-9207. [DOI] [PubMed] [Google Scholar]