

Summary
Protein C is a vitamin K-dependent serine protease zymogen in plasma which upon activation to activated protein C (APC) by thrombin down-regulates the clotting cascade by limited proteolysis of the procoagulant cofactors Va and VIIIa. In addition to its anticoagulant activity, APC also exhibits potent cytoprotective and anti-inflammatory activities. While the anticoagulant activity of APC is enhanced by the cofactor function of protein S on membrane phospholipids, the cytoprotective intracellular signaling activity of APC requires complex formation with endothelial protein C receptor (EPCR) expressed on the vascular endothelium. Two natural variants of APC [Arg-147 to Trp substitution (R147W) and Lys-150 deletion (K150del)] have been identified in the Chinese population as hotspot mutants occurring with high frequencies of 27.8% and 13.9%, respectively, among 36 protein C deficient subjects. The affected individuals exhibit variable thrombotic tendencies. To understand the underlying cause of the thrombotic phenotype in these patients, we expressed these two protein C variants in mammalian cells and characterized their anticoagulant and anti-inflammatory properties using established in vitro and cellular assays. Our results suggest that both R147W and K150del variants have normal amidolytic and proteolytic activities in the absence of cofactors. However, the R147W mutant exhibits ~3 times lower affinity for binding to EPCR and the K150del variant has ~2–3-fold impaired anticoagulant activity in the presence of protein S. These results provide some insight into the possible pathogenic mechanism of protein C deficiency in Chinese patients carrying these mutations.
Introduction
Protein C is a vitamin K-dependent serine protease zymogen in plasma playing a critical role in regulation of thrombin generation (1, 2). The mature protein C polypeptide, synthesized by the liver, has a multi-domain structure comprised of an N-terminal γ-carboxyglutamic acid (Gla) domain (residues 1–45), two epidermal growth factor (EGF)-like domains (residues 46–136), a linking peptide (residues 137–157) between the light chain and the heavy chain, an activation peptide (residues 158–169), and a C-terminal serine protease domain (residues 170–419) which contains the active-site catalytic triad residues Ser-360, Asp-257 and His-211 (Ser-195, Asp-102 and His-57 in the chymotrypsinogen numbering system) (3–5). Protein C is activated by the thrombin-thrombomodulin (TM) complex on the surface of endothelial cells to activated protein C (APC) (1). APC in complex with its cofactor protein S inactivates procoagulant cofactors Va (FVa) and VIIIa (FVIIIa) by limited proteolysis, thereby down-regulating thrombin generation during the blood coagulation process (1, 2). In addition to its anticoagulant activity, APC also has direct cytoprotective and anti-inflammatory activities (6). The latter properties of APC are mediated by the protease forming a complex with endothelial protein C/APC receptor (EPCR) expressed on the vascular endothelium (6–8). EPCR is a type 1 transmembrane receptor, which also promotes the activation of protein C by the thrombin-TM complex (9). Both the zymogen and activated forms of protein C bind to EPCR via their Gla domains with identical affinities (10, 11). When APC is bound to EPCR, it activates protease-activated receptor 1 (PAR-1) to elicit cytoprotective and anti-inflammatory signaling responses in vascular endothelial cells (6).
Protein C deficiency exhibits an autosomal dominant pattern of inheritance and is associated with an increased risk of venous thromboembolism (VTE) (12). Results from several studies have indicated that protein C is one of the most important risk factors for venous thrombophilia in Asian patients (13–15). The prevalence of protein C deficiency in Japanese patients with VTE has been found to be three times higher than that of Caucasian patients (13). We recently identified 25 mutations of the protein C gene (PROC) in 36 unrelated probands with protein C deficiency in the Chinese population (16–19). Out of the 25 mutations, the two R147W and K150del variants were found to be the most common, occurring with frequencies of 27.8% and 13.9%, respectively (16). However, inconsistent results have been reported about the prevalence of the two mutations in the general population. One study did not find any R147W variant among 3493 healthy Chinese adults (17), yet another study reported a prevalence rate 0.87% (9 out of 1031 control subjects) for the same mutation (20). The result of the latter study is consistent with another study reporting a prevalence rate of 0.85% (11 out 1292 healthy subjects) for R147W in the Taiwanese Chinese (21). A very low prevalence 0.028% has been reported for K150del in general Chinese population (17).
Both Arg147 and Lys150 are located on a 21-residue peptide (residues 137–157) in the C terminus of the protein C light chain, which connects the EGF2 domain to the activation peptide (residues 158–169) of the zymogen and it is also linked to the heavy chain through a disulfide bond between Cys141 and Cys277 (22). This peptide contains 8 basic Lys and Arg residues but only one acidic residue (Glu149), rendering it highly basic. The peptide fragment from the C-terminal region of the light chain (residues 147–157) is missing in the x-ray crystal structure of human APC due to scattered electron density (22). The functional importance of this region of APC can be gleaned from a previous study showing that a protein C synthetic peptide corresponding to residues 142 to 155 blocks the anticoagulant activity of APC, possibly suggesting that this region constitutes an interactive-site for FVa in the anticoagulant pathway (23). In another study, Mosnier et al. reported that a Glu149 to Ala substitution (E149A) mutant of human APC, while exhibiting increased anticoagulant activity, has lost its cytoprotective function which suggests a role for this region of the protease in the anti-inflammatory pathway (24).
In this study, we investigated the molecular basis of the thrombophilic phenotype of Chinese patients carrying the C-terminal light chain R147W and K150del hotspot mutants by expressing both mutants in mammalian cells. Following purification to homogeneity, we characterized the zymogenic and enzymatic properties of both mutants using established in vitro and cell-based assay systems. The results demonstrated that both mutant proteases retain their intact active-sites and thus catalytic activities, however, the capacity of the R147W and K150del mutants for interaction with the cofactors EPCR and protein S, respectively, had been compromised. The cofactor-dependent impairment in the activity of these mutants may contribute to the thrombophilic phenotype of patients carrying these mutations.
Materials and methods
Construction, expression, and purification of recombinant proteins
The expression and purification of recombinant wild-type protein C in human embryonic kidney (HEK-293) cells have been described previously (25). The Arg147 to Trp substitution (R147W) and the Lys150 deletion (K150del) mutants of protein C were constructed by PCR site-directed mutagenesis methods and the mutant proteins were expressed in HEK-293 cells using the same expression/purification vector system. The protein C derivatives were isolated from 20–60 L cell culture supernatants by a combination of immunoaffinity and ion exchange chromatography using the HPC4 monoclonal antibody immobilized on Affi-gel 10 (Bio-Rad) and a Mono Q column (Pharmacia), respectively, as described (25). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed on a 10% polyacrylamide gel and stained with Coomassie Blue R-250 to verify homogeneity and quality of all protein C derivatives under non-reducing conditions. The zymogens (0.5–1.0 mg) were converted to activated protein C (APC) by thrombin (25 μg) in 0.1 M NaCl, 0.02 M Tris-HCl, pH 7.4 (TBS) containing 5 mM EDTA for 2 h at 37°C as described (25). The APC derivatives were separated from thrombin using an FPLC Mono Q column developed with a 40 mL linear gradient from 0.1 to 1.0 M NaCl. The properly γ-carboxylated APC derivatives were eluted at ~0.45 M NaCl as described (25). The concentrations of APC derivatives were determined from the absorbance at 280 nm assuming a molecular mass of 56 kDa and an extinction coefficient (E1%1cm) of 14.5, and by stoichiometric titration of enzymes with known concentrations of recombinant protein C inhibitor (PCI) as described (25). The recombinant proteins including PCI (25) and soluble TM (26) were expressed and purified as described by the cited methods.
Human plasma proteins factor Xa (FXa), prothrombin, FVa and antithrombin (AT) were purchased from Hematologic Technologies Inc. (Essex Junction, VT). Human protein S was purchased from Enzyme Research Laboratories (South Bend, IN). Phospholipid vesicles containing 80% phosphatidylcholine and 20% phosphatidylserine (PC/PS) were prepared as described (27). The aPTT reagent (Alexin) was purchased from Trinity Biotech (St. Louis, MO). Normal pooled plasma was purchased from George King Bio-Medical, Inc. (Overland Park, KS). Human protein S deficient plasma was purchased from Affinity Biological Inc. (Ontario, Canada). Factor Va Leiden, purified from the plasma of a homozygous patient was a generous gift from Dr. Charles Esmon (Oklahoma Medical Research Foundation, Oklahoma City, OK). The chromogenic substrates, Spectrozyme PCa (SpPCa) and Spectrozyme FXa (SpFXa) were purchased from American Diagnostica (Greenwich, CT) and S2238 was purchased from Kabi Pharmacia/Chromogenix (Franklin, OH).
Protein C activation
The initial rate of wild-type and mutant protein C activation by thrombin was measured in both the absence and presence of TM as described (28). In the absence of TM, the time course of protein C activation by thrombin (10 nM) was studied at room temperature in TBS buffer containing 1 mg/mL bovine serum albumin (BSA), 0.1% polyethylene glycol (PEG) 8000 in the presence of 1 mM EDTA in 96-well assay plates. At different time intervals, thrombin activity was quenched by 0.5 μM AT and the rate of protein C activation was measured from the cleavage rate of SpPCa at 405 nm by a Vmax Kinetic Microplate Reader (Molecular Devices, Menlo Park, CA) as described (28). The experimental conditions in the presence of TM were the same with the exception that the activation by thrombin (1.0 nM) was carried out in the presence of a saturating concentration of TM (50 nM) and 2.5 mM Ca2+. The concentration of APC in reaction mixtures was determined by reference to a standard curve which was prepared by the total activation of protein C derivatives with excess thrombin at the time of experiments.
Cleavage of chromogenic substrates
The steady-state kinetics of hydrolysis of SpPCa (7.8–1000 μM) by APC derivatives (4 nM) was measured in TBS containing 2.5mM Ca2+, 1.0 mg/mL BSA and 0.1% PEG 8000 (TBS/Ca2+) at 405 nm at room temperature in a Vmax Kinetic Microplate Reader as described above. The Km and kcat values for the substrate hydrolysis were calculated from the Michaelis-Menten equation and the catalytic efficiencies were expressed as ratios of kcat/Km as described (25).
Inhibition by plasma inhibitors
The reactivity of the APC derivatives with plasma inhibitors was assessed by incubating the proteases at a final concentration of 20 nM APC with normal plasma at 37 °C. At indicated time points, aliquots were removed, and the residual amidolytic activity was determined using the chromogenic substrate SpPCa as described above. The half-life of each protease was calculated based on the time required to observe 50% decline in the amidolytic activity. The reactivity of APC derivatives with the plasma serpins antithrombin, PCI, and α1-antithrypsin was also analyzed in the purified system using an amidolytic activity assay as described (29).
Anticoagulant activity
The anticoagulant activity of APC derivatives was evaluated in both purified and plasma-based assay systems as described (25). Briefly, FVa (2.5 nM) was incubated with increasing concentration of either wild-type or mutant APC (0–5 nM) on 25 μM PC/PS vesicles in TBS/Ca2+. Following 10 min incubation at room temperature, the remaining FVa activity was determined in a prothrombinase assay from the FVa-catalyzed prothrombin activation by FXa as described (25). The prothrombinase assay was carried out for 30 s with 1 μM prothrombin and 1 nM FXa. The remaining activity of FVa was determined from the decrease of the FVa-dependent rate of thrombin generation as monitored by an amidolytic activity assay using 100 μM S2238. The inactivation of FVa by increasing concentrations of APC was also monitored in the presence of protein S (110 nM) as described above with the exception that the time of incubation was decreased to 1 min. The FVa inactivation assay was also carried out with a fixed concentration of APC (1 nM) in the presence of varying concentrations of protein S (0–60 nM) using the same methods. The time course of inactivation of FVa and FVa Leiden (1 nM) in both the absence and presence of protein S (110 nM) was monitored by both wild-type APC and APC-K150del (0.2 nM each) using the same experimental methods.
The anticoagulant activity of wild-type APC and APC-K150del toward FVIIIa was evaluated using purified human FVIIIa as described (30). Briefly, FVIIIa (1 nM) was incubated with wild-type or mutant APC (2.5 nM) in the absence or presence of protein S (110 nM) on PC/PS vesicles (25 μM) at room temperature in TBS/Ca2+. At different time intervals (0–30 min), the remaining FVIIIa activity was determined in an intrinsic Tenase assay from the FVIIIa-dependent factor X activation by factor IXa as described (30). Factor X activation with the intrinsic Tenase complex was carried out for 1 min with excess factor X (0.5 μM) and saturating factor IXa (10 nM) on PC/PS (25 μM) vesicles at room temperature. Finally, the remaining activity of FVIIIa was determined from a decrease in the rate of factor Xa generation as monitored by an amidolytic activity assay using 200 μM SpFXa.
The anticoagulant activities were also evaluated in plasma by an aPTT assay using a STart 4 fibrinometer (Diagnostica/Stago, Asnieres, France). Briefly, 0.05 ml TBS containing 0–20 nM final concentrations of APC derivatives were incubated with a mixture of 0.05 mL of normal pooled plasma plus 0.05 mL of the aPTT reagent (Alexin) for 5 min before the initiation of clotting by the addition of 0.05 mL of 35 mM CaCl2 at 37 °C as described (25). The same aPTT assay was used to evaluate the anticoagulant activity of the APC-K150del mutant using protein S deficient plasma with or without supplementing it with a near physiological concentration of purified human protein S (10 μg/mL).
Interaction with EPCR
The affinity of protein C derivatives for interaction with EPCR was evaluated by an ELISA-based binding assay using the HPC4-tagged recombinant soluble EPCR (sEPCR) (obtained from Dr. Esmon from Oklahoma Medical Research Foundation) as described (31). Briefly, 96-well flat bottom microtiter plates were coated with the HPC4 monoclonal antibody in TBS containing 1 mM CaCl2 overnight at 4 °C. After washing the plates three times with the same TBS buffer containing 0.05% Tween 20, they were incubated with 2% BSA in TBS/Ca2+ for 2 h at room temperature. The plates were washed again as described above and then incubated with sEPCR (0.5 μM in TBS/Ca2+ containing 0.1% BSA) for 1 h. The plates were rinsed and incubated with wild-type or mutant APC (7.8–500 nM) diluted in the same Ca2+ containing TBS buffer for 1 h. After the plates were rinsed again, they were incubated with a goat anti-protein C polyclonal antibody (1 μg/mL) for 1 h. The plates were then washed and incubated with rabbit anti-goat IgG (KPL, MD, 1:1000) for 1 h. The plates were developed with 2, 2-azino-di (3-ethylbenzthiazoline-6-sulfonate) (KPL, Gaithersburg, MD). Colorimetric analysis was performed by measuring the absorbance values at 405 nm as described above. All treatments were performed in duplicate and repeated at least twice.
Endothelial cell permeability assay
The EPCR-dependent capacity of APC derivatives to elicit cytoprotective signaling activity was evaluated by a permeability assay using transformed human umbilical vein endothelial cells (EA.hy926) which were obtained from Dr. C. Edgell (University of North Carolina at Chapel Hill, NC). These cells are commonly used for the analysis of the EPCR-dependent signaling function of APC (31). The endothelial cell permeability in response to thrombin (10 nM for 10 min) following treatment with APC derivatives (10–20 nM plus 1 units/mL hirudin for 3 h) was quantitated by spectrophotometric measurement of the flux of Evans blue-bound albumin across functional cell monolayers using a modified 2-compartment chamber model as described (31). Results were expressed as mean ±SD and all experiments were repeated at least three times.
A variation of the same permeability assay was used to evaluate the interaction of the protein C derivatives with EPCR on endothelial cells. In this case, endothelial cells were initially incubated with increasing concentrations of protein C derivatives (to occupy the cell surface EPCR) followed by their incubation with thrombin (1 nM) for three hours. It has been demonstrated that under these experimental conditions, the occupancy of EPCR confers a PAR-1-dependent protective activity for 1–2 nM thrombin (31). The endothelial cell permeability was then induced by a high concentration of thrombin (10 nM for 10 min) as described above and the EPCR affinities of protein C derivatives were evaluated.
Statistical analysis
Results are expressed as mean ±SD, and t-Tests (paired or independent) were used to assess data. Differences were considered statistically significant at p values of <0.05. All experiments were repeated at least three times.
Results
Expression and purification of protein C derivatives
All protein C derivatives were expressed in HEK-293 cells and purified to homogeneity on the HPC4 immunoaffinity column as described (25). The fully γ-carboxylated zymogens were isolated using a Mono Q ion exchange column as described under “Materials and methods”. For the preparation of the activated forms of recombinant protein C derivatives, the HPC4 immunoaffinity purified zymogens were treated with thrombin before applying the proteins on the Mono Q column as described (25). SDS-PAGE analysis indicated that the recombinant proteins have been purified to homogeneity and that both wild-type and variants migrate with expected apparent molecular masses of ~55–60 kDa under non-reducing conditions (data not shown). The active-site concentrations of APC derivatives were calculated by an amidolytic activity assay and active-site titrations with known concentrations of PCI as described (25). These concentrations were within 90–100% of those expected based on the zymogen concentrations as determined from the absorbance at 280 nm, further confirming that the recombinant proteins have been purified to homogeneity.
Protein C activation
The time course of the initial rate of activation of protein C derivatives by thrombin in both the absence and presence of thrombomodulin (TM) was evaluated. As presented in Fig. 1, thrombin activated both R147W and K150del variants with a rate that was essentially identical to the rate of wild-type protein C activation by the protease in both the absence (Fig. 1A) and presence of TM and Ca2+ (Fig. 1B). Similarly, thrombin (1 nM) activated a physiological concentration of both zymogen mutants (~80 nM) with a normal rate on HUVECs which express both TM and EPCR on their surfaces (data not shown). These results suggest that an altered rate of protein C activation by thrombin is not an explanation for the higher incidence of thrombosis observed in selected Chinese patients carrying these mutations.
Figure 1.

Initial rate of protein C activation by thrombin. (A) In the absence of TM, protein C (1 μM) was incubated with 10 nM thrombin in TBS containing 1.0 mM EDTA. At indicated time intervals, the activity of thrombin was inhibited by antithrombin and the rate of APC generation was determined by an amidolytic activity as described in “Materials and methods”. (B) In the presence of soluble TM (50 nM), the time course of protein C activation by 1 nM thrombin was carried out in TBS/Ca2+ and the rate of APC generation was determined as described above. The symbols in both panels are: wild-type protein C (○), protein C R147W (●), protein C K150del (□). Data are derived from at least three independent measurements (±SD).
Amidolytic activity
The kinetic parameters for the hydrolysis of the chromogenic substrate SpPCa by the APC derivatives are presented in Table 1. Relative to cleavage by the wild-type APC, a similar amidolytic activity was observed for both R147W and K150del variants of APC, indicating that the mutagenesis of these residues did not adversely affect the folding and/or the reactivity of catalytic residues. These results were not surprising since neither one of the mutated residues is located on the catalytic domain of the protease.
Table 1.
Kinetic constants for the cleavages of Spectrozyme PCa by the APC derivatives.
| kcat (s−1) | Km (μM) | kcat/Km (μM−1 s−1) | |
|---|---|---|---|
| APC-WT | 28.7 ± 2.2 | 173.2 ± 14.4 | 0.16 ± 0.04 |
| APC-R147W | 24.4 ± 1.0 | 184.0 ± 15.6 | 0.13 ± 0.03 |
| APC-K150del | 28.9 ± 0.59 | 176.7 ± 9.4 | 0.16 ± 0.03 |
The steady-state hydrolysis of Spectrozyme PCa (15–2000 μM) by the APC derivatives (4 nM) was measured in TBS containing 2.5 mM Ca2+, 1.0 mg/ml BSA and 0.1% PEG 8000 as described under “Materials and methods”.
Anticoagulant activity
The anticoagulant activity of APC derivatives was evaluated in both the absence and presence of protein S. As presented in Fig. 2, the APC concentration dependence of FVa inactivation in the absence (Fig. 2A) and presence of protein S (Fig. 2B) indicated that the R147W mutant has a normal anticoagulant activity toward FVa in both the absence and presence of a saturating concentration of protein S. However, while the K150del mutant exhibited a normal proteolytic activity toward FVa in the absence of protein S, the mutant had a relatively low activity in the presence of protein S. Relative to wild-type APC, the activity of the K150del mutant toward FVa was impaired ~2-fold in the presence of protein S (Fig. 2B).
Figure 2.

Factor Va degradation by wild-type and mutant APC derivatives in the absence and presence of protein S. (A) The degradation of human FVa (2.5 nM) by increasing concentrations of APC wild-type (○), APC-R147W (●) and APC-K150del (□) was carried out on PC/PS vesicles (25 μM) in TBS containing 1.0 mg/mL BSA, 0.1% PEG 8000 and 2.5 mM Ca2+ in a 96-well assay plate. Following 10 min incubation at room temperature, the remaining cofactor activity of FVa was determined by a prothrombinase assay as described under “Materials and methods”. (B) The same as A, except that the APC concentration dependence of FVa degradation was carried out in the presence of protein S (110 nM) for 1 min. The data in both panels are averages of at least three independent measurements (±SD) with *p<0.05 in panel B.
APC must sequentially cleave at least two peptide bonds after Arg506 and Arg306 to inactivate FVa (32). It has been demonstrated that the cleavage at the Arg306 site of FVa requires membrane and occurs at a slower rate than the cleavage at the Arg506 site (32). Noting that the Arg506 site is mutated to a Gln in the FVa Leiden, we decided to compare the cleavage of FVa Leiden with that of wild-type FVa in order to determine the basis for the protein S-dependent decrease in the anticoagulant activity of the APC-K150del mutant. The time course of the inactivation of wild-type FVa and FVa Leiden by APC and APC-K150del is presented in Fig. 3. In agreement with results presented above, a statistically significant protein S-dependent decrease in the anti-FVa activity of the APC-K150del mutant was observed with wild-type FVa (Fig. 3A), however, both wild-type and mutant APC inactivated FVa Leiden in the presence of protein S with essentially identical rates (Fig. 3B), suggesting that the protein S-dependent decrease in the anticoagulant activity of the APC-K150del mutant is solely due its poorer recognition of the Arg506 cleavage site.
Figure 3.

Time course of wild-type FVa and FVa Leiden degradation by wild-type APC and APC-K150del in the presence of protein S. (A) The time course of degradation of human FVa (1 nM) by wild-type APC (○) and APC-K150del (●) (0.2 nM each) was carried out in the presence of protein S (110 nM) on PC/PS vesicles (25 μM) in TBS containing 1.0 mg/mL BSA, 0.1% PEG 8000 and 2.5 mM Ca2+ in a 96-well assay plate at room temperature. The remaining cofactor activity of FVa was determined by a prothrombinase assay as described under “Materials and methods”. (B) The same as A, except that instead of wild-type FVa, FVa Leiden was used in the cofactor degradation study. The data in both panels are averages of at least three independent measurements (±SD). Unpaired t-test in panel A suggests statistical significance with *p<0.05.
Next, we evaluated the anticoagulant activity of APC derivatives toward FVIIIa in the purified system (Fig. 4). Interestingly, similar to results with FVa, the APC-K150del mutant exhibited a statistically significant lower activity toward FVIIIa only in the presence of protein S (Fig. 4B), supporting our hypothesis that the anticoagulant activity of the APC mutant is only impaired in the presence of the cofactor.
Figure 4.
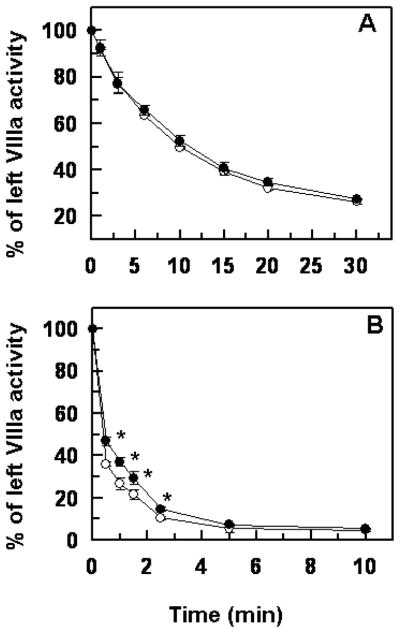
Time course of degradation of FVIIIa by wild-type APC and APC-K150del in the absence and presence of protein S. (A) The time course of degradation of human FVIIIa (1 nM) by wild-type APC (○) and APC-K150del (●) (2.5 nM each) was carried out on PC/PS vesicles (25 μM) in TBS containing 1.0 mg/mL BSA, 0.1% PEG 8000 and 2.5 mM Ca2+ in a 96-well assay plate at room temperature. The remaining cofactor activity of FVIIIa was determined by an intrinsic Tenase assay as described under “Materials and methods”. (B) The same as A, except that the time course of FVIIIa degradation by APC derivatives was carried out in the presence of protein S (110 nM). The data in both panels are averages of at least three independent measurements (±SD). Unpaired t-test in panel B suggests statistical significance with *p<0.05.
Consistent with results in the purified system, the R147W APC mutant exhibited normal anticoagulation activity in the plasma-based aPTT assay, thus prolonging the clotting time of normal plasma with an efficiency that was essentially identical to that observed with wild-type APC (Fig. 5). However, similar to results in the purified system, the K150del mutant showed significantly reduced anticoagulant activity in the aPTT assay (Fig. 5). To provide further support for the hypothesis that the protein S-dependent anticoagulant activity of the APC-K150del mutant has been impaired, the activity of the APC-K150del mutant was evaluated in a clotting assay using human protein S deficient plasma. The results presented in Fig. 6 demonstrate that both wild-type and APC-K150del exhibit identical anticoagulant activities in the protein S deficient plasma in the absence of added protein S (Fig. 6A). In agreement with results presented above, the APC-K150del mutant exhibited significantly lower clotting activity if the protein S deficient plasma was supplemented with purified protein S (Fig. 6B). Taken together, these results clearly suggest that Lys150 may play a protein S cofactor-dependent role in the anticoagulant function of APC.
Figure 5.

Plasma clotting activity of wild-type and mutant APC derivatives. The clotting activities of wild-type APC (○), APC-R147W (●) and APC-K150del (□) were determined as a function of increasing concentrations of proteases (0–20 nM) at 37 °C as described under “Materials and methods”. Data are derived from at least three independent measurements (±SD).
Figure 6.

Clotting activity of APC and APC-K150del in protein S deficient plasma. (A) The clotting activities of wild-type APC (○) and APC-K150del (●) in protein S deficient plasma were determined as a function of increasing concentrations of proteases (0–20 nM) at 37 °C as described under “Materials and methods”. (B) The same as A except that the plasma was supplemented with purified human protein S (10 μg/mL). Data are derived from at least three independent measurements (±SD). Unpaired t-test in panel B suggests statistical significance with *p<0.05.
Inhibition by plasma inhibitors
To evaluate the extent of the reactivity of APC derivatives with plasma inhibitors, the variant proteases were added to pre-warmed plasma at a final concentration of 20 nM and incubated at 37 °C followed by the analysis of the time course of the residual APC amidolytic activities using the chromogenic substrate SpPCa. As presented in Fig. 7, wild-type APC and both variants exhibited essentially identical reactivity with plasma inhibitors at all time points examined. In agreement with these results, inhibition studies with the major APC-specific plasma inhibitors in plasma (PCI and α1-proteinase inhibitor) in the purified system suggested that the reactivity of the APC variants with plasma inhibitors has not been altered (data not presented).
Figure 7.

Time course of inhibition of APC derivatives by human plasma. APC derivatives (20 nM each) were incubated with normal plasma at 37 °C. At indicated time points, aliquots were removed, and the residual amidolytic activity was determined by the chromogenic substrate SpPCa as described in “Materials and methods”. The symbols are: APC wild-type (○), APC-R147W (●), and APC-K150del (□). Data are derived from at least three independent measurements (±SD).
Interaction with EPCR
The capacity of APC derivatives to interact with EPCR was evaluated by an ELISA-based binding assay. As presented in Fig. 8A, both wild-type and the K150del mutant interacted with EPCR with similar affinity, however, the R147W mutant exhibited ~3-fold lower affinity for EPCR. Thus, in contrast to a dissociation constant of ~30 nM for both wild-type and K150del APC, this value was increased to ~100 nM for the R147W mutant. Previous studies have reported a similar affinity for the interaction of APC with EPCR using other binding approaches (10, 11), supporting the reliability of our results.
Figure 8.

Interaction with EPCR and signaling activities of APC derivatives. (A) An ELISA-based binding assay using the monoclonal antibody, HPC4 (1 μg/mL), and a goat polyclonal anti-protein C antibody was used to measure the affinity of APC derivatives (APC wild-type (○), APC-R147W (●), and APC-K150del (□)) for interaction with the HPC4-tagged soluble EPCR (0.5 μM). (B) The inhibition of thrombin-induced permeability (10 nM for 10 min) by APC derivatives (20 nM each) was monitored from the flux of Evans blue-bound albumin across endothelial cells as described under “Materials and methods”. Cont (−) represents negative control not incubated with thrombin. *p< 0.05 vs. thrombin (Th).
Intracellular signaling activity
It is known that APC elicits cytoprotective signaling responses in endothelial cells in response to thrombin and proinflammatory cytokines (6). The endothelial cell permeability assay has been frequently used to analyze the intracellular signaling activity of APC (33). Thus, we evaluated the signaling activity of wild-type APC and the R147W and K150del derivatives in a thrombin-mediated permeability assay as described (31, 33). The results presented in Fig. 8B suggest that both APC variants exhibit normal signaling activities in the permeability assay. The minimum concentration of APC which was required to elicit an optimal protective response in this assay was determined to be ~10 nM as demonstrated in past studies. The protective activity of APC in this assay required interaction of the protease with EPCR since a function-blocking anti-EPCR antibody blocked the protective activity of APC (data not shown). In light of the findings that this endothelial cell permeability assay was not sensitive to observing the loss of interaction of APC-R147W with EPCR as was observed in the direct ELISA-based binding assay, we developed a more sensitive assay to evaluate the interaction of APC derivatives with the endothelial cell surface EPCR. We took advantage of the observation that the occupancy of EPCR on vascular endothelial cells by the zymogen protein C switches the PAR-1-dependent permeability-enhancing effect of thrombin to a protective permeability-lowering effect (30). Thus, we incubated endothelial cells with increasing concentrations of the protein C zymogen derivatives before activating the cell surface PAR-1 with 1 nM thrombin. As shown in Fig. 9A, in the absence of EPCR occupancy, thrombin alone (1 nM) potently enhanced the permeability of endothelial cells. However, prior treatment of the endothelial cell monolayer with protein C derivatives resulted in a thrombin-mediated protective effect that reached its optimal level at 50 nM protein C with all three variants. However, while the K150del variant exhibited a concentration-dependent activity similar to that observed for wild-type protein C, the R147W variant was less effective in switching the EPCR-dependent protective activity of thrombin (Fig. 9A). Thus, instead of ~80% relative protective effect with 20 nM of both wild-type and K150del protein C zymogens, ~50% protective effect was observed with the same concentration of protein C R147W (Fig. 9B). These results support the direct ELISA-based binding data that the affinity of the APC-R147W variant for binding to EPCR has been impaired (see Discussion for more details).
Figure 9.

Cytoprotective signaling activity of protein C derivatives in the permeability assay. (A) Confluent endothelial cell monolayers were treated with increasing concentrations of the zymogen protein C derivatives for 30 min followed by incubation of the cells with 1 nM thrombin. The permeability was induced with 10 nM thrombin (Th) for 10 min and the inhibition of cell permeability was monitored from the flux of Evans blue-bound albumin across endothelial cells as described under “Materials and methods”. Cont (−) represents negative control not incubated with thrombin. (B) The plot of maximal protection (inhibition of permeability) as a function of different concentrations of protein C derivatives derived from the data in panel A. The symbols in panel B are: wild-type protein C (○), protein C R147W (●), protein C K150del (□). Data are derived from two independent measurements (±SD).
Discussion
There is growing evidence that protein C deficiency is one of the most important risk factors for VTE in the Asian population (13–15). A study in Japan indicated that the frequency of mutations in the PROC gene was almost three times higher than that of Caucasian patients (13). Moreover, the mutation profile of the PROC gene is characterized by a founder effect in a given population (34, 35). In previous studies, we have identified two significant ethnicity-specific mutations: Arg147Trp and Lys150del which occurred with 10 times (27.8%) and 5 times (13.9%) higher frequency, respectively, in 36 unrelated subjects with protein C deficiency in the Chinese population (16–19) (see the Supplemental Table).
The Arg147Trp mutation in the PROC gene was first identified in an American of Hawaiian descent (36) and two studies have reported to be the most common mutation in both the normal Chinese population and patients with DVT (20, 21), though another study screening healthy individuals for protein C polymorphism did not find any R147W mutation among 3493 subjects (17). The basis for the discrepancy between these studies is not known. In our studies, all ten of the subjects with the Arg147Trp mutation suffered from DVT (Suppl. Table). Both protein C antigen and functional levels in the six heterozygotes (patients 1–6, Suppl. Table) were lower than the normal levels, suggesting that the mutation may partly impair protein C synthesis (16). However, 60% of these patients (6/10) had the mutation in combination with other protein C deficiencies (3 patients), protein S deficiency (one patient) or acquired precipitating factors (2 patients), indicating that the Arg147Trp mutation is associated with DVT but is a relatively weak risk factor and that the lower antigen and activity levels may not be solely due to Arg147Trp mutation (16) (Suppl. Table). A recent study reported a relatively normal antigen level for subjects carrying solely this mutation in the heterozygous state, suggesting that R147W may primarily be associated with type II protein C deficiency (20). The characterization of the recombinant form of this mutant in the purified system suggests that the Arg147 to Trp substitution has no adverse effect on the folding and/or the reactivity of the catalytic pocket of the enzyme. Moreover, the mutant zymogen was activated normally by the thrombin-TM complex and its activated form also exhibited a wild-type like anticoagulant activity in both plasma-based and purified assay systems in both the absence and presence of protein S. The only defect that could be identified in the R147W mutant was its ~3-fold loss of binding affinity for EPCR (Fig. 8A). Thus, the molecular basis for the defect due to this mutation may have an inflammatory origin in the affected DVT patients. Nevertheless, our attempt to establish this concept for the APC R147W mutant in an assay reporting the anti-inflammatory signaling activity of the protease in a thrombin-induced permeability assay was not successful since the cytoprotective signaling activity of the mutant was normal when compared to recombinant wild-type APC on HUVECs (Fig. 8B). It should however be noted that HUVECs express large quantities of EPCR on their surfaces and thus they may not be sensitive for assessing a modest EPCR binding defect using the permeability assay which requires both EPCR binding and PAR-1 cleavage (6). In the in vitro cell culture systems, the optimal signaling effect of APC requires 10–20 nM APC to bind EPCR and to cleave threshold levels of PAR-1 in order to observe the signaling response. However, the in vivo anti-inflammatory effect of APC is mediated by sub-nanomolar concentrations of the protease on the capillary endothelial cells which abundantly express TM, but much lower levels of EPCR (37). Thus, the anti-inflammatory activity of APC-R147W on capillary endothelial cells with limited EPCR may be compromised in particular if patients have underlying inflammation which can also lead to down-regulation of EPCR on these cells (38). In addition to APC, the occupancy of EPCR by the zymogen protein C is also known to elicit anti-inflammatory responses (31, 39). Indeed in another assay reporting the anti-inflammatory effect of EPCR occupancy by the protein C zymogen, the R147W variant was much less effective than wild-type protein C (50% vs. 80% at 20 nM) in eliciting a protective effect by thrombin (Fig. 9). Taken together, these results predict a defect in the EPCR-dependent anti-inflammatory mechanism of the APC-R147W mutant in patients carrying this mutation. The observation that a Glu149 to Ala substitution (E149A) mutant of human APC has lost its cytoprotective activity while retaining its normal anticoagulant activity (24) supports the hypothesis that the C-terminal light chain residues of APC may be critical for the anti-inflammatory function of APC. It should, however, be noted that although the activation of the R147W mutant by thrombin was found to be normal on HUVECs, the possibility that the activation of this protein C mutant on capillary endothelial cells has been impaired cannot be ruled out.
The Lys150del mutation was first reported in a Japanese patient and it was also one of the most prevalent mutations in the Japanese population (40, 41). The mutation is thought to be associated with type II protein C deficiency and causes the loss of anticoagulant activity but not amidolytic activity (41). In our clinical studies, Lys150del was identified as the second hotspot mutation in five subjects, three of whom experienced VTE, and had only the heterozygous mutation without any other identifiable genetic or environmental risk factors; one VTE patient had the mutation combined with two other heterozygous protein C mutations, and the last one with the heterozygous mutation was a normal carrier (Suppl. Table). In our study, the five patients with the Lys150del mutation did not show a typical type II protein C deficiency. Similar phenomena have been found for the Arg169Trp and Arg15Trp mutations which are sometimes associated with type II but also with type I deficiency. The reason for the discrepancy is unknown (42, 43). It is of interest to note that our multiple transfection attempts consistently yielded lower expression levels for this protein C mutant. Thus, the possibility that a mild type I deficiency also contributes to the thrombotic phenotype in these patients cannot be ruled out at this time. Nevertheless, in agreement with the K150del mutation as being primarily a type II protein C deficiency, the characterization of the recombinant K150del mutant in the purified system identified a defect in the protein S-dependent anticoagulant function of this protein C variant. This was evidenced by the observation that the mutant exhibited a loss of activity toward both FVa and FVIIIa in the purified system only in the presence but not in the absence of protein S. Consistent with these results the activity of this mutant was also impaired in the plasma-based clotting assay using normal plasma but not protein S deficient plasma. All other activities of this mutant were normal in various protein C/APC assays as described above. The FVa degradation studies in the purified system suggest that the cofactor effect of protein S in promoting the cleavage of the Arg506 site with the K150del mutant has been eliminated. This is derived from the observation that the APC-K150del mutant exhibited normal anticoagulant activity toward FVa Leiden in the presence of protein S.
In summary, our results demonstrate that residues of the C-terminal light chain of APC including Arg147 and Lys150 are important for the physiological function of the protease in the anti-inflammatory and anticoagulant pathways, respectively. In addition to these residues other natural variants including Phe139, Cys141, Arg143, and Arg152 residues located in the C-terminus of the protein C light chain have been reported in the protein C database (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=PROC). Thus, it appears that the exposed and hydrophilic nature of these residues has rendered them suitable for constituting functional exosites, thereby facilitating the interaction of protein C/APC with its various macromolecular substrates and cofactors in plasma or on vascular endothelial cells. Nevertheless, both R147W and K150del hotspot mutants, found in the Chinese population, are low risk factors for thrombosis and may exert their phenotypic defects in combination with other mutations and/or under physiological conditions where additional underlying risk factors (i.e., inflammation) are present.
Supplementary Material
Acknowledgments
The research discussed herein was supported by grants awarded by the National Heart, Lung, and Blood Institute of the National Institute of Health (HL 101917 and HL 62565 to ARR).
We would like to thank Dr. Philip Fay for human FVIIIa and Audrey Rezaie for proofreading the manuscript.
References
- 1.Esmon CT. Molecular events that control the protein C anticoagulant pathway. Thromb Haemost. 1993;70:29–35. [PubMed] [Google Scholar]
- 2.Walker FJ, Fay PJ. Regulation of blood coagulation by the protein C system. FASEB J. 1992;6:2561–7. doi: 10.1096/fasebj.6.8.1317308. [DOI] [PubMed] [Google Scholar]
- 3.Stenflo J. Structure-function relationships of epidermal growth factor modules in vitamin K-dependent clotting factors. Blood. 1991;78:1637–51. [PubMed] [Google Scholar]
- 4.Mather T, Oganessyan V, Hof P, et al. The 2. 8 Å crystal structure of Gla-domainless activated protein C. EMBO J. 1996;15:6822–31. [PMC free article] [PubMed] [Google Scholar]
- 5.Bode W, Mayr I, Baumann U, et al. The refined 1. 9 Å crystal structure of human _-thrombin: interaction with D-Phe-Pro-Arg chlorometheylketone and significance of the Tyr-Pro-Pro-Trp insertion segment. EMBO J. 1989;8:3467–75. doi: 10.1002/j.1460-2075.1989.tb08511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mosnier LO, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–72. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 7.Fukudome K, Esmon CT. Identification, cloning and regulation of a novel endothelial cell protein C/activated protein C receptor. J Biol Chem. 1994;269:26486–91. [PubMed] [Google Scholar]
- 8.Taylor FB, Stearns-Kurosawa DJ, Kurosawa S, et al. The endothelial cell protein C receptor aids in host defense against Escherichia coli sepsis. Blood. 2000;95:1680–6. [PubMed] [Google Scholar]
- 9.Stearns-Kurosawa DJ, Kurosawa S, Mollica JS, et al. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc Natl Acad Sci (USA) 1996;93:10212–6. doi: 10.1073/pnas.93.19.10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukudome K, Kurosawa S, Stearns-Kurosawa DJ, et al. The endothelial cell protein C receptor. Cell surface expression and direct ligand binding by the soluble receptor. J Biol Chem. 1996;271:17491–8. doi: 10.1074/jbc.271.29.17491. [DOI] [PubMed] [Google Scholar]
- 11.Regan LM, Mollica JS, Rezaie AR, et al. The interaction between the endothelial cell protein C receptor and protein C is dictated by the gamma-carboxyglutamic acid domain of protein C. J Biol Chem. 1997;272:26279–84. doi: 10.1074/jbc.272.42.26279. [DOI] [PubMed] [Google Scholar]
- 12.Dahlbäck B. The protein C anticoagulant system: inherited defects as basis for venous thrombosis. Thromb Res. 1995;77:1–43. doi: 10.1016/0049-3848(94)00138-4. [DOI] [PubMed] [Google Scholar]
- 13.Kinoshita S, Iida H, Inoue S, et al. Protein S and protein C gene mutations in Japanese deep vein thrombosis patients. Clin Biochem. 2005;38:908–15. doi: 10.1016/j.clinbiochem.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 14.Shen MC, Lin JS, Tsay W. High prevalence of antithrombin III, protein C and protein S deficiency, but no factor V Leiden mutation in venous thrombophilic Chinese patients in Taiwan. Thromb Res. 1997;87:377–85. doi: 10.1016/s0049-3848(97)00141-2. [DOI] [PubMed] [Google Scholar]
- 15.Shen MC, Lin JS, Tsay W. Protein C and protein S deficiencies are the most important risk factors associated with thrombosis in Chinese venous thrombophilic patients in Taiwan. Thromb Res. 2000;99:447–52. doi: 10.1016/s0049-3848(00)00265-6. [DOI] [PubMed] [Google Scholar]
- 16.Ding Q, Shen W, Ye X, et al. Clinical and genetic features of protein C deficiency in 23 unrelated Chinese patients. Blood Cells Mol Dis. 2012 Aug 27; doi: 10.1016/j.bcmd.2012.08.004. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 17.Zhu T, Ding Q, Bai X, et al. Normal ranges and genetic variants of antithrombin, protein C and protein S in the general Chinese population. Results of the Chinese Hemostasis Investigation on Natural Anticoagulants Study I Group. Haematologica. 2011;96:1033–40. doi: 10.3324/haematol.2010.037515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu T, Dai J, Liu H, et al. Homozygous protein C deficiency with late onset venous thrombosis: identification and in vitro expression study of a novel Pro275Ser mutation. Pathology. 2012;44:348–53. doi: 10.1097/PAT.0b013e328353a218. [DOI] [PubMed] [Google Scholar]
- 19.Zhou RF, Cai XH, Xie S, et al. Molecular mechanism for hereditary protein C deficiency in two Chinese families with thrombosis. J Thromb Haemost. 2006;4:1154–6. doi: 10.1111/j.1538-7836.2006.01913.x. [DOI] [PubMed] [Google Scholar]
- 20.Tang L, Guo T, Yang R, et al. Genetic background analysis of protein C deficiency demonstrates a recurrent mutation associated with venous thrombosis in Chinese population. PLos One. 7:e35773. doi: 10.1371/journal.pone.0035773. Epub 2012 Apr 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsay W, Shen MC. R147W mutation of PROC gene is common in venous thrombotic patients in Taiwanese Chinese. Am J Hematol. 2004;76:8–13. doi: 10.1002/ajh.20043. [DOI] [PubMed] [Google Scholar]
- 22.Mather T, Oganessyan V, Hof P, et al. The 2. 8 A crystal structure of Gla-domainless activated protein C. EMBO J. 1996;15:6822–31. [PMC free article] [PubMed] [Google Scholar]
- 23.Mesters RM, Heeb MJ, Griffin JH. A novel exosite in the light chain of human activated protein C essential for interaction with blood coagulation factor Va. Biochemistry. 1993;32:12656–63. doi: 10.1021/bi00210a014. [DOI] [PubMed] [Google Scholar]
- 24.Mosnier LO, Zampolli A, Kerschen EJ, et al. Hyperantithrombotic, noncytoprotective Glu149Ala-activated protein C mutant. Blood. 2009;113:5970–8. doi: 10.1182/blood-2008-10-183327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang L, Manithody C, Rezaie AR. Contribution of basic residues of the 70–80-loop to heparin binding and anticoagulant function of activated protein C. Biochemistry. 2002;41:6149–57. doi: 10.1021/bi015899r. [DOI] [PubMed] [Google Scholar]
- 26.Yang L, Rezaie AR. Calcium-binding sites of the thrombin-thrombomodulin-protein C complex: possible implications for the effect of platelet factor 4 on the activation of vitamin K-dependent coagulation factors. Thromb Haemost. 2007;97:899–906. doi: 10.1160/th06-12-0697. [DOI] [PubMed] [Google Scholar]
- 27.Smirnov MD, Esmon CT. Phosphatidylethanolamine incorporation into vesicles selectively enhances factor Va inactivation by activated protein C. J Biol Chem. 1994;269:816–9. [PubMed] [Google Scholar]
- 28.Yang L, Manithody C, Rezaie AR. Activation of protein C by the thrombin-thrombomodulin complex: cooperative roles of Arg-35 of thrombin and Arg-67 of protein C. Proc Natl Acad Sci (USA) 2006;103:879–84. doi: 10.1073/pnas.0507700103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang L, Manithody C, Walston TD, et al. Thrombomodulin enhances the reactivity of thrombin with protein C inhibitor by providing both a binding site for the serpin and allosterically modulating the activity of thrombin. J Biol Chem. 2003;278:37465–70. doi: 10.1074/jbc.M307243200. [DOI] [PubMed] [Google Scholar]
- 30.Manithody C, Fay PJ, Rezaie AR. Exosite-dependent regulation of factor VIIIa by activated protein C. Blood. 2003;101:4802–7. doi: 10.1182/blood-2003-01-0126. [DOI] [PubMed] [Google Scholar]
- 31.Bae J-S, Yang L, Manithody C, et al. The ligand occupancy of endothelial protein C receptor switches the PAR-1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells. Blood. 2007;110:3909–16. doi: 10.1182/blood-2007-06-096651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalafatis M, Mann KG. Role of the membrane in the inactivation of factor Va by activated protein C. J Biol Chem. 1993;268:27246–57. [PubMed] [Google Scholar]
- 33.Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005;105:3178–84. doi: 10.1182/blood-2004-10-3985. [DOI] [PubMed] [Google Scholar]
- 34.Reitsma PH, Poort SR, Allaart CF, et al. The spectrum of genetic defects in a panel of 40 Dutch families with symptomatic protein C deficiency type I: heterogeneity and founder effects. Blood. 1991;78:890–4. [PubMed] [Google Scholar]
- 35.Gandrille S, Aiach M. Identification of mutations in 90 of 121 consecutive symptomatic French patients with a type I protein C deficiency. The French INSERM Network on Molecular Abnormalities Responsible for Protein C and Protein S deficiencies. Blood. 1995;86:2598–605. [PubMed] [Google Scholar]
- 36.Tsay W, Greengard JS, Montgomery RR, et al. Genetic mutations in ten unrelated American patients with symptomatic type 1 protein C deficiency. Blood Coagul Fibrinolysis. 1993;4:791–6. [PubMed] [Google Scholar]
- 37.Laszik Z, Mitro A, Taylor FB, Jr, et al. Human protein C receptor is present primarily on endothelium of large blood vessels: implications for the control of the protein C pathway. Circulation. 1997;96:3633–40. doi: 10.1161/01.cir.96.10.3633. [DOI] [PubMed] [Google Scholar]
- 38.Xu J, Qu D, Esmon NL, et al. Metalloproteolytic release of endothelial cell protein C receptor. J Biol Chem. 2000;275:6038–44. doi: 10.1074/jbc.275.8.6038. [DOI] [PubMed] [Google Scholar]
- 39.Zheng X, Li W, Song Y, et al. Non-hematopoietic EPCR regulates the coagulation and inflammatory responses during endotoxemia. J Thromb Haemost. 2007;5:1394–400. doi: 10.1111/j.1538-7836.2007.02592.x. [DOI] [PubMed] [Google Scholar]
- 40.Miyata T, Sakata T, Yasumuro Y, et al. Genetic analysis of protein C deficiency in nineteen Japanese families: five recurrent defects can explain half of the deficiencies. Thromb Res. 1998;92:181–7. doi: 10.1016/s0049-3848(98)00131-5. [DOI] [PubMed] [Google Scholar]
- 41.Miyata T, Sato Y, Ishikawa J, et al. Prevalence of genetic mutations in protein S, protein C and antithrombin genes in Japanese patients with deep vein thrombosis. Thromb Res. 2009;124:14–8. doi: 10.1016/j.thromres.2008.08.020. [DOI] [PubMed] [Google Scholar]
- 42.Reitsma PH. Protein C deficiency: from gene defects to disease. Thromb Haemost. 1997;78:344–50. [PubMed] [Google Scholar]
- 43.Steinkamp M, Geva A, Joffe S, et al. Chronic disseminated intravascular coagulation and childhood-onset skin necrosis resulting from homozygosity for a protein C Gla domain mutation, Arg15Trp. J Pediatr Hematol Oncol. 2002;24:685–8. doi: 10.1097/00043426-200211000-00018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.