

Abstract
Introduction
Recent in vitro studies suggest that CAPN3 deficiency leads initially to accelerated myofiber formation followed by depletion of satellite cells (SC). In normal muscle, upregulation of miR-1 and miR-206 facilitates transition from proliferating SCs to differentiating myogenic progenitors.
Methods
We examined the histopathological stages, Pax7 SC content, and muscle specific microRNA expression in biopsy specimens from well-characterized LGMD 2A patients to gain insight into disease pathogenesis.
Results
Three distinct stages of pathological changes were identified that represented the continuum of the dystrophic process from prominent inflammation with necrosis and regeneration to prominent fibrosis, which correlated with age and disease duration. Pax7-positive SCs were highest in fibrotic group and correlated with down-regulation of miR-1, miR-133a, and miR-206.
Conclusions
These observations, and other published reports, are consistent with microRNA dysregulation leading to inability of Pax7-positive SCs to transit from proliferation to differentiation. This results in impaired regeneration and fibrosis.
Keywords: LGMD2A, Fibrosis, microRNA, Pax, muscle regeneration
INTRODUCTION
The limb-girdle muscular dystrophies (LGMDs) are a clinically and genetically heterogeneous group of disorders characterized by progressive weakness and wasting of the proximal limb girdle muscles. LGMD type 2A (calpainopathy) is one of the most common forms of LGMD worldwide1–5 and is caused by mutations in the CAPN3 gene, which encodes a skeletal muscle specific Ca2+ - activated intracellular cysteine protease, calpain-3 (CAPN3)6. As a protease, CAPN3 potentially has a broad range of substrates, and the anchorage of CAPN3 to the muscle-specific titin protein is thought to serve a stabilizing role to prevent autolysis.7 A series of studies have demonstrated the role of CAPN3 in sarcomere assembly, turnover, and maintenance.6,8 It has also been observed in other cellular compartments, which suggests multiple functions in skeletal muscle.9–12 Of particular interest, in vitro evidence was presented that CAPN3 plays a role in myogenic differentiation, more specifically in the maintenance of the pool of reserve satellite cells (SC) by down-regulating myoD.13 It has been speculated that a calpain deficiency state would lead to an overall perturbed regeneration response via accelerated myofiber neoformation, followed by a gradual exhaustion of the SC pool. An important missing piece of understanding of the pathogenic process in CAPN3 deficiency is muscle fibrosis, a shared fate with other dystrophies, probably best characterized in Duchenne muscular dystrophy (DMD). Endomysial fibrosis is an endstage consequence of muscle fiber loss. It is often explained by a putative failure of muscle fiber regeneration that requires a stepwise process that includes activation of SCs with transition to proliferation and differentiation, followed by lateral fusion of myotubes with each other and a subsequent union to surviving muscle stumps. Recent literature has shown that certain muscle-specific microRNAs (miRs) are known to play an important post-transcriptional regulatory role in this process.14–17 For example, miR-1 and miR-206 facilitate SC differentiation by restricting their proliferative potential. Conversely, down-regulation or inhibition of miRNA-1 and miRNA-206 enhances SC proliferation and increases Pax7 protein levels in vivo.16–18 This is well-illustrated in mice that lack miR-206, where muscle regeneration was delayed in response to cardiotoxin injury due to inefficient SC differentiation.19 Furthermore, deletion of miR-206 from the mdx model for DMD exacerbated the dystrophic phenotype resulting from failure of SC differentiation.19 The results from the mdx model demonstrate an inverse relationship between miR-206 and SC differentiation DMD, a form of dystrophy with extensive fibrosis. These findings further link miR dysregulation with connective tissue proliferation.16,17,20,21 Another muscle specific microRNA, miR-133a, has been shown to play an essential role in the maintenance of adult skeletal muscle structure, function, bioenergetics, and myofiber identity.22
We hypothesized that the severity of LGMD2A is a consequence of muscle fibrosis related to dysregulation of miR-1, miR-206, and miR-133, muscle specific microRNAs. We first characterized the muscle biopsy findings from our well characterized LGMD2A population and identified 3 distinct stages of pathological changes that represent the continuum of the disease process. A significant increase of Pax7-positive SCs were found in all samples in comparison with control muscle, similar to those observations in DMD biopsies.20 A Pax7-positive SC increase correlated with down-regulation of miR-1, miR-133a, and miR-206 in the fibrotic group. This suggests inefficient regeneration resulting from inability of Pax7-positive cells to differentiate into competent SCs. The fact that these same microRNAs are similarly dysregulated in DMD16,21,23 and in X-linked canine muscular dystrophy 17 provides a potential therapeutic target for delaying the onset of skeletal muscle fibrosis not only in CAPN3 deficiency but other muscular dystrophies as well.
METHODS
Subjects
Forty five patients were evaluated through an NIH supported LGMD characterization study (NIAMS U54 AR050733-05). Ethics approval was given by the Institutional Review Board of Ohio State University and Nationwide Children’s Hospital. Written informed consent was obtained at the time of enrollment. Consistent information was obtained for each patient to establish ethnic and geographical origin, family history, consanguinity, age at onset, initial distribution of symptoms, pattern of muscle involvement, ambulatory status, disease progression, and serum creatine kinase (CK) levels.
Histopathological Analysis of Muscle Biopsies
Thirty-eight biopsy specimens from a well-characterized cohort of 45 LGMD2A patients based on DNA testing (see supplementary data; patients 2 and 4 had 2 biopsies, 13 and 14 years apart, respectively) were available for histopathological analysis. These were archived biopsy specimens obtained between 1994 and 2010. All were mounted in gum tragacanth and initially frozen in isopentane cooled in liquid nitrogen. A standard battery of stains included H&E, modified Gomori trichrome, succinic dehydrogenase, NADH dehydrogenase, and ATPase (pH 4.2, 4.6, and 9.4). Where necessary, considering the age of the biopsies, some stains were repeated.
The histopathological features of each case were categorized according to the following criteria: inflammation [(a) none, (b) minimal with small mononuclear cell (MNC) collections restricted to perivascular regions in the perimysium, or (c) prominent multifocal areas of MNC collections extending beyond perivascular regions with or without eosinophils]; necrosis/phagocytosis or regeneration (present in > 10 fibers per section); lobulated fibers (present in >50% of fibers per section); and fibrosis (grade 0, absent; grade 1, scattered, non-contiguous, focal endomysial connective tissue deposits related to muscle fiber loss; grade 2, contiguous endomysial connective deposits throughout the muscle section; grade 3, contiguous endomysial connective deposits throughout the muscle with fibrosis and fat replacing focal regions of muscle fiber loss; grade 4, diffuse fibro-fatty changes with marked dropout in the muscle fibers).
Fiber type specific identification of PAx7-positive satellite cell number
Based upon tissue preservation from the archived tissue, 13 biceps muscle biopsy specimens were available for Pax7 staining that included group 1 (n = 1), group 2 (n =3), and group 3 (n=9). Five of the group 3 biopsies showed lobulated fibers. Three biceps muscle biopsies from patients with myalgias without histological changes were used as controls. Ten-12 μm serial cryosections were properly aligned on plus glass slides for cross-sectional fiber analysis. Pax-7 positive satellite cells were identified with mouse Pax-7 IgG1 antibody (R & D systems) by an immunohistochemistry protocol of Super Sensitive polymer-HRP detection kit using i6000™ Automated Staining System from Biogenex®. Briefly, cryosections were fixed in 2% paraformaldehyde for 10 min at 4°C and incubated in Pax-7 antibody (1:100 dilutions) for 30 min after blocking with peroxide, and Power Block™ for 10 min. Slides were washed 5 times with IHC supersensitive™ wash buffer. Finally, 3, 3′-Diaminobenzidine (DAB) was used as a substrate with Mayor hematoxylin used as a counter stain. The adjacent sections were stained for ATPase activity after acid pre-incubation at pH 4.2 followed by a series of dehydration steps to determine the muscle fiber type (in biceps muscle, type 1 muscle fibers constitute approximately 42 of total fibers).24
From the Pax7-stained slides, randomly selected images were captured at x270 or x540 magnification to allow unambiguous determination of SC localization. Fiber type specific localization was determined by matching the serial ATPase stained sections at pH 4.2. The data were used to determine the number of SCs per type 1 and type 2 muscle fibers. The number of fibers analyzed per case (mean ± SEM) included type 1 = 228.08 ± 50.78 and type 2 = 80.85 ± 18.48, well above the numbers needed for sufficient estimation of SCs for each fiber type.25 The overrepresentation of type 1 fibers is related to the type 1 fiber predominance characteristic of CAPN3 deficiency.
Identification of muscle specific microRNA expression
For these studies, the same biceps biopsy materials used for PAx7-positive SC analysis were grouped representing inflammatory (Group 1 and 2; n=3) or fibrotic spectrum [Group 3 with lobulated fibers (n=3) and Group 3 with non-lobulated fibers (n=3)] and controls (n=3). Total RNA was isolated from the specimens using a mirVana miRNA isolation kit (Life Technologies®). Reverse transcription was performed by using a Taqman microRNA reverse transcription kit (Applied Biosystems®). Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) for miR-1, miR-206, miR-133a and U6snRNA was performed using predesigned Taqman primers and probes. Each miRNA expression was normalized to U6snRNA expression. Expression data were given as means of relative expression values obtained from 3 samples in conjunction with standard error of means and presented in a graph format.
Statistical Analyses
For all comparisons with summary statistics indicating practical significant difference, two-sample t tests were used. Significance level was set at 0.05.
RESULTS
Figure 1 shows the localization of the CAPN3 mutations in our cohort of 45 LGMD2A patients. Clinical severity was not strictly correlated with disease duration, and although variability of disease course could not be attributed to the genotype, most of the moderate to severe phenotypes were associated with nonsense mutations clustered in the proteolytic domain. See supplementary data for further details of clinical, histopathological and genotypic features (Tables S1, S2 and S3).
Figure 1.

Localization of the mutations in the Calpain-3 gene. The exons with light blue background represent the missense mutations; a green background color was used for the frameshift mutations, purple for deletions, red for stop codon, and yellow for splice site mutations. The circle shape indicates the patients with a mild phenotype. The exons encircled in a rectangular shape represent the group with a moderate clinical course, and the diamond shape for the more severe phenotype. Although the genotype-phenotype correlation is difficult to interpret in cases of double heterozygosity, most of the moderate to severe phenotypes correlated with nonsense mutations clustered in the proteolytic domain.
Histopathological studies
Histopathological criteria detailed in Methods, permitted us to place each of the 36 biopsies into one of the 3 groups (Table S1): Group 1 (n=7), prominent multifocal inflammation with minimal fibrosis (grade 0–1); Group 2 (n=8), minimal inflammation and grade 1–2 fibrosis; Group 3 (n=23), no inflammation accompanied by grade 2–4 fibrosis.
Group 1 with minimal or no fibrosis and with multifocal prominent inflammation, necrosis and regeneration also displayed conspicuous eosinophilic infiltrates in 5 cases (patients 1, 2, 3, 4, and 6) (Figure 2A). The patients with eosinophilic infiltrates were younger (ages of 4 – 25, mean 3.4 ± 7.1 years) at the time the biopsies were performed. The duration of symptoms (the time between the reported age of onset of clinical symptoms and patient age at the time of biopsy) varied from 1 to 11 years, mean 4.6 ± 3.7 years. Fibrosis grade was 0 in 3 cases and 1 in 2 cases. Immunoblot analysis showed severely reduced to complete absence of the CAPN3 protein in all available cases (not shown). Two biopsies [ patients 1 and 3, see Tables S1, S2 and S3] were from patients who had an unusual initial presentation in the first decade of life consisting of sporadic muscle pain, marked CK elevation (11,000 U/L) and peripheral blood eosinophilia. One of the 5 patients [n=3] is homozygous for a genetic mutation [c.2306G>A] previously associated with an eosinophilic response.26
Figure 2.
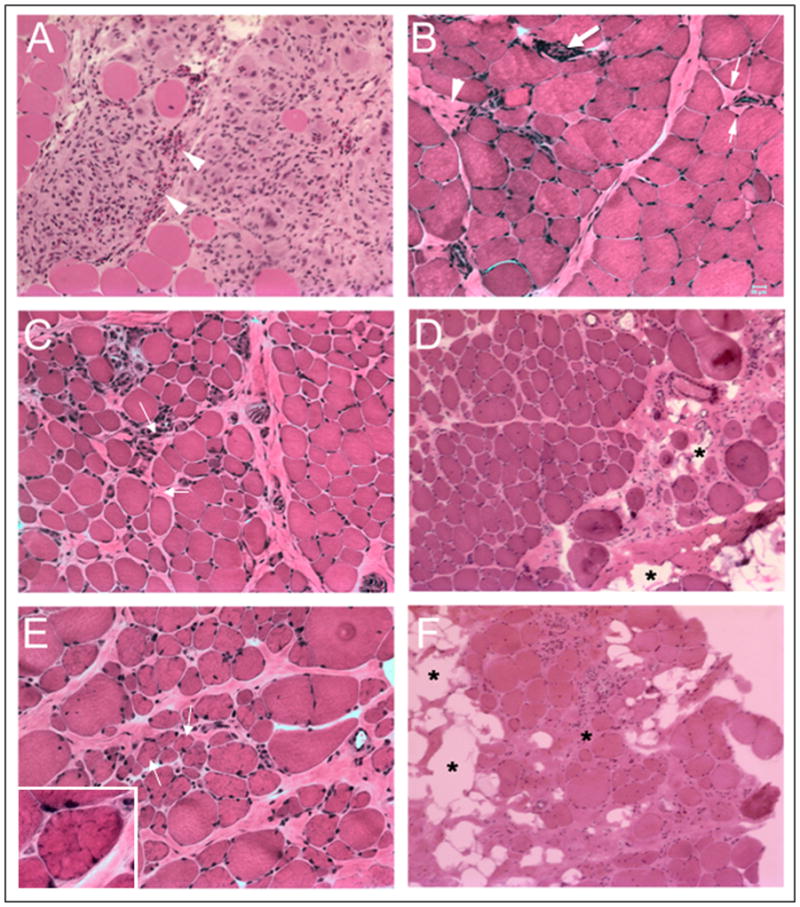
A large area of mononuclear cell collections including eosinophils (arrow heads) are seen in a representative case from group 1 (A). A representative area of grade 1 fibrosis illustrating pericapillary fibrosis (small arrows) and an area of muscle fiber drop out replaced by fibrosis (arrow head) from a biopsy classified as group 2 (group 2 represents minimal inflammation with grade 1–2 fibrosis) is seen in (B) with a large arrow pointing to a small area of endomysial perivascular inflammation. Diffuse mild endomysial fibrosis (grade 2 fibrosis, arrows) with scarce mononuclear cells is seen in a specimen classified as group 2 (C). Moderately severe diffuse fibrosis, perimysium greater than endomysium (grade 3) is seen with fatty tissue replacement in some areas of the perimysium (asterisks) in a representative specimen from group 3 (D). Small and medium size lobulated fibers (arrows and inset) are shown in a specimen with diffuse endomysial and perimysial fibrosis from a group 3A biopsy (E). Diffuse fibrofatty changes (asterisks) with marked drop out in the muscle fibers from a biopsy in group 3B representing grade 4 fibrosis in (F). Bar = 20 μm for all panels.
Eight cases fit criteria for Group 2. These cases showed minimal inflammation and fiber necrosis or regeneration occurring scattered or in small clusters and grade 1 or 2 fibrosis (Figure 2 B, C). These patients represented the intermediate age group (mean age 25.9 ± 9.1 years, symptom duration 10.9 ± 5.6 years). Grade 2 fibrosis was present in 2 patients (n= 8, 13), one of the youngest patients and an older patient in this group with the longest duration of symptoms. Group 3, representing the oldest group (33.9 ± 11.8 years) with the longest symptom-duration (17.6 ± 9.3 years) was composed of 23 cases with prominent fibrosis (grade 2–4), and no inflammatory infiltrates (Figure 2D–F). Interestingly, in this group (Table S1), 11 cases (Group 3A) showed a prominent lobulated pattern of cytoarchitectural change in small and medium fibers (Figure 2E). The mean age (33.7 ± 7.7 years) and the degree of fibrosis (2.7 ± 0.4) in the lobulated group was similar to those in the remaining 12 patients (Group 3B) who had a non-lobulated pattern (34.1 ± 14.2 and 2.8 ± 1.4 respectively), while the symptom duration was slightly shorter in the lobulated group without reaching statistical significance (16.5 ± 7.8 vs. 18.5 ± 10.1 years).
Overall, considering the entire cohort there was a good correlation between age, duration of symptoms and degree of fibrosis (correlation coefficient of 0.604 for the age and 0.583 for the symptoms duration and fibrosis) (Figures 3A–D).
Figure 3.

Relationship between age and fibrosis (A, B), and duration of symptoms and fibrosis (C, D). In B, mean age of groups is plotted against mean fibrosis grade; in D, mean duration of symptoms is plotted against mean fibrosis grade for each histopathological group.
PAx7-positive satellite cell number and the disease stage in biopsies
Recent in vitro studies have reported that down-regulation of MyoD by CAPN3 promotes generation of reserve cells in C2C12 myoblasts, suggesting a role for CAPN3 as a potential new player involved in muscle regeneration by promoting renewal of the SC compartment.13 In order to explore whether or not CAPN3 mutations have a negative effect on the SC pool, we quantified the number of Pax7-positive nuclei in type 1 and type 2 muscle fibers in biopsies from 3 distinct histopathological groups representing the entire spectrum of calpain histopathology. Contrary to the expectation of a gradual decrease of the SC pool with disease duration (as proposed by Stuelsatz et al) 13, we observed a significant increase of Pax7-positive SCs in all samples in comparison with control muscle specimens, similar to the observations in DMD biopsies 20. For the entire cohort, the number of Pax7-positive cells per muscle fiber was significantly greater than controls (0.192 ± 0.037, n=13 vs. 0.065 ± 0.006, n=3; p=0.006). In addition, the number of SC per type 1 fibers was less than the number per type 2 fibers (0.171 ± 0.038 vs. 0.275 ± 0.061, n=13; p=0.01), a reversal of findings in our control specimens. These findings in our controls are in close agreement with the previously reported normal values using the same SC marker.25 This fiber type specific change of SC content was maintained in all samples from each group. Moreover, the number of Pax 7-positive nuclei per muscle fiber showed a positive correlation with fibrosis grade when 3 histopathological groups were compared (Figure 4A). Group 3 (n=9) with no inflammation, necrosis, or regeneration and a mean fibrosis grade of 3.1 showed the highest number of SCs per both fiber types in comparison with 1 sample from group 1 with prominent inflammation, and 3 samples from group 2 with mild inflammation and grade 1 fibrosis. The biopsy from group 1 with prominent inflammation and eosinophilia (patient 3 in Figure 1, Tables S1, S2, and S3) had the lowest number of SC per both type 1 and type 2 fibers accompanied by prominent regeneration (Figure 4B). Fiber type specific localization of SCs is shown in Figure 4 D and E.
Figure 4.

Table (A) shows fiber type specific distribution of satellite cells in LGMD2A biopsies representing histopathological groups, 1, 2 and 3. DD=disease duration, FG=fibrosis grade, SC=satellite cell. Group 3 with the highest mean FG of 3.1 has the highest number of SCs (B). SC number in lobulated and non-lobulated biopsies is shown in (C). Non-lobulated biopsies (n=4) with mean FG of 3.5 displays significantly high SC number per type 1 fibers (*p=0.035) and per total fibers compared to lobulated biopsies (n=5) with FG of 2.8 (+p=0.026). Serial cross sections of a biceps muscle from a LGMD patient illustrate Pax7 positive SCs (arrows) using immunohistochemistry (D) and fiber types with ATPase at pH 4.2 (E). Fiber type specific localization of SCs (see methods) was determined by matching the serial ATPase stained sections at pH 4.2. Some fibers with Pax7 positive nuclei marked as a, b and c in D match with type 1 fibers (darkly stained) in E. II, marks a type 2 fiber with Pax7 positive SC. Bar = 50 μm.
Quantifying muscle fiber types revealed type 1 fiber predominance with increasing age and duration of disease process in calpainopathy. The percentage of type 1 fibers in group 3 was significantly higher, 80.9 ± 6.8 % (n=9) compared to group 2 with mild inflammation (50.3 ± 6.4, n=3; p=0.03). Within group 3, type 1 fiber predominance was slightly higher in the subgroup with a lobulated pattern (83.9 ± 8.2, n=5) compared to those without lobulation (77.0 ± 19.3, n=4) while type 1 fiber SC content was significantly higher in the non-lobulated group (p=0.03) correlating with higher degree of fibrosis (Figure 3C). For the entire cohort of group 3, increases in SC number per muscle fiber showed a positive correlation with the fibrosis grade (r = 0.658). This strong correlation was present only for type 1 fibers (r = 0.698). These findings clearly show that in calpainopathy, Pax7-positive SCs are not depleted but instead are increased significantly, correlating with age and disease progression.
Muscle specific microRNA expression and PAx7-positive satellite cells in calpainopathy
We analyzed the expression profiles miR-1, miR-206 and miR-133a by qRT-PCR in muscle biopsies representing different histopathological stages of the dystrophic process in calpainopathy. We speculated that increased Pax 7-positive nuclei are a morphologic correlate of decreased miR-1 and miR-206 levels, reflecting inefficient regeneration, and that type 1 fiber predominance is a manifestation of miR-133a down-regulation. As shown in Figure 5, miR-1 expression was slightly reduced in the specimens with inflammation, down to 80% of control levels; however, a significant down-regulation was observed in the group 3 cohort (0.54 ± 0.10, n=6 vs. 1.00 ± 0.19, n=3; p= 0.039) more notably in the non-lobulated fibrotic biopsies (38% of control level; −0.6 fold change) similar to those observations in DMD biopsies and the canine model for DMD.17,23 miR-133a expression levels showed a similar pattern of changes to miR-1 with the lowest expression in the non-lobulated fibrotic biopsies from group 3 compared to controls (45% of control level; −0.55 fold change).
Figure 5.

Expression levels of miR-1, miR-133a and miR-206 in biceps muscles of LGMD2A patients grouped as inflammatory and non-inflammatory (with non-lobulated and lobulated fibers), and of controls. All expression levels were determined by real time RT-PCR. Results are presented as mean relative expression ± MSE; n=3. p=0.05 for control vs. lobulated group; p=0.039 for control (n=3) vs. the entire non-inflammatory cohort (lobulated + non-lobulated, n=6).
miR-206 was previously shown to increase dramatically (more than 10-fold) in regenerating muscle fibers following cardiotoxin (CTX)-induced injury related to SC proliferation. Similar findings are seen in mdx mice, a model associated with robust muscle fiber necrosis, regeneration, and inflammation.17 We observed only a small, 1.6- fold increase of miR-206 expression in the biopsies with inflammation and necrosis/regeneration from younger patients (Figure. 4). Specimens from group 3 with prominent lobulated fibers also showed a modest, 2-fold increase in miR-206 expression. However, in group 3, fibrotic biopsies without lobulated fibers, miR-206 expression was reduced to levels less than control muscles. Overall, these results suggest that miR-206 is not abundantly expressed in calpainopathy muscles even in the early stages with inflammation, necrosis, and regeneration, and declined with age and progression of the dystrophic process.
DISCUSSION
We performed detailed correlative clinical and histopathological studies on a large cohort of a well-characterized LGMD 2A patient population collected over a 10-year period. In our cohort of 45 subjects, 46 different CAPN3 gene variances were identified including 12 novel mutations. A hot spot in exons 4–5 of the catalytic domain II of CAPN3 was demonstrated with an interesting correlation of clinical severity and nonsense mutations clustered in the proteolytic domain (see supplementary data).
Analysis of biopsies based on histopathological criteria led to identification of 3 groups of patients who were clustered by age, symptom duration, and clinical severity. The youngest patients with the shortest disease duration at the time of biopsy were clustered in group 1 and were characterized by prominent inflammation; the oldest patients with the longest duration of symptoms were typically in group 3. They had no inflammation but did have high grade fibrosis. The biopsies from group 2 with minimal inflammation, necrosis, and regeneration were from patients with intermediate disease severity. This suggests that groups 1, 2 and 3 represent a continuum of disease in calpainopathy. In our cohort, the age and symptom duration show a strong correlation with fibrosis grade, supporting the conclusion that biopsies from patients with longstanding disease are characterized by a minimally active necrotic process and prominent interstitial fibrosis. Furthermore, we had the opportunity to examine biopsy materials from patients who were biopsied twice (patients 2 and 4; they were re-biopsied 13 and 14 years later; see supplementary data). We found that the first biopsy at a younger age had prominent inflammation with eosinophilia, and the second biopsy showed no inflammation but significant fibrosis, providing direct evidence for the presence of the 2 ends of the disease spectrum in the same individual.
In this series, the presence of lobulated, predominantly small and medium fibers was observed in about one-fourth (11 out of 38) of all biopsies, and it was markedly overrepresented (48%) in group 3 from patients with a long clinical course. Lobulated fibers expressed type 1 fiber histochemical markers as previously reported.27 A lobulated pattern of cytoskeletal change, although nonspecific, is frequently observed in muscle biopsies of patients with calpainopathy. Gene expression profiles in LGMD 2A muscle biopsies with lobulated fibers have shown an upregulation of genes that express actin-filament binding proteins consistent with the structural changes of the intermyofibrillary network.28 Zebra fish studies show that down-regulation of miR-1 and miR-133 disrupts actin filament organization during sarcomere assembly.29 Moreover, studies have shown that miR-133a suppresses the type 1 fiber genetic program, and mice that lack miR-133a have alterations in the intermyofibrillary network, ring-like fibers, and lobulated-like fibers and interestingly, fast to slow myofiber conversion.22 As we predicted, miR-133a expression was down-regulated in biopsies from group 3, where we see type 1 fiber predominance. No previous studies have demonstrated the relationship between the CAPN3 mutations and a specific mRNA leading to type 1 fiber predominance.
The analysis of Pax7-positive nuclei in the representative biopsies from each group showed a significant increase of SCs that correlated with the duration of disease and the degree of fibrosis. We observed the highest number of Pax7 positive SCs in the group with the highest grade of fibrosis but no inflammation, necrosis, or regeneration. Our findings are similar to observations made in DMD muscle biopsies, that, despite increasing fibrosis, Pax7-positive nuclei per muscle fiber were elevated. This highlights the discrepancy between histological and cell culture studies.13,20,30–32 Moreover, we see an inverse relationship between the number of Pax7-positive cells and miR-1, miR-133a and miR-206 expression levels, indicating that increased Pax 7-positive nuclei is a morphologic correlate of inefficient regeneration leading to increased fibrosis. In fact, a recent study using a three-dimensional muscle tissue engineering paradigm provides proof of concept for this direct relationship between Pax7 expressing cells and miR-1. In that study, the number of Pax7-positive cells was reduced by transfection with miR-1 compared to microRNA-scrambled controls.33
MicroRNAs are small, non-coding RNA molecules that post-transcriptionally regulate gene expression in a tissue specific manner. The 3 muscle-specific microRNAs, miR-1, miR-206 and miR-133 have been shown to play important roles in muscle development and regeneration.14–17 Their potential roles in human myopathic disorders have now begun to gain interest. In our biopsy specimens, we observed a reduction in miR-1 and miR-133a, most notable in the fibrotic group similar to DMD biopsies.23 We found miR-206 was modestly up-regulated (1.6 to 2 fold change) in the inflammatory and lobulated groups, but down-regulated below control levels in the fibrotic group representing an advanced stage of the calpainopathy. Moreover, this same pattern of miR-1 and miR-206 reduction is associated with a significant increase of Pax7-positive SCs in both calpainopathy and DMD biopsies.20 Taken together, these observations strongly indicate that miR-1 and miR-206 participate in a regulatory manner that allows transition of SCs from proliferation to differentiation and that the absence of this transition results in an excessive number of Pax7-positive SCs, impaired myofiber repair/regeneration, and consequent increased fibrosis.
It should be noted that the level of miR-206 up-regulation seen prior to the very late phase of calpainopathy is modest compared to examples of robust regeneration as seen in wild type cardiotoxin-induced injury, where miR-206 increases dramatically (more than 10-fold) within regenerating muscle fibers. This quantitative analysis as well as the results of in situ hybridization indicates that miR-206 is highly expressed in myotubes newly formed from SCs in a paradigm with robust regenerative capacity.17 On the other hand, the changes in the expression levels of these microRNAs in calpainopathy or DMD are more comparable to those reported in the canine dystrophy model, which exhibits a more severe clinical phenotype accompanied by progressive muscle degeneration.17
We believe that our findings are important for illustrating the presence of a similar pattern of microRNA dysregulation in calpainopathy and DMD, both of which show inefficient regeneration leading to fibrosis, even though the underlying gene defects are uniquely different. The link to the understanding of this similar pattern of microRNA dysregulation associated with markedly disparate gene defects requires further study to unravel a common feature of the dystrophic process. The importance cannot be overlooked in the evolution of novel therapeutic strategies.
Supplementary Material
Acknowledgments
This work was supported by NIH NIAMS U54 AR050733-05, Jesse’s Journey, and the muscular Dystrophy Association
Abbreviations
- CAPN3
Calpain 3
- CK
Creatine kinase
- CTX
Cardiotoxin
- DMD
Duchenne muscular dystrophy
- LGMD
Limb girdle muscular dystrophy
- miR
MicroRNA
- MNC
Mononuclear cell
- SC
Satellite cell
References
- 1.Moore SA, Shilling CJ, Westra S, Wall C, Wicklund MP, Stolle C, et al. Limb-girdle muscular dystrophy in the United States. J Neuropathol Exp Neurol. 2006;65:995–1003. doi: 10.1097/01.jnen.0000235854.77716.6c. [DOI] [PubMed] [Google Scholar]
- 2.Fanin M, Nascimbeni AC, Fulizio L, Angelini C. The frequency of limb girdle muscular dystrophy 2A in northeastern Italy. Neuromuscul Disord. 2005;15:218–224. doi: 10.1016/j.nmd.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Piluso G, Politano L, Aurino S, Fanin M, Ricci E, Ventriglia VM, et al. Extensive scanning of the calpain-3 gene broadens the spectrum of LGMD2A phenotypes. J Med Genet. 2005;42:686–693. doi: 10.1136/jmg.2004.028738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Minami N, Nishino I, Kobayashi O, Ikezoe K, Goto Y, Nonaka I. Mutations of calpain 3 gene in patients with sporadic limb-girdle muscular dystrophy in Japan. J Neurol Sci. 1999;171:31–37. doi: 10.1016/s0022-510x(99)00245-2. [DOI] [PubMed] [Google Scholar]
- 5.Richard I, Brenguier L, Dincer P, Roudaut C, Bady B, Burgunder JM, et al. Multiple independent molecular etiology for limb-girdle muscular dystrophy type 2A patients from various geographical origins. Am J Hum Genet. 1997;60:1128–1138. [PMC free article] [PubMed] [Google Scholar]
- 6.Kramerova I, Kudryashova E, Venkatraman G, Spencer MJ. Calpain 3 participates in sarcomere remodeling by acting upstream of the ubiquitin-proteasome pathway. Hum Mol Genet. 2005;14:2125–2134. doi: 10.1093/hmg/ddi217. [DOI] [PubMed] [Google Scholar]
- 7.Ermolova N, Kudryashova E, DiFranco M, Vergara J, Kramerova I, Spencer MJ. Pathogenity of some limb girdle muscular dystrophy mutations can result from reduced anchorage to myofibrils and altered stability of calpain 3. Hum Mol Genet. 2011;20:3331–3345. doi: 10.1093/hmg/ddr239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beckmann JS, Spencer M. Calpain 3, the “gatekeeper” of proper sarcomere assembly, turnover and maintenance. Neuromuscul Disord. 2008;18:913–921. doi: 10.1016/j.nmd.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kramerova I, Kudryashova E, Wu B, Ottenheijm C, Granzier H, Spencer MJ. Novel role of calpain-3 in the triad-associated protein complex regulating calcium release in skeletal muscle. Hum Mol Genet. 2008;17:3271–3280. doi: 10.1093/hmg/ddn223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ojima K, Ono Y, Ottenheijm C, Hata S, Suzuki H, Granzier H, et al. Non-proteolytic functions of calpain-3 in sarcoplasmic reticulum in skeletal muscles. J Mol Biol. 2011;407:439–449. doi: 10.1016/j.jmb.2011.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang Y, de Morree A, van Remoortere A, Bushby K, Frants RR, Dunnen JT, et al. Calpain 3 is a modulator of the dysferlin protein complex in skeletal muscle. Hum Mol Genet. 2008;17:1855–1866. doi: 10.1093/hmg/ddn081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baghdiguian S, Martin M, Richard I, Pons F, Astier C, Bourg N, et al. Calpain 3 deficiency is associated with myonuclear apoptosis and profound perturbation of the IkappaB alpha/NF-kappaB pathway in limb-girdle muscular dystrophy type 2A. Nat Med. 1999;5:503–511. doi: 10.1038/8385. [DOI] [PubMed] [Google Scholar]
- 13.Stuelsatz P, Pouzoulet F, Lamarre Y, Dargelos E, Poussard S, Leibovitch S, et al. Down-regulation of MyoD by calpain 3 promotes generation of reserve cells in C2C12 myoblasts. J Biol Chem. 2010;285:12670–12683. doi: 10.1074/jbc.M109.063966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Rooij E, Liu N, Olson EN. MicroRNAs flex their muscles. Trends Genet. 2008;24:159–166. doi: 10.1016/j.tig.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 15.Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38:228–233. doi: 10.1038/ng1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen JF, Tao Y, Li J, Deng Z, Yan Z, Xiao X, et al. microRNA-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J Cell Biol. 2010;190:867–879. doi: 10.1083/jcb.200911036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuasa K, Hagiwara Y, Ando M, Nakamura A, Takeda S, Hijikata T. MicroRNA-206 is highly expressed in newly formed muscle fibers: implications regarding potential for muscle regeneration and maturation in muscular dystrophy. Cell Struct Funct. 2008;33:163–169. doi: 10.1247/csf.08022. [DOI] [PubMed] [Google Scholar]
- 18.Dey BK, Gagan J, Dutta A. miR-206 and -486 induce myoblast differentiation by downregulating Pax7. Mol Cell Biol. 2011;31:203–214. doi: 10.1128/MCB.01009-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu N, Williams AH, Maxeiner JM, Bezprozvannaya S, Shelton JM, Richardson JA, et al. microRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. J Clin Invest. 2012;122:2054–2065. doi: 10.1172/JCI62656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kottlors M, Kirschner J. Elevated satellite cell number in Duchenne muscular dystrophy. Cell Tissue Res. 2010;340:541–548. doi: 10.1007/s00441-010-0976-6. [DOI] [PubMed] [Google Scholar]
- 21.Cacchiarelli D, Martone J, Girardi E, Cesana M, Incitti T, Morlando M, et al. MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab. 2010;12:341–351. doi: 10.1016/j.cmet.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 22.Liu N, Bezprozvannaya S, Shelton JM, Frisard MI, Hulver MW, McMillan RP, et al. Mice lacking microRNA 133a develop dynamin 2-dependent centronuclear myopathy. J Clin Invest. 2011;121:3258–3268. doi: 10.1172/JCI46267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greco S, De Simone M, Colussi C, Zaccagnini G, Fasanaro P, Pescatori M, et al. Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J. 2009;23:3335–3346. doi: 10.1096/fj.08-128579. [DOI] [PubMed] [Google Scholar]
- 24.Johnson MA, Polgar J, Weightman D, Appleton D. Data on the distribution of fibre types in thirty-six human muscles. An autopsy study. J Neurol Sci. 1973;18:111–129. doi: 10.1016/0022-510x(73)90023-3. [DOI] [PubMed] [Google Scholar]
- 25.Mackey AL, Kjaer M, Charifi N, Henriksson J, Bojsen-Moller J, Holm L, et al. Assessment of satellite cell number and activity status in human skeletal muscle biopsies. Muscle Nerve. 2009;40:455–465. doi: 10.1002/mus.21369. [DOI] [PubMed] [Google Scholar]
- 26.Krahn M, Lopez de Munain A, Streichenberger N, Bernard R, Pecheux C, Testard H, et al. CAPN3 mutations in patients with idiopathic eosinophilic myositis. Ann Neurol. 2006;59:905–911. doi: 10.1002/ana.20833. [DOI] [PubMed] [Google Scholar]
- 27.Hermanova M, Zapletalova E, Sedlackova J, Chrobakova T, Letocha O, Kroupova I, et al. Analysis of histopathologic and molecular pathologic findings in Czech LGMD2A patients. Muscle Nerve. 2006;33:424–432. doi: 10.1002/mus.20480. [DOI] [PubMed] [Google Scholar]
- 28.Keira Y, Noguchi S, Kurokawa R, Fujita M, Minami N, Hayashi YK, et al. Characterization of lobulated fibers in limb girdle muscular dystrophy type 2A by gene expression profiling. Neurosci Res. 2007;57:513–521. doi: 10.1016/j.neures.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 29.Mishima Y, Abreu-Goodger C, Staton AA, Stahlhut C, Shou C, Cheng C, et al. Zebrafish miR-1 and miR-133 shape muscle gene expression and regulate sarcomeric actin organization. Genes Dev. 2009;23:619–632. doi: 10.1101/gad.1760209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishimoto S, Goto I, Ohta M, Kuroiwa Y. A quantitative study of the muscle satellite cells in various neuromuscular disorders. J Neurol Sci. 1983;62:303–314. doi: 10.1016/0022-510x(83)90207-1. [DOI] [PubMed] [Google Scholar]
- 31.Delaporte C, Dehaupas M, Fardeau M. Comparison between the growth pattern of cell cultures from normal and Duchenne dystrophy muscle. J Neurol Sci. 1984;64:149–160. doi: 10.1016/0022-510x(84)90033-9. [DOI] [PubMed] [Google Scholar]
- 32.Luz MA, Marques MJ, Santo Neto H. Impaired regeneration of dystrophin-deficient muscle fibers is caused by exhaustion of myogenic cells. Braz J Med Biol Res. 2002;35:691–695. doi: 10.1590/s0100-879x2002000600009. [DOI] [PubMed] [Google Scholar]
- 33.Koning M, Werker PM, van der Schaft DW, Bank RA, Harmsen MC. MicroRNA-1 and microRNA-206 improve differentiation potential of human satellite cells: a novel approach for tissue engineering of skeletal muscle. Tissue Eng Part A. 2012;18:889–898. doi: 10.1089/ten.TEA.2011.0191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.