

Abstract
This letter describes the further chemical optimization of the M5 PAM MLPCN probes ML129 and ML172. A multi-dimensional iterative parallel synthesis effort quickly explored isatin replacements and a number of southern heterobiaryl variations with no improvement over ML129 and ML172. An HTS campaign identified several weak M5 PAMs (M5 EC50 >10 μM) with a structurally related isatin core that possessed a southern phenethyl ether linkage. While SAR within the HTS series was very shallow and unable to be optimized, grafting the phenethyl ether linkage onto the ML129/ML172 cores led to the first sub-micromolar M5 PAM, ML326 (VU0467903), (human and rat M5 EC50s of 409 nM and 480 nM, respectively) with excellent mAChR selectivity (M1-M4 EC50s <30 μM) and a robust 20-fold leftward shift of the ACh CRC.
Keywords: Muscarinic acetylcholine receptors, M5, Positive allosteric modulator (PAM), ML326
There are five muscarinic acetylcholine receptor (mAChR) subtypes (M1 – M5) widely expressed in both the central nervous system (CNS) and periphery of mammals.1–5 These receptors, whose endogenous agonist is acetylcholine (ACh), play critical roles in regulating a variety of diverse physiological processes. Within the CNS, the M1, M4 and M5 subtypes are believed to be the most important with respect to normal neuronal functioning.1–5 Of these three, M5 is the least studied as a combined result of its lower expression levels6 (< 2% of total muscarinic receptor population within the brain) and, until recently, a near absence of highly selective M5 receptor ligands. Much of our current understanding surrounding the function of M5 has come from M5 receptor localization, M5 knock-out mice7 and experiments conducted with non-selective muscarinic ligands.8 It is intriguing to note that the M5 receptor is the only muscarinic receptor observed the substantia nigra pars compacta (based on mRNA detection),9,10 leading to the prediction that M5 functions in addiction/reward mechanisms. This hypothesis was supported in man through the clinically observed correlation between a specific M5 gene mutation and an increase in cigarette consumption (+ 27%), as well as, an increased risk for cannabis dependence (+ 290%).11 Additionally, M5 receptors have been localized on the cerebrovascular system and shown to be critical in the ACh-induced dilation necessary for normal blood perfusion.12 In a broader sense, M5 KO mice show decreased prepulse inhibition,13 CNS neuronal abnormalities and cognitive deficits,14 thus supporting the potential for M5 ligands in the treatment of numerous CNS disorders including schizophrenia, Alzheimer's disease, ischemia and migraine.1–5
To date, few M5 PAMs have been reported,15–17 and none of these display the potency and efficacy necessary to definitively probe the role of selective M5 activation (Fig. 1); therefore, in this Letter, we detail the further optimization of these ligand, and the discovery of the first submicromolar and selective M5 PAMs.
Figure 1.
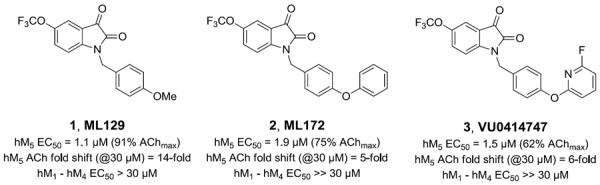
Structures and activities of the reported M5 PAMs 1–3. All three were derived from a pan-M1, M3, M5-PAM that afforded both M1 selective PAMs and, by virtue of the 5-OCF3 moiety, M5 selective PAMs.
Within the isatin scaffold, alternate substitution afforded a highly selective M1 PAM, ML137, 4.18 We recently reported on a number of replacements (tertiary hydroxyls and spirocyclic replacements, etc.) for the isatin moiety that maintained M1 PAM activity (Fig. 2).19,20 Therefore, our optimization plan for ML129 consisted of evaluating both spirocyclic and other replacements for the isatin moiety, while continuing to investigate alternatives for the southern p-OMe benzyl motif in an effort to improve M5 PAM potency to below 1 μM (Fig. 3).
Figure 2.
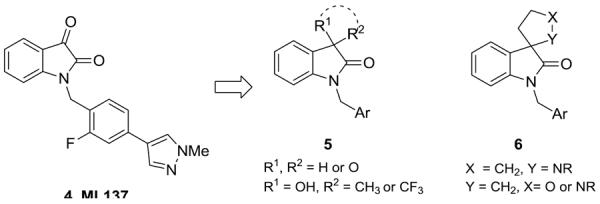
Structures and activities of the reported M1 PAMs 4–6. Mulitple productive replacements (tertiary hydroxyls, dioxalanes and spiro furans/pyrroles) were identified for the isatin moiety of ML137 that maintained selective M1 PAM activity.
Figure 3.
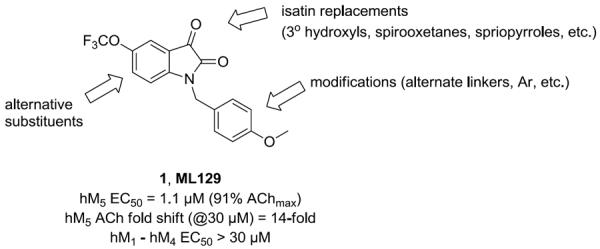
Multi-dimenisional chemical optimization plan for ML129 (1).
First, we explored alternatives to the 5-OCF3 moiety on the isatin core, and despite evaluating other positions and alternate functionalities, all were devoid of M5 PAM activity. Employing the chemistry previously reported for analogs 5 of ML137,19 we prepared the corresponding 3° hydroxyl and dioxalane analogs of ML129, and while there was a slight elevation in the ACh max, all of the analogs were >10 μM as M5 PAMs, indicating a disconnect in the SAR between M1 and M5. We next explored novel 3° hydroxyl analogs with diverse functionalities, which proved to be more productive. Treatment of ML129 with DABCO in nitromethane afforded the 3° alcohol 7 in quantitative yield (Scheme 1);21 importantly, 7 was of comparable potency and efficacy to ML129 (M5 EC50 = 1.4 μM, 75% ACh Max), but the nitro moiety was not attractive as an in vivo probe.
Scheme 1.
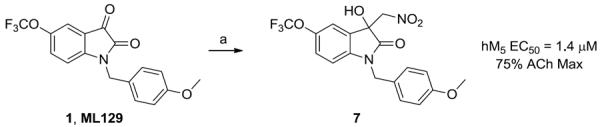
Synthesis of 3° hydroxyl analog 7. Reagents and conditions: (a) DABCO, nitromethane (neat), 100%.
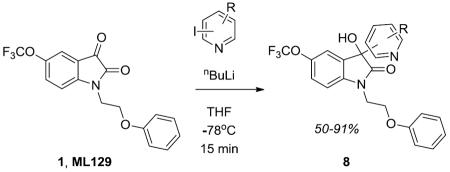
This led us to explore other 3° alcohols possessing heterocycles, and most notably pyridines. Here, we employed two routes to survey pyridyl analogs 8 and pyridyl methyl homologs 11 (Scheme 2). Again, utilizing ML129 as starting material, exposure to a functionalized pyridyl boronic acid 9 under Cu(OTf)2 catalysis, affords analogs 8 in 25–70% yields.22 We also employed direct lithiation/addition in some instances.23 The homologated congeners 11 were easily accessed following the Li protocol,24 employing picolines under Bronsted acid catalysis. SAR again proved shallow, with all analogs 11 showing no activity as M5 PAMs; however, one of the 3-pyridyl-based 3° hydroxyls 8a proved to be a moderately active M5 PAM (M5 EC50 = 2.2 μM, 74% ACh Max), but SAR was steep.3
Scheme 2.
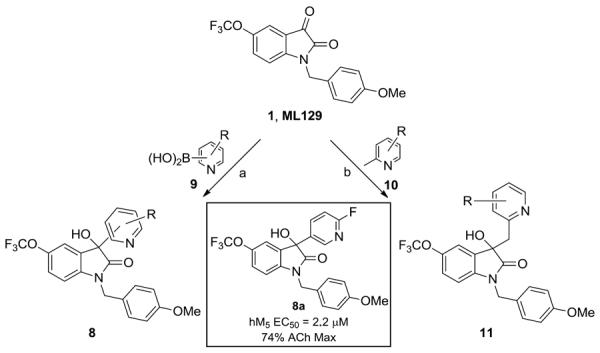
Synthesis of pyridyl-based 3° hydroxyl analogs 8 and 11. Reagents and conditions: (a) Cu(OTf)2, 1,10-phenathroline, LiOH, DCE, reflux, 48 hours, 5–45%; (b) TfOH, dioxane, 180 °C, mw, 45 min, 22–74%.
We therefore elected to introduce a more subtle variant of the istain carbonyl in the form of a spiro-oxetane, and evaluate this carbonyl isostere for M5 activity (Scheme 3). A Wittig reaction with ML129 provided 12, which was smoothly converted into the diol 13 in 57% overall yield. Mono-triflation of 13 and nucelophilic displacement proceeded in low yield (~6%) to the spioroxetane 14.25 Interestingly, 14 was inactive as an M5 PAM, and only afforded a marginal base-line increase in the ACh Max.
Scheme 3.
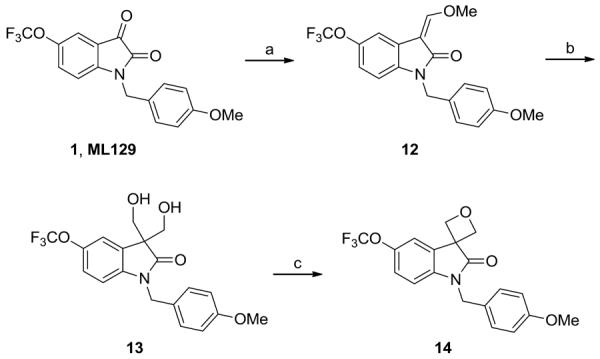
Synthesis of a spiro-oxetane analog 14. Reagents and conditions: (a) methyl methoxyphosphonium bromide, LDA, THF, −78 °C to rt, 70%; (b) PTSA, aq. formaldehyde, Na2CO3, THF:H2O, rt, 82%; (c) Tf2O, lutidine, DCM, - 78 °C to rt, 6%.
Other modifications, such as spiro-pyrrolidines and dioxalanes, which were well tolerated for M1 PAM activity in the ML137 series,19,20 were uniformly inactive on the ML129 scaffold. Clearly, SAR was steep and did not cross-over between M1 and M5. Moreover, despite the synthesis and screening of hundreds of analogs of ML129, surveying multiple regions, we were unable to develop a submicromolar M5 PAM.
At this point, we pursued a new high throughput screen (HTS) for M5 run in triple-add mode performed on the MLPCN screening deck (~360,000 compounds, PubChem AID 624103). Early in the process, and prior to official `hit' confirmation (Fig. 4), we noticed a structurally related, weak single-point (31%@3 μM) hit, CID2145491 (15). This `hit' possessed a phenethyl ether linkage, a moiety we had not yet explored on the ML129 core, and we were very surprised to find an isatin core without the 5-OCF3 moiety, and a 7-methyl group)15 with activity at M5. We then reviewed the preliminary HTS data, and found a number of related compounds 16 with diverse functionality on the phenyl ring of the phenethyl ether, as well as either a 5- or 7-methyl group on the isatin core with weak M5 activity (23–60% ACh max at 30 μM). Based on these data, we immediately questioned if the juxtaposition of ML129 with 15, leading to analog 17 would lead to an improved M5 PAM.
Figure 4.
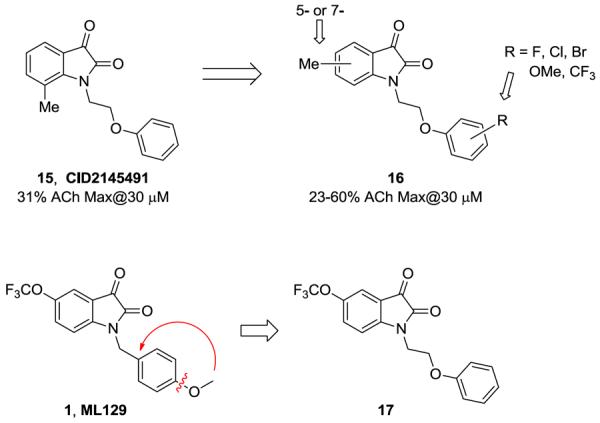
M5 Triple-add HTS PAM screening hit 15 and related analogs 16. Could juxtaposition of ML129 into 17 afford an improved M5 PAM?
To evaluate this possibility, we prepared 17 by alkylating the 5-OCF3 isatin 18 with commercial (2-bromoethyoxy)benzene under microwave-assisted conditions in 82% yield (Scheme 4).26
Scheme 4.
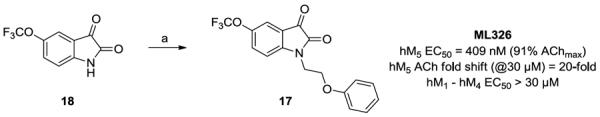
Synthesis of phenethyl ether analog 17 (ML326). Reagents and conditions: (a) (2-bromoethoxy)benzene, K2CO3, KI, ACN, mw, 160 °C, 10 min, 82%.
Gratifyingly, this structural modification led to one of the most potent M5 PAM we have ever identified. 17 (VU0467903), later declared an MLPCN probe and given the designation ML326, was a potent M5 PAM (Fig. 5) on both human (EC50 = 409 nM, 91% ACh Max, pEC50 = 6.39±0.04) and rat M5 (EC50 = 500 nM, 59% ACh Max). In fold-shift assays, ML326 afforded a robust 20-fold leftward shift of the ACh CRC, and was found to highly selective versus M1-M4 (EC50s >30 μM). Very encouraged by the initial in vitro potency and selectivity profile for ML326 we prepared gram quantities and set about characterizing it through our in-house tier 1 DMPK assays (PPB, rat/human microsomal stability with predicted intrinsic clearance, P450 inhibition, rat brain homogenate binding, etc.) and single dose PK/CNS exposure in rat. Many of these experiments were initiated in parallel, including the collection of rat plasma/brain samples; however we encountered insurmountable LCMS/MS analytical quantization issues due to poor ionization of ML326 using ESI, APPI, and APCI ionization probes, which prevented the determination of routine tier 1 DMPK parameters and in vivo rat exposure. Alternative methods including chemical derivatization and UV absorbance also failed to provide the requisite sensitivity for detection of ML326 thus preventing the completion of numerous studies. However, we were able to assess that ML326 had relatively clean CYP and ancillary pharmacology profiles.
Figure 5.
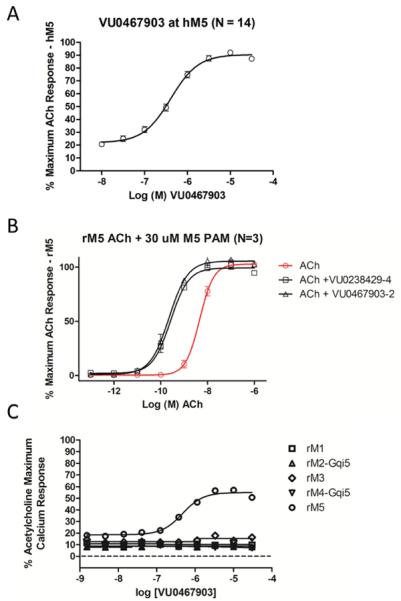
In vitro pharmacological characterization of 17 (VU0467903, ML326). A) M5 PAM concentration-response-curve (CRC) afforded an EC50 of 409 nM, 91% ACh Max; B) rat M5 fold-shift assay with 1 (VU0238429, ML129) and 17, demonstrating a robust 20-fold shift (human fold-shift data is the same). ACh EC50 = 4.58 nM, pEC50 = 8.34±0.04, ACh + 429 EC50 = 263.4 pM, pEC50 = 9.58±0.06, ACh + 903 EC50 = 225.4 pM, pEC50 = 9.65±0.06; C) rat mAChR selectivity data with 17, indicating >30 μM versus M1-M4 (human data is the same).
Based on this promising data, we prepared other analogs 19 (Table 1) with substituents in the phenyl ring of 17, following the route depicted in scheme 4. While this afforded a number of potent and selective M5 PAMs, they all suffered from the same poor ionization profiles which precluded extensive DMPK profiling. Based on the ionization issue and relatively flat SAR, this series is no longer the subject of chemical optimization.
Table 1.
Structures and activities of analogs 19.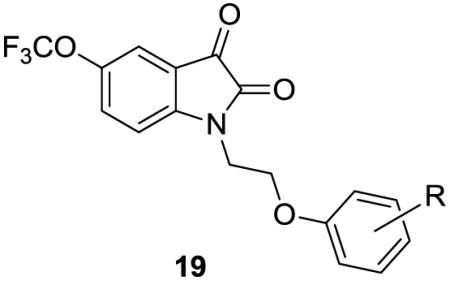
| Cmpd | R | M5 EC50 (μM) | pEC50±SEM | %ACh Max |
|---|---|---|---|---|
| ML326 | H | 0.41 | 6.39±0.04 | 91±1.6 |
| 19a | 4-OPh | 1.05 | 5.98±0.04 | 93±1.9 |
| 19b | 4-Cl | 0.45 | 6.35±0.04 | 92±1.7 |
| 19c | 4-OMe | 0.59 | 6.23±0.04 | 89±1.7 |
| 19d | 3-Me | 0.53 | 6.28±0.03 | 91±1.3 |
All values are the average of three idependent experiments.
In summary, further elaboration of the ML129 M5 PAM structure has been explored, and SAR was particularly steep. Insight from a weak M5 PAM HTS hit led to the hybridization with ML129 to afford ML326 (VU0467903), the first highly selective (>30 μM versus M1-M4) and sub-micromolar (human (EC50 = 409 nM, 91% ACh Max) and rat M5 (EC50 = 500 nM, 59% ACh Max)) M5 PAM. Interestingly, the preliminary HTS hits that inspired the structural modifications to ML129, leading to ML326 did not confirm upon re-order and full CRC. Finally, the poor ionization of ML326 precluded in-depth DMPK profiling; however, ML3276 is a valuable in vitro probe and is serving as an important probe in electrophysiology studies, which will be reported shortly.
Acknowledgments
The authors thank the NIH (U54MH084659) and William K. Warren, Jr. who funded the William K. Warren, Jr. Chair in Medicine (to CWL). Vanderbilt University is a Specialized Chemistry Center within the MLPCN and all the ML# probes are freely available upon request. Scripps was supported by the National Institute of Health Molecular Library Probe Production Center grant U54 MH084512. We thank Lina DeLuca (Lead Identification Division, Scripps Florida) for compound management.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bonner TI, Buckley NJ, Young AC, Brann MR. Science. 1987;237:527–532. doi: 10.1126/science.3037705. [DOI] [PubMed] [Google Scholar]
- 2.Dencker D, Thomsen M, Wörtwein G, Weikop P, Cui Y, Jeon J, Wess J, Fink-Jensen A. ACS Chem. Neurosci. 2012;3:80–89. doi: 10.1021/cn200110q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW. J. Med. Chem. 2012;55:1445–1464. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bridges TM, LeBois EP, Hopkins CR, Wood MR, Jones JK, Conn PJ, Lindsley CW. Drug News & Perspect. 2010;23:229–240. doi: 10.1358/dnp.2010.23.4.1416977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conn PJ, Jones C, Lindsley CW. Trends in Pharm. Sci. 2009;30:148–156. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yasuda RP, Ciesla W, Flores LR, Wall SJ, Li M, Satkus SA, Weisstein JS, Spagnola BV, Wolfe BB. Mol. Pharmacol. 1993;43:149–157. [PubMed] [Google Scholar]
- 7.Wess J, Eglen RM, Gautam D. Nat. Rev. Drug Discov. 2007;6:721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- 8.Steidl S, Miller AD, Blaha CD, Yeomans PLoS ONE. 2011;6(11):e27538. doi: 10.1371/journal.pone.0027538. doi:10.1371/journal.pone.0027538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vilaro MT, Palacios JM, Mengod G. Neurosci. Lett. 1990;114:154–159. doi: 10.1016/0304-3940(90)90064-g. [DOI] [PubMed] [Google Scholar]
- 10.Weiner DM, Levey AI, Brann MR. Proc. Natl. Acad. Sci. USA. 1990;87:7050–7054. doi: 10.1073/pnas.87.18.7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anney RJL, Lotfi-Miri M, Olsson CA, Reid SC, Hemphill SA, Patton GC. BMC Genetics. 2007;8(46) doi: 10.1186/1471-2156-8-46. doi:10.1186/1471-2156-8-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamada M, Lamping KG, Duttaroy A, Zhang W, Cui Y, Bymaster FP, McKinzie DL, Felder CC, Deng C-X, Faraci FM, Wess J. Proc. Natl. Acad. Sci. USA. 2001;98:14096–14101. doi: 10.1073/pnas.251542998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomsen M, Wörtwein G, Fink-Jessen A, Woldbye DPD, Wess J, Caine SB. Psychopharmacology. 2007;192:97–110. doi: 10.1007/s00213-006-0682-y. [DOI] [PubMed] [Google Scholar]
- 14.Araya R, Noguchi T, Yuhki M, Kitamura N, Higuchi M, Saido TC, Seki K, Itohara S, Kawano M, Tanemura K, Takashima A, Yamada K, Kondoh Y, Kanno I, Wess J, Yamada M. Neurobiol. Dis. 2006;24:334–344. doi: 10.1016/j.nbd.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 15.Bridges TM, Marlo JE, Niswender CM, Jones CK, Jadhav SB, Gentry PR, Plumley HC, Weaver CD, Conn PJ, Lindsley CW. J. Med. Chem. 2009;52:3445–3448. doi: 10.1021/jm900286j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bridges TM, Kennedy JP, Cho HP, Breininger ML, Gentry PR, Hopkins CR, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2010;20:558–562. doi: 10.1016/j.bmcl.2009.11.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bridges TM, Kennedy JP, Hopkins CR, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2010;20:5617–5622. doi: 10.1016/j.bmcl.2010.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bridges TM, Kennedy JP, Cho HP, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2010;20:1972–1975. doi: 10.1016/j.bmcl.2010.01.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melancon BJ, Poslunsey MS, Gentry PR, Tarr JC, Mattmann ME, Bridges TM, Utley TJ, Sheffler DJ, Daniels JS, Niswender CM, Conn PJ, Lindsley CW, Wood MR. Bioorg. Med. Chem. Lett. 2013;23:412–416. doi: 10.1016/j.bmcl.2012.11.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poslunsey MS, Melancon BJ, Gentry PR, Bridges TM, Utley TJ, Sheffler DJ, Daniels JS, Niswender CM, Conn PJ, Lindsley CW, Wood MR. Bioorg. Med. Chem. Lett. 2013 doi: 10.1016/j.bmcl.2013.01.017. DOI: http://dx.doi.org/10.1016/j.bmcl.2013.01.017. [DOI] [PMC free article] [PubMed]
- 21.Meshram HM, Ramesh P, Kumar S, Swetha A. Tetrahedron Lett. 2011;52:5862–5864. [Google Scholar]
- 22.Zhang J, Chen J, Ding J, Liu M, Wu H. Tetrahedron. 2011;67:9347–9351. [Google Scholar]
-
23.Scheme to access analogs 8 via lithiation/addition sequence.
- 24.Niu R, Xiao J, Liang T, Li X. Org. Lett. 2012;14:676–679. doi: 10.1021/ol2030982. [DOI] [PubMed] [Google Scholar]
- 25.Wuitschik G, Carreira EM, Wagner B, Fischer H, Parrilla I, Schuler F, Rogers-Evans M, Muller K. J. Med. Chem. 2010;53:3227–3246. doi: 10.1021/jm9018788. [DOI] [PubMed] [Google Scholar]
- 26.1-(2-phenoxyethyl)-5-(trifluoromethoxy)indoline-2,3-dione, ML326: The title compound was synthesized in one step from commercially available starting materials according to the following procedure. Into a 20 mL microwave reaction vial, containing a magnetic stir bar, were weighed 5-(trifluoromethoxy)isatin (460 mg, 2.0 mmol), K2CO3 (550 mg, 4.0 mmol), KI (33 mg, 0.20 mmol), followed by acetonitrile (20 mL, 0.1 M) and 2-bromoethyl phenyl ether (480 mg, 2.4 mmol). After being sealed with a crimp cap, the vessel was placed in a microwave reactor and heated to 160 °C for 10 minutes, with magnetic stirring. After cooling to ambient temperature, the reaction was diluted with CH2Cl2 (~20 mL) and washed with brine. The organic layer was separated and dried over Na2SO4. Solvent was removed under reduced pressure and the crude product was purified via flash column chromatography (silica gel, hexane/ethyl acetate, 0% to 50% ethyl acetate gradient). Product containing fractions were combined and the solvents removed under reduced pressure to obtain 583 mg of ML326 (83% yield) as a red-orange powder. TLC Rf = 0.79 (hexane/ethyl acetate 1:1); 1H NMR (400 MHz, CDCl3 calibrated to 7.26) δ7.52–7.46 (m, 2H), 7.31–7.25 (m, 3H), 6.98 (t, J = 7.4 Hz, 1H), 6.82 (d, 2H), 4.28 (t, J = 5.0 Hz, 2H), 4.17 (t, J = 5.0 Hz, 2H); 13C NMR (125 MHz, CDCl3 calibrated to 77.16) δ182.25, 158.27, 157.93, 150.01, 145.44, 131.02, 129.78, 121.75, 118.32, 114.39, 112.89, 65.94, 40.62; HRMS calcd for C17H13NO4F3[M+H+]; 352.0797 found 352.0795;.