

Abstract
Recent evidence suggests atypical protein kinase C (aPKC) isoforms are required for both TNF- and VEGF-induced breakdown of the blood-retinal barrier (BRB) and endothelial permeability to 70kDa dextran or albumin. A chemical library screen revealed a series of novel small molecule phenylthiophene based inhibitors of aPKC isoforms that effectively block permeability in cell culture and in vivo. In an effort to further elucidate the structural requirements of this series of inhibitors, we detail in this study a structure-activity relationship (SAR) built on screening hit 1, which expands on our initial pharmacophore model. The biological activity of our analogues was evaluated in models of bona fide aPKC-dependent signaling including NFκB driven-gene transcription as a marker for an inflammatory response and VEGF/TNF-induced vascular endothelial permeability. The EC50 for the most efficacious inhibitors (6, 32) was in the low nanomolar range in these two cellular assays. Our study demonstrates the key structural elements that confer inhibitory activity and highlights the requirement for electron-donating moieties off the C-4 aryl moiety of the 2-amino-3-carboxy-4-phenylthiophene backbone. These studies suggest that this class has potential for further development into small molecule aPKC inhibitors with therapeutic efficacy in a host of diseases involving increased vascular permeability and inflammation.
Keywords: phenylthiophenes, vascular permeability, macular edema, inflammation, protein kinase C, vascular endothelial growth factor, tumor necrosis factor
Retinal vascular permeability and subsequent macular edema (ME) contribute to the loss of vision in a host of blinding diseases. Breakdown of the BRB in response to elevated VEGF or inflammatory cytokines contributes to the pathology of diseases such as diabetic retinopathy, uveitis, and retinal vein occlusions and is associated with vision loss.1 Biological therapies targeting and binding VEGF have demonstrated excellent results in reducing macular edema and preserving visual function in recent clinical trials for many patients.2 Recently, aPKC has been identified as a common signaling molecule that mediates VEGF, TNF, CCL2 and thrombin induced permeability and BRB dysfunction.3–6 Drugs targeting aPKC may provide an effective means to control vascular permeability and prevent edema induced by multiple vasoactive cytokines.
Atypical PKC isoforms are a distinct subfamily of the PKC isoforms that do not require Ca2+ and diacylglycerol (DAG) for activation. The aPKC isoforms consist of two isozymes, aPKCζ and aPKCι (also called λ in mouse), both containing a unique regulatory C1 domain, a Phox and Bem 1 (PB1) protein-protein interacting motif, and a pseudosubstrate domain in the amine terminus with the kinase domain residing within the carboxy terminus.7 Genetic loss-of-function experiments for aPKCι reveal this kinase is intimately involved in the formation of cellular polarity evident from early embryonic lethality.8 Importantly, once polarity has been established, depletion of aPKC does not appear to induce cell polarity defects or disorganization of the junctional complex.9 Gene deletion of aPKCζ exhibits only minor defects in immune system development and alterations in NFκB signaling during the innate immune response.10,11 Numerous studies implicate aberrant aPKC signaling associated with diseases involving inflammation and proliferation; therefore, aPKC isoforms have emerged as novel therapeutic targets. Animal studies targeting aPKC isoforms have demonstrated effectiveness in models of type II diabetes, cancer, and inflammation.12–14 Hence, targeting aPKC may be an effective treatment for a variety of diseases including macular edema and inflammation.
Screening a Chembridge small molecule library, we identified a series of phenylthiophenes as potent and non-toxic aPKC inhibitors that are effective at preventing both VEGF and TNF-induced retinal permeability in the rodent retina without inducing measurable cell death or observable retinal pathology.5,15 These hits exhibit a non-competitive kinetic mechanism of action and maintain selectivity in both kinase screens and in cellular based assays supporting phenylthiophenes as a viable chemotype for aPKC inhibition.15 Several recent publications have attempted to design small molecule inhibitors of atypical PKCs and some progress has been made with isoform specificity and potency. One report describes a series of allosteric aPKCζ inhibitors that exhibits micromolar efficacy in the prevention of TNF-induced NFκB activity.13 However this class of inhibitors still exhibit robust activation of PDK1, an upstream activator of aPKCζ,17 which may lead to a less than optimal cellular response as is evident from their efficacy in cell-based assays compared to the phenylthiophene-based inhibitors.
In this manuscript, we report on medicinal chemistry efforts to further characterize and elucidate the inhibitory activity of phenylthiophene based aPKC inhibitors. Based on the screening hit 1 (Table 1), we synthesized a series of analogues to develop and explore structure-activity relationships (SAR) against an aPKC. A subset of the most potent compounds was then evaluated in cellular assays to determine their efficacy as inhibitors of retinal endothelial permeability and NFκB activation.
Table 1.
Initial phenylthiophene SAR vs. aPKCζa
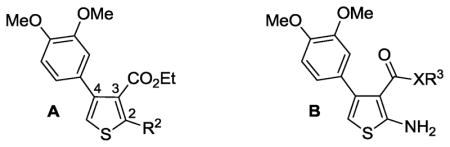
| |||
|---|---|---|---|
| No. | Str. | R2 or XR3 | Inh (%)a |
| 123 | A | NH2 | 100 |
| 2 | A | H | 3 |
| 3 | A | NHMe | 32 |
| 4 | A | NMe2 | 60 |
| 5 | A | NHCONH2 | 14 |
| 6 | B | O-i-Pr | 100 |
| 7 | B |
|
57 |
| 8 | B |
|
49 |
| 9 | B | OPh | 48 |
| 10 | B | OCH2Ph | 100 |
| 11 | B | OCH2Ph-2-Me | 64 |
| 12 | B | OCH2Ph-3-Me | 67 |
| 13 | B | OCH2Ph-4-Me | 50 |
| 14 | B | OCH2Ph-3-OMe | 100 |
| 15 | B | OCH2Ph-2-F | 100 |
| 16 | B | OCH2Ph-4-F | 2 |
| 17 | B | OCH2Ph-3-CN | 70 |
| 18 | B | OCH2Ph-4-CN | 10 |
| 19 | B | OCH2-4-pyridyl | 53 |
| 20 | B | NHEt | 50 |
| 21 | B |
|
55 |
| 22 | B | NHCH2Ph | 43 |
| 23 | B | NHCH2-4-pyridyl | 3 |
| 24a24 | B | COXR3 = CN | 87 |
| 24b | B | COXR3 = CO2H | 65 |
Inhibition determined from ADP Quest assay with 30 μM of inhibitor using aPKCζ (500 ng/ml). Values are the mean of at least n≥5 and were repeated in independent experiments. Standard error of the mean <20%. The experimental conditions are reported in the Supplementary Material.
Str. = chemical structure.
The heterocyclic 2-aminothiophene 3-carboxylic ethyl ester analogues (1, 25–28, 31–39, Table 2) were prepared in two steps by the Gewald reaction18 from aryl-substituted acetophenones and ethyl cyanoacetate (Scheme 1a). To make substructure B derivatives shown in Table 1, a two-step sequence of N-2 Boc protection and saponification of 1 gave acid 51, which was further derivatized to intermediate esters 53–66 or amides 67–70 (Table S1, Supplementary Materials) using standard coupling conditions.19 Boc deprotection with trifluoroacetic acid then provided target analogues 6–20. This same two-step sequence was applied to selected ethyl esters with different C-4 aromatic substitution patterns (Table 2) to make a subseries of benzyl ester derivatives (Scheme 1b). Analogues 24a and 24b (Table 1) were made (Methods A and C, respectively, Supplementary Materials) to compare the kinase activity of 1 with its C-3 nitrile and carboxylic acid congeners.
Table 2.
4-Aryl SAR of phenylthiophenes vs. aPKCζa
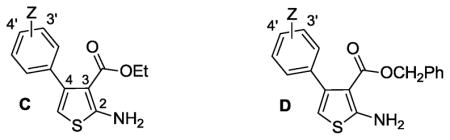
| ||||||
|---|---|---|---|---|---|---|
| Z | No. | Str. | Inh (%)a | No. | Str. | Inh |
| 3′,4′-(OMe)2 | 1 | C | 100 | 10 | D | 100 |
| 3′-Me | 2525, 26 | C | 77 | 40 | D | 68 |
| 4′-Me | 2624, 26–28, 30 | C | 80 | 41 | D | 60 |
| 3′-OMe | 2724, 26–29 | C | 82 | 42 | D | 57 |
| 4′-OMe | 2823, 28–31 | C | 100 | 43 | D | 86 |
| 4′-NH2 | 2931, 32 | C | 100 | ---- | D | n.d. |
| 4′-NMe2 | 30 | C | 100 | ---- | D | n.d. |
| 3′-NHAc | 3132 | C | 89 | ---- | D | n.d. |
| 3′,4′-OCH2O- | 3232 | C | 100 | 44 | D | 85 |
| 2′-F,4′-OMe | 33 | C | 100 | 45 | D | 78 |
| 3′-F,4′-OMe | 34 | C | 100 | D | n.d. | |
| 3′-CF3 | 3533–35 | C | 30 | 46 | D | 29 |
| 4′-CF3 | 3632 | C | 16 | 47 | D | 21 |
| 2′-F | 37 | C | 62 | 48 | D | 57 |
| 3′-F | 3832 | C | 38 | 49 | D | 53 |
| 3′-CN | 3926, 31 | C | 32 | 50 | D | 25 |
See footnote a, Table 1.
Scheme 1a.

a Reagents and conditions: (i) methyl cyanoacetate, NH4OAc, AcOH, toluene, reflux, 18–48 h; (ii) sulfur powder, Et2NH, EtOH, 50 °C, 3 h (iii) Boc2O, DMAP, pyridine, 55 °C, 3 h; (iv) KOH, aq. EtOH, reflux, 5 h; (v) for 52: R3OH, CDI, DCM or R3OH, DCC, THF or R3OH, Ph3P, DEAD, THF; (vi) TFA, DCM; (vii) for 52: R3NH2, EDC, HOBt, NEt3, DMF, 24 h.
To make C-4 anilino congeners (29, 30; Table 2) of 1 we started with nitro precursor 94 (Scheme 2). The N-2 amino function was bis-Boc protected followed by iron/acetic acid reduction to 96. Standard alkylation conditions provided N,N-dimethyl intermediate 97. Boc deprotection of 96 and 97 afforded the target compounds.
Scheme 2a.

a Reagents and conditions: (i) Boc2O, DMAP, pyridine, 55 °C, 5 h; (ii) for 95: Fe powder, AcOH, 60 °C, 1 h; (iii) MeI, K2CO3, acetone, 60 °C; (iv) TFA, DCM, rt, 16 h; (v) EtOH, 130 °C, 48 h.
To explore the effect of modifying the 2-NH2 on kinase activity, a small series of derivatives was synthesized (Scheme 3). Deamination of 1 under Sandmeyer conditions provided the C-2 protio analogue 2, whereas simple alkylation gave the mono- and dimethylamino derivatives 3 and 4, respectively. The urea 5 was prepared in two steps by reaction of 1 with trichloroacetyl isocynate followed by trichloroacetyl cleavage with ammonia.20
Scheme 3a.

a Reagents and conditions: (i) t-BuONO, CuCl2, EtOH, 1 h, then aq. NH4Cl; (ii) MeI, K2CO3, acetone 60 °C, 16 h; (iii) MeI, NaH, DMF, rt, 1 h; (iv) trichloroacetyl isocyanate, THF, 0 °C, 3 h; (v) NH3, MeOH, 0 °C, 1 h.
Isosteric replacement of the central thiophene ring of 1 to provide the pyrazole (100) and isoxazole (101) congeners was achieved by utilizing synthetic procedures for closely related compounds. Thus, condensation of enol ether 99 with hydrazine21 or hydroxylamine22 gave the respective target compounds (Scheme 4).
Scheme 4a.
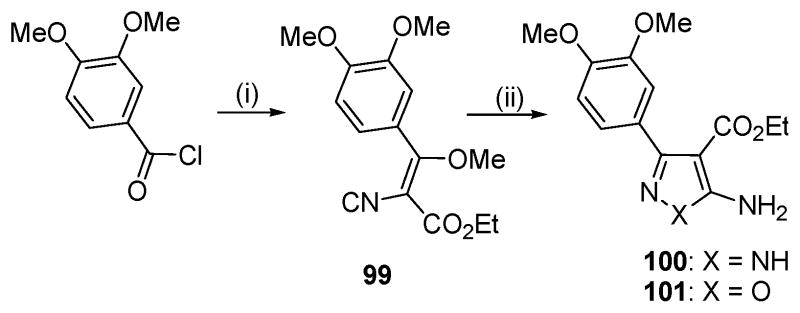
a Reagents and conditions: (i) ethyl cyanoacetate, NaH, THF, then Me2SO4, NaHCO3, aq. dioxane; (ii) for 100: NH2NH2 · HCl, NEt3, EtOH, reflux; for 101: NH2OH · HCl, NEt3, EtOH, reflux.
All compounds were rigorously purified by flash chromatography or crystallization, and their structural assignments were supported by diagnostic peaks in the 1H NMR spectra and by mass spectrometry. Several of the compounds of Tables 1 and 2 are known, but were made to help delineate an SAR for this series. References to prior preparations for each known compound are cited.
SAR for phenylthiophene analogues in an isolated kinase assay
We initially screened the phenylthiophene analogues at, 30 μM against aPKCζ in an in vitro kinase assay.
Our initial SAR efforts were directed toward exploring variations at the C-2 and C-3 positions of a series of initial phenylthiophene hits, which we had previously characterized.4 Two subseries of analogues (A and B, Table 1) were synthesized with modifications made at R2 and R3. For A, it is clear that C-2 amine substitution is required with maximal potency associated with a primary amine (1 vs 3 vs 4). Modification to R2 = H (2) or urea (5) essentially abolishes activity
Having established NH2 as an optimal substituent for R2, we then studied a number of ester variations around the C-3 position of the phenylthiophene core (subseries B, Table 1). After confirming earlier results showing significant inhibitory activity (100%) for the ethyl (1) and the 2-propyl (6) esters,4 we synthesized a series of additional ester analogues (7–19) with R3 moieties representing a range of physical, electronic and steric properties. The installation of solubilizing groups (7, 8) as well as phenyl (9) decreased inhibitory activity towards aPKCζ compared to 1. However, simple homologation of phenyl to benzyl (10) restored full inhibition so additional benzyl esters were investigated. Analogues with a range of electron donating and withdrawing substituents at open positions on the phenyl ring were synthesized (11 – 18) as well as a single example of a heterocycle (19) (Table 1). Here, substituents off the 2- and 3-positions of the phenyl ring (14, 15, 17) were well tolerated regardless of the electronics whereas 4-position (16, 18) were not. Noteworthy is the remarkable difference in activity due to positioning of fluorine (15 vs 16). This, along with the 4-cyano analogue (18), suggests that strongly electron-withdrawing moieties at the 4-position are unfavorable, which is reinforced by data for the 4-pyridyl congener (19). The mildly electron-donating methyl substituent contributes to modest inhibition regardless of positioning (11 vs 12 vs 13). Overall, the ester SAR suggests that the C-3 position of the thiophene core can tolerate lipophilic ester functionality, but with some electronic and geometric constraints.
Selected amide congeners (20 – 23, Table 1) were also prepared to evaluate the effect of an ester to amide modification at C-3. XR3 substituents of varying size and lipophilicity showed significantly lower activity suggesting a possible detrimental effect due to a different orientation of the amide substituent relative to ester. Interestingly, a nitrile substituent at R3 (24a) demonstrated significant inhibitory activity (87%), whereas the C-3 carboxylic acid analogue (24b) exhibited only moderate activity (65% inhibition).
Table 2 expands upon our initial SAR survey by looking at a number of analogues with variable substitution (Z) off the C-4 aryl moiety while retaining optimized substituents established for C-2 and C-3 as shown in Table 1. Analogues with a C-3 ethyl or benzyl ester moiety are shown in subseries C and D, respectively. A series of electron donating and withdrawing substituents were evaluated at variable positions on the C-4 aryl substituent.
Robust inhibition of aPKCζ was observed for ethyl ester analogues (subseries C) having electron-donating groups in the 3′- and 4′-positions on the phenyl ring (25 – 34). Furthermore, this inhibition was generally maintained for corresponding benzyl ester analogues (subseries D) where pairwise comparisons can be made (1 vs 10, 26 vs 41, 28 vs 43, 32 vs 44, 33 vs 45). Conversely, electron withdrawing Z-groups for ethyl ester (35 – 39) or benzyl ester (46 – 50) analogues generally significantly decreased inhibitory activity. In summary, the data of Table 2 suggest that kinase inhibition within the 2-amino-3-carboxy-4-phenylthiophene core is optimized by electron-donating Z-substituents off the C-4 aryl moiety.
The SAR established in Tables 1 and 2 suggests that a generally electron-rich heterocycle is required for optimal kinase activity. To test this hypothesis, we made two congeners of 1 in which the thiophene core was replaced by electron deficient heterocyles pyrazole (100) and isoxazole (101). Neither of these central core modifications was effective at inhibiting aPKCζ (30% and 22% inhibition, respectively, at 30 μM) (Table S2, Supplemental Materials).
Having completed our initial screening, we determined IC50 values for several of our most potent compounds from Tables 1 and 2. These range from IC50 = 1–6 μM with electron-donating groups at 3′- and 4′-positions on the phenyl ring being the most potent and with analogues 29, 30, and 32 demonstrating the highest potency (Table 3).
Table 3.
Cellular assays for selected phenylthiophenes
| No. | aPKC kinase assay | TNF-induced NFκB activation | TNF/VEGF-induced endothelial permeability | ||
|---|---|---|---|---|---|
| Inh. (%)a | IC50 (μM)a | Inh. (%)b | EC50 (μM)b | EC50 (μM)c | |
| 6 | 100 | 6 | 90 | 0.003 | 0.001 |
| 10 | 100 | 6 | 64 | 0.002 | n.d. |
| 14 | 100 | 5 | 80 | 0.001 | >0.1 |
| 29 | 100 | 2 | 72 | 0.001 | 0.27 |
| 30 | 100 | 1 | 37 | 0.001 | 0.28 |
| 32 | 100 | 2 | 34 | 0.002 | 0.02 |
| 33 | 100 | 4 | 65 | 0.01 | n.d. |
Inhibition determined from ADP Quest assay with 30 μM (reported in Tables 1 & 2) or a 5 point dose-response curve to determine IC50 against aPKCζ (500 ng/ml). Values are the mean of at least n≥5 and were repeated in independent experiments. Standard error of the mean <20%.
Inhibition determined from TNF-induced NFκB activity assay with 0.1 μM or a 5 point dose-response curve to determine EC50. Values are the mean of at least n>4 and were repeated in independent experiments. Standard error of the mean <35%.
EC50 determined from VEGF/TNF-induced retinal endothelial permeability assay using a 5 point dose response curve. Values are the mean of at least n≥8 and were repeated in independent experiments. Standard error of the mean <30%. All experimental conditions are reported in the supplementary methods. IC50 and EC50 values were calculated using a variable slope dose-response curve in GraphPad (SM methods).
Evaluation of phenylthiophene inhibitors in cell-based assays
Following IC50 determinations, we evaluated the compounds of Table 3 in two cell-based assays. A single dose of 0.1 μM was used in an initial screen to determine the ability of a compound to prevent TNF-induced NFκB transcriptional activation in a stable cell line (HEK293) expressing a NFκB reporter system. This assay is a surrogate for downstream signaling of aPKCζ as this kinase has been shown to regulate NFκB activation in multiple cell types as evidenced from an animal knockout model.36 Importantly, this assay has been used previously to determine aPKCζ inhibitor cellular efficacy.13 A robust increase of NFκB by TNF was demonstrated and complete inhibition of TNF-induced NFκB activation was verified using an IκB kinase inhibitor VII (IKK-I) (Figure S1, Supplemental Materials). Using this assay, six analogues (6, 10, 14, 29, 30, 32) were tested and all demonstrated significant inhibition of TNF-induced NFκB activity and were further analyzed using dose-response curves to determine EC50 values as depicted for compound 6 (Figure 1A). Each compound demonstrated low nanomolar potency toward suppressing NFκB activity (Table 3), demonstrating that these analogues exhibit potent anti-inflammatory effects. Importantly, this assay is a bona fide marker of aPKC activity and downstream signaling, and cellular data correlates with in vitro enzyme data supporting aPKC as the cellular target.
Figure 1.
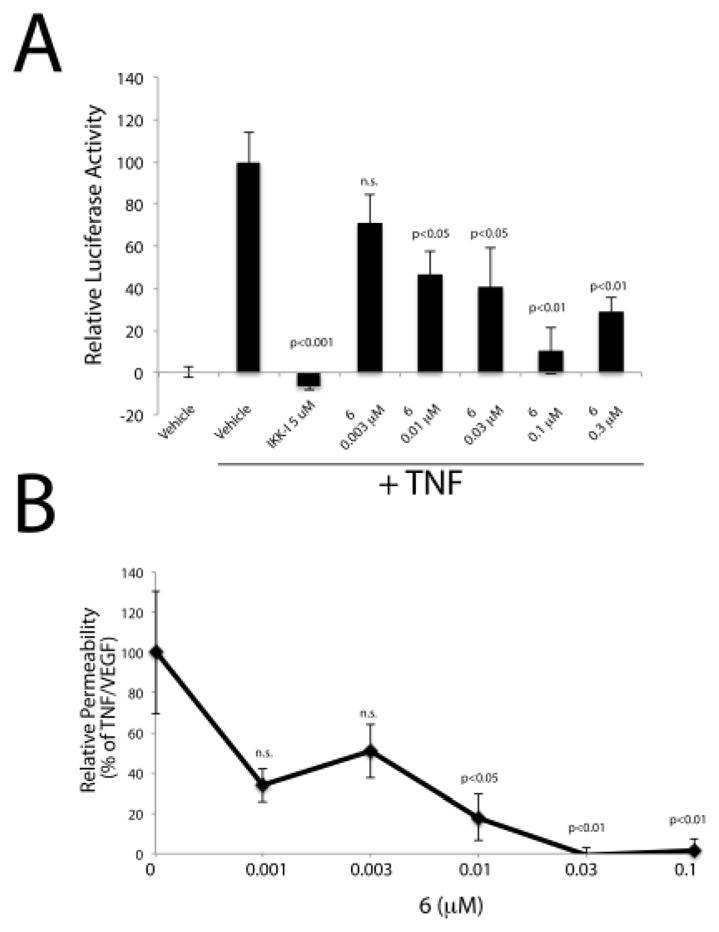
Examples of cell-based efficacy of phenylthiophenes
A) HEK293pNFkBluc cells were treated where indicated with 5 ng/ml TNF for 5 h. A 3 h pre-treatment of a dose-response of 6 was performed where indicated and a Bright-Glo luciferase reaction was performed (SM methods); n>5. B) Permeability assays were performed (SM methods) following a dose-response of compound 6 and the addition of VEGF/TNF for 1.5 h; n≥7 combined from multiple independent experiments. Statistics were performed using One way ANOVA with Neuman-Keuls posthoc test in GraphPad.
We further evaluated compounds of Table 3 in an assay to assess their capability to prevent retinal endothelial permeability, a hallmark of ME. This is a permeability assay that was performed using retinal endothelial cells and monitoring the flux of 70kDa RITC-dextran across an endothelial monolayer. This assay has been used extensively to monitor the permeabilizing effects of growth factors and inflammatory cytokines implicated in ocular disease37 and requires aPKCι.15 Dose response curves were generated with 6, 14, 29, 30 and 32 with the results shown in Table 3 along with a representative dose response curve for 6 (Figure 1B). Four compounds, (6, 29, 30, 32) showed good efficacy in preventing TNF/VEGF-induced endothelial permeability, with compound 6 exhibiting an EC50 of 1 nM.
The cell based efficacy data is supported with a favorable specificity profile for our better candidates. Importantly, 6 has been shown to exhibit a high degree of specificity towards the aPKC isoforms as compared to its closest AGC family members and does not block VEGF-induced Akt activation or Erk activation in endothelial cells.15 Compounds 6, 14, 30 and 32-display low micromolar to high nanomolar potency against aPKC isoforms without discrimination for PKCζ or PKCι (Table S3, Supplemental Materials). A preliminary in silico calculation of physico-chemical parameters (Table S4, Supplemental Materials) for selected compounds of Table 3 suggests favorable biopharmaceutical properties for this class compounds but clearly further stability, pharmacokinetic and toxicity analyses are required. cLogP values for compounds 6, 14, 30 and 32 are 4.256, 5.165, 4.459, and 3.854. In addition, topological polar surface are (TPSA) for compounds 6, 14, 30 and 32 are 70.78, 80.00, 55.55, and 70.78, respectively. Overall, the enzyme and cellular data support this class of phenylthiophenes as potent and specific inhibitors of aPKC isoforms with significant efficacy in preventing vascular permeability and NFκB-induced gene transcription.
Previously, we reported a group of phenylthiophene based molecules as the first biologically active aPKC inhibitors with high enzyme specificity and a non-competitive mode of action with nanomolar efficacy in cells.15 In this paper, we have expanded the screening for aPKC inhibitors by developing an SAR to further delineate structural requirements and refine a pharmacophore for this new class of aPKC inhibitors. The addition of electron donating groups (Z) to the C-4 aryl substituent, principally in the 3′- and 4′-positions, combined with a C-2 primary amine and C-3 ester moiety confer robust inhibitory activity for this class of compounds. The reason for the discrepancy between kinase potency (IC50 low micromolar) and cellular potency (EC50 low nanomolar) is unclear but does not appear to be due to off-target effects as 6 has been shown to possess excellent selectivity in a panel of 20 AGC kinases and this inhibitor class has been demonstrated to not affect canonical VEGF signaling cascades.15
Our series of phenythiophenes are the first compounds to exhibit low nanomolar activity against aPKC activity in cells with biomarkers of both NFκB dependent gene expression and TNF/VEGF-induced vascular permeability. Our data suggest that the phenylthiophene pharmacophore can be further optimized to provide therapeutic agents with the potential to treat diseases, such as macular edema, that involve vascular permeability induced by growth factors such as VEGF and inflammatory cytokines such as TNF. More broadly we believe that expansion of this class can lead to agents that target acute inflammatory responses.
Future SAR will focus on molecular modifications that further expand upon the current series to optimize drug-like properties while maintaining aPKC selectivity and improving potency. These will entail modifications of the phenylthiophene core, which will include exploration of the C-5 position. Our current studies provide a good starting point to achieve this goal.
Supplementary Material
Acknowledgments
This research was supported by research grants from JDRF, NIH (R01 EY012021), and support from Research to Prevent Blindness (DAA). HDHS and YJ acknowledge generous support by the University of Michigan College of Pharmacy Ella and Hans Vahlteich Research Fund.
Footnotes
Details of the chemical syntheses and assays performed can be found in Supplemental Materials.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Pan PS, Vasko RC, Lapera SA, Johnson VA, Sellers RP, Lin CC, Pan CM, Davis MR, Ardi VC, McAlpine SR. Bioorg Med Chem. 2009;17:5806. doi: 10.1016/j.bmc.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nguyen QD, Brown DM, Marcus DM, Boyer DS, Patel S, Feiner L, Gibson A, Sy J, Rundle AC, Hopkins JJ, Rubio RG, Ehrlich JS. Ophthalmology. 2012;119:789. doi: 10.1016/j.ophtha.2011.12.039. [DOI] [PubMed] [Google Scholar]
- 3.Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2006;26:797. doi: 10.1038/sj.jcbfm.9600229. [DOI] [PubMed] [Google Scholar]
- 4.Titchenell PM, Lin CM, Keil JM, Sundstrom JM, Smith CD, Antonetti DA. Biochem J. 2012;446:455. doi: 10.1042/BJ20111961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aveleira CA, Lin CM, Abcouwer SF, Ambrosio AF, Antonetti DA. Diabetes. 2010;59:2872. doi: 10.2337/db09-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minshall RD, Vandenbroucke EE, Holinstat M, Place AT, Tiruppathi C, Vogel SM, van Nieuw Amerongen GP, Mehta D, Malik AB. Microvasc Res. 2010;80:240. doi: 10.1016/j.mvr.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steinberg SF. Physiol Rev. 2008;88:1341. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moscat J, Rennert P, Diaz-Meco MT. Cell Death and Differentiation. 2006;13:702. doi: 10.1038/sj.cdd.4401823. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki A, Yamanaka T, Hirose T, Manabe N, Mizuno K, Shimizu M, Akimoto K, Izumi Y, Ohnishi T, Ohno S. J Cell Biol. 2001;152:1183. doi: 10.1083/jcb.152.6.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leitges M, Sanz L, Martin P, Duran A, Braun U, Garcia JF, Camacho F, Diaz-Meco MT, Rennert PD, Moscat J. Mol Cell. 2001;8:771. doi: 10.1016/s1097-2765(01)00361-6. [DOI] [PubMed] [Google Scholar]
- 11.Bandyopadhyay G, Standaert ML, Sajan MP, Kanoh Y, Miura A, Braun U, Kruse F, Leitges M, Farese RV. Molecular Endocrinology. 2004;18:373. doi: 10.1210/me.2003-0087. [DOI] [PubMed] [Google Scholar]
- 12.Sajan MP, Nimal S, Mastorides S, Acevedo-Duncan M, Kahn CR, Fields AP, Braun U, Leitges M, Farese RV. Metabolism: Clinical and Experimental. 2012;61:459. doi: 10.1016/j.metabol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frohner W, Lopez-Garcia LA, Neimanis S, Weber N, Navratil J, Maurer F, Stroba A, Zhang H, Biondi RM, Engel M. Journal of Medicinal Chemistry. 2011;54:6714. doi: 10.1021/jm2005892. [DOI] [PubMed] [Google Scholar]
- 14.Murray NR, Kalari KR, Fields AP. Journal of Cellular Physiology. 2011;226:879. doi: 10.1002/jcp.22463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Titchenell PM, Lin CM, Keil JM, Sundstrom JM, Smith CD, Antonetti DA. The Biochemical journal. 2012;446:455. doi: 10.1042/BJ20111961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baell JB, Holloway GA. J Med Chem. 2010;53:2719. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 17.Balendran A, Biondi RM, Cheung PC, Casamayor A, Deak M, Alessi DR. J Biol Chem. 2000;275:20806. doi: 10.1074/jbc.M000421200. [DOI] [PubMed] [Google Scholar]
- 18.Buchstaller HP, Siebert CD, Lyssy RH, Frank I, Duran A, Gottschlich R, Noe CR. Monatsh Chem. 2001;132:279. [Google Scholar]
- 19.Montalbetti CAGN, Falque V. Tetrahedron. 2005;61:10827. [Google Scholar]
- 20.Janetka JW, Almeida L, Ashwell S, Brassil PJ, Daly K, Deng C, Gero T, Glynn RE, Horn CL, Ioannidis S, Lyne P, Newcombe NJ, Oza VB, Pass M, Springer SK, Su M, Toader D, Vasbinder MM, Yu D, Yu Y, Zabludoff SD. Bioorg Med Chem Lett. 2008;18:4242. doi: 10.1016/j.bmcl.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 21.Velicelebi G, Stauderman KA, King F, Pei Y, Whitten JP. 2009076454A2. PCT Int Appl WO.
- 22.Breinlinger EC, Cusack KP, Hobson AD, Li B, Gordon TD, Stoffel RH, Wallace GA, Gronsgaard P, Wang L. 2009011850A2. PCT Int Appl WO.
- 23.Shvedov VI, Ryzhkova VK, Grinev AN. Khim Geterotsikl Soedin. 1967:239. [Google Scholar]
- 24.Chen D, Lee KC, Terrell LR. 2011075559A1. PCT Int Appl WO.
- 25.Moorman AR, Romagnoli R, Baraldi PG. 2002083083A2. PCT Int Appl WO.
- 26.Nara H, Kaieda A, Sato K, Terauchi J. 2007049820A1. PCT Int Appl WO.
- 27.Cravo D, Hallakou-Bozec S, Bolze S, Lepifre F, Faveriel L, Durand J-D, Charon C. 2011080277A1. PCT Int Appl WO.
- 28.Stein RL, Case A, Yeh L-A, Cuny G, Duval E. 2006060702A1. PCT Int Appl WO.
- 29.Duval E, Case A, Stein RL, Cuny GD. Bioorg Med Chem Lett. 2005;15:1885. doi: 10.1016/j.bmcl.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Kathiravan MK, Shishoo CJ, Chitre TS, Mahadik KR, Jain KS. Synth Commun. 2007;37:4273. [Google Scholar]
- 31.Tormyshev VM, Trukhin DV, Rogozhnikova OY, Mikhalina TV, Troitskaya TI, Flinn A. Synlett. 2006:2559. [Google Scholar]
- 32.Hodge CN, Janzen WP, Williams KP, Cheatham LA. 2005033102A2. PCT Int Appl WO.
- 33.Wang Y, Yang T. 2012100732A1. PCT Int Appl WO.
- 34.Scammells PJ, Aurelio L, Christopoulos A, Sexton PM, Valant C, Flynn BL, Linden JM. 2009049362A1. PCT Int Appl WO.
- 35.Aurelio L, Figler H, Flynn BL, Linden J, Scammells PJ. Bioorg Med Chem. 2008;16:1319. doi: 10.1016/j.bmc.2007.10.065. [DOI] [PubMed] [Google Scholar]
- 36.Diaz-Meco MT, Moscat J. Immunological Reviews. 2012;246:154. doi: 10.1111/j.1600-065X.2012.01093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Antonetti DA, Wolpert EB. Methods Mol Med. 2003;89:365. doi: 10.1385/1-59259-419-0:365. [DOI] [PubMed] [Google Scholar]
- 38.Huth JR, Mendoza R, Olejniczak ET, Johnson RW, Cothron DA, Liu Y, Lerner CG, Chen J, Hajduk PJ. Journal of the American Chemical Society. 2005;127:217. doi: 10.1021/ja0455547. [DOI] [PubMed] [Google Scholar]
- 39.Mansuy D. Journal of hepatology. 1997;26(Suppl 2):22. doi: 10.1016/s0168-8278(97)80493-x. [DOI] [PubMed] [Google Scholar]
- 40.Fouda HG, Avery MJ, Dalvie D, Falkner FC, Melvin LS, Ronfeld RA. Drug Metab Dispos. 1997;25:140. [PubMed] [Google Scholar]
- 41.Medower C, Wen L, Johnson WW. Chem Res Toxicol. 2008;21:1570. doi: 10.1021/tx700430n. [DOI] [PubMed] [Google Scholar]
- 42.Dalvie DK, Kalgutkar AS, Khojasteh-Bakht SC, Obach RS, O’Donnell JP. Chem Res Toxicol. 2002;15:269. doi: 10.1021/tx015574b. [DOI] [PubMed] [Google Scholar]
- 43.Pereillo JM, Maftouh M, Andrieu A, Uzabiaga MF, Fedeli O, Savi P, Pascal M, Herbert JM, Maffrand JP, Picard C. Drug Metab Dispos. 2002;30:1288. doi: 10.1124/dmd.30.11.1288. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.