

Abstract
The mammalian target of rapamycin (mTOR) is a conserved protein kinase involved in a multitude of cellular processes including cell growth. Increased mTOR activation is observed in multiple human cancers and inhibition of mTOR has proven efficacious in numerous clinical trials. mTOR comprises two complexes, termed mTORC1 and mTORC2. Both complexes respond to growth factors, whereas only mTORC1 is controlled by nutrients, such as glucose and amino acids. Since the discovery of mTOR, extensive studies have intricately detailed the molecular mechanisms by which mTORC1 is regulated. Somewhat paradoxically, amino acid induced mTORC1 activation—arguably the most essential stimulus leading to mTORC1 activation—is the least understood. Here we review the current knowledge of nutrient dependent regulation of mTORC1.
TOR signaling pathway
The target of rapamycin (TOR, mTOR in mammals or also referred to as mechanistic TOR) is a conserved atypical serine/threonine protein kinase that belongs to the phosphatidylinositol 3-kinase-related kinase (PIKK) family; however, it is a protein kinase. As its name implies, it is the target of rapamycin, a natural compound first isolated 37 years ago from the bacterium Streptomyces hygroscopicus and soon afterwards discovered to have antiproliferative properties. 16 years later, genetic screening in Saccharomyces cerevisiae identified that mutated TOR1 and TOR2 genes conferred rapamycin resistance[1, 2]. Subsequent studies in mammals uncovered mTOR as the target of rapamycin[3-5]. Analogues of rapamycin are currently in clinical trials for the treatment of various cancers. Everolimus and Temsirolimus have recently been approved for late stage renal cancer[6].
Early studies in yeast revealed TOR as a multi-functional protein kinase and not all functions of TOR were sensitive to inhibition by rapamycin. This led to the discovery of two distinct TOR complexes, TORC1 and TORC2[7]. The two TOR complexes are conserved in mammals, referred to as mTORC1 and mTORC2, the former of which is potently inhibited by rapamycin[8, 9]. Later studies demonstrated that prolonged rapamycin treatment also inhibits mTORC2 assembly by sequestering mTOR in some cell types[10, 11]. Growth factors control both mTOR complexes and mTORC1 is also regulated by stress and nutrients, such as amino acids (AAs) and glucose (Figure 1) [12]. This review focuses on nutrient regulation of mTORC1; additional information on mTORC2 is reviewed elsewhere [13]. mTOR is the catalytic subunit of mTORC1. Other components of mTORC1 include: Regulatory-associated protein of mTOR (Raptor), which is involved in substrate recognition; mTORC1 inhibitory modulators proline-rich AKT/PKB substrate 40 kDa (PRAS40) and Dep-domain mTOR interacting protein (Deptor); and the positive mTORC1 regulator mammalian lethal with sec-13 protein 8 (mLST8, also known as GβL)[14]. Three independent groups recently showed that mTOR controls its own activation by degrading Deptor through SCF(βTrCP) E3 ligase, although the mechanistic details of these studies differ significantly[15-17].
Figure 1. The mTOR signaling cascade.
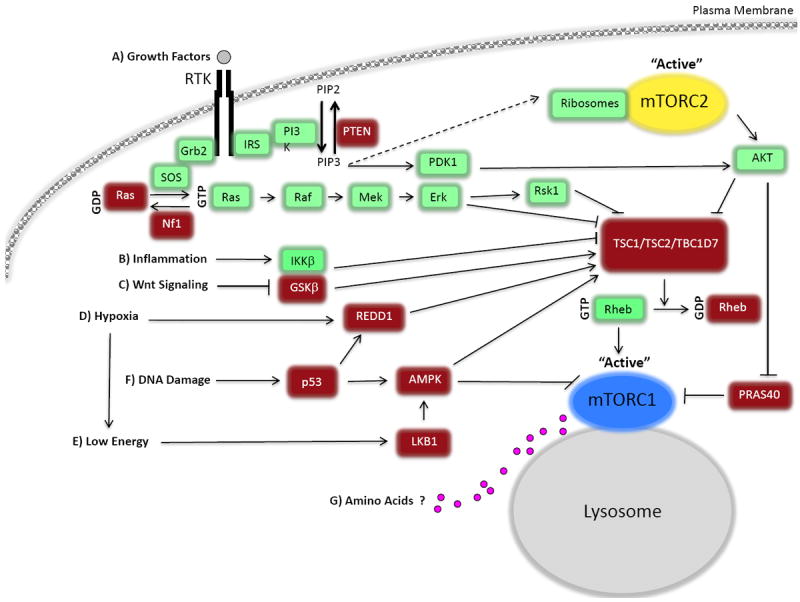
Mammalian target of rapamycin complex 1 (mTORC1) and 2 (mTORC2) are both controlled by growth factors, whereas mTORC1 is also regulated by cellular energy status, oxygen, stress, and amino acids (AAs). A) mTOR is controlled by growth factors through the classical phosphatidylinositide 3-kinase-protein kinase B (also known as AKT), PI3K-AKT, pathway and through the Ras signaling cascade. PI3K is activated and recruited to the membrane by insulin receptor substrate (IRS-1), where it catalyzes the phosphorylation of PtdIns(4,5)P2 (PIP2) to PtdIns(3,4,5)P3 (PIP3). AKT is recruited via its pleckstrin homology (PH) domain binding to PIP3, and is phosphorylated and activated by the phosphoinositide-dependent kinase-1 (PDK1). mTORC2 is thought to be activated downstream of PI3K, possibly by ribosome binding, although many unanswered questions still remain. Dashed arrow represents missing components possibly involved that have not been identified. Full activation of AKT requires the phosphorylation of T308 (by PDK1) and S473 (by mTORC2). Although, the requirement of AKT phosphorylation at T308 or S473 depends on its substrate. AKT kinase activation can promote mTORC1 signaling in two ways: by phosphorylating and inhibiting tuberous sclerosis complex 2 (TSC2) GTPase activating protein (GAP) activity, thus activating Rheb, and by phosphorylating proline-rich AKT/PKB substrate 40 kDa (PRAS40), which increases PRAS40-14-3-3 binding and relieves PRAS40 inhibition on mTORC1. In addition to AKT, the Ras pathway regulates mTORC1 through ERK and RSK1, which phosphorylate and inhibit TSC1/2. TBC1D7, a third subunit of TSC, promotes TSC1-TSC2 interaction and Rheb-GAP activity. RTK, denotes receptor tyrosine kinase. B) IKKβ can also inhibit TSC1/TSC2 in response to inflammation. C) GSK3-β, which is regulated by Wnt signaling, phosphorylates TSC2 and increases its GAP activity in an AMP-activated dependent kinase (AMPK)-priming phosphorylating manner. D) Hypoxia promotes expression of the regulated in development and DNA damage responses 1 (REDD1), which activates TSC1/TSC2, and hypoxia activates AMPK due to the failure to generate sufficient ATP. E) When cellular energy is low AMPK is activated and regulates mTORC1 by phosphorylating and activating TSC2. Also, AMPK can phosphorylate Raptor and inhibit mTORC1 function. F) DNA damage results in the inhibition of mTORC1 activity through the p53-dependent up-regulation of REDD1 and AMPK. G) AA signaling activates mTORC1 (see Figure 3). Pink circles represent AAs.
Ribosomal S6 kinase (S6K) and eIF4E binding protein (4EBP, also known as PHAS-1) are the two best-characterized substrates of mTORC1, which promote protein synthesis (reviewed in [18]). Although mTORC1 has been shown to regulate translation in numerous studies, the overall translational program controlled by mTORC1 has been uncertain until recently. Two global profiling studies have identified specific mRNAs whose translation is strongly stimulated by mTORC1; they encode proteins involved in translation, cell proliferation, invasion, and metabolism [19, 20]. In addition to controlling protein synthesis, mTORC1 has been shown to target and control components involved in autophagy, lipid synthesis, insulin action, and ribosome biosynthesis (reviewed in [14]). High mTORC1 activation suppresses autophagy under nutrient sufficiency. Recent studies have demonstrated that phosphorylation of ULK1 and ATG13, both subunits of the autophagy initiating kinase ULK1 complex, may represent an underlying mechanism of autophagy inhibition by mTORC1 [21-24].
Multiple upstream signals including growth factors, stress, and nutrients control mTORC1 (Figure 1) [12]. Growth factors activate the PI3K-AKT-TSC signaling cascade. TSC1 and TSC2 form a physical and functional TSC complex. TSC1 stabilizes TSC2 [25, 26], and TSC2 acts as a GTPase activating protein (GAP) to promote the inherent GTP hydrolysis activity of the small GTPase Rheb [27-32]. A third component of TSC, TBC1D7, has recently been identified; it is thought to promote TSC1-TSC2 interaction and Rheb-GAP activity[33]. Activated GTP-bound Rheb, under TSC inhibition, binds to and potently activates mTORC1 through an unknown mode of action. In addition to growth factors, additional stimuli such as inflammation, Wnt signaling, hypoxia, low energy status, and DNA damage unite at the TSC complex in order to control mTORC1 (reviewed in [34]). Therefore, the TSC complex represents a crucial upstream regulator of mTORC1 (Figure 1).
Nutrient availability is fundamental for cell growth and the survival of all organisms. Cells respond to the amount of nutrients by triggering either energy-consuming anabolic pathways under nutrient sufficiency, or energy-producing catabolic pathways under stress and starvation conditions. mTOR orchestrates these processes; it is activated under nutrient rich conditions and its function blocked under nutrient limiting conditions. mTORC1 carefully integrates these signals to control many fundamental processes involved in cellular metabolism and growth. Glucose availability and the fluctuation of energy is translated to mTOR by AMP-activated protein kinase (AMPK), which directly senses energy fluctuation[35]. By contrast, the identification of the AA sensor or sensors and their location – extracellular, intracellular, or within the lysosome -- remain unclear. Many intermediates in controlling AA mTORC1 activation have been identified including the best-characterized Rag GTPases (Table 1). However, the discovery of some components significantly conflict with others, obscuring a clear pathway and possibly implicating more than one. In this review, we outline recent advancements made in deciphering the molecular mechanisms involved in the nutrient-mTORC1 signaling cascade, including newly recognized components. Furthermore, we highlight unanswered questions and what future directions should entail.
Table 1.
Components reported to be involved in amino acid signaling to mTORC1
| Component | Function | mTORC1 Activation | References |
|---|---|---|---|
| Arf | Small GTPase, vesicle transport mediator | ↓ | [70] |
| C7orf59 | Component of the Ragulator complex, Ragulator is GEF for Rag A/B | ↑ | [55] |
| GCN2 | Kinase, binds uncharged tRNA | ↓ | [79-80] |
| HBXIP | Component of the Ragulator complex, Ragulator is GEF for Rag A/B | ↑ | [55] |
| IPMK | Inositol and lipid kinase, binds mTOR & Raptor | ↑ | [75] |
| LeuRS | Enzyme, charges leucine to cognate tRNA, binds to Rags, reported as a RagD GAP | ↑ | [71-72] |
| MAP4K3 | Kinase, thought to be activated downstream AA but upstream of mTORC1 | ↑ | [91-92] |
| mLST8 (GβL) | Binds both mTOR complexes, necessary for mTOR-Raptor AA sensitive interaction | ↑ | [73-74] |
| MP1 | Component of Ragulator complex, Ragulator is GEF for RagA/B | ↑ | [51,55] |
| mTOR | Kinase, catalytic subunit of mTORC1 | ↑ | [93] |
| p14 | Component of Ragulator complex, Ragulator is GEF for Rag A/B | ↑ | [51,55] |
| p18 | Component of Ragulator complex, anchors complex to lysosome, Ragulator is GEF for Rag A/B | ↑ | [51,55] |
| p62 | Targets cargo for autophagy, binds to Rag Complex | ↑ | [94] |
| PAT1 | Transporter at the lysosome involved in AA mTORC1 activation | ↓ | [62,65-67] |
| PLD | Enzyme, catalyzes the hydrolysis of PC to form PA, binds & activates Rheb | ↑ | [81-82] |
| Rab5 | Small GTPase, vesicle transport mediator, inhibition of mTORC1 dependent on Rags | ↓ | [70] |
| RalA | Small GTPase, partially rescues mTORC1 when Rheb is absent | ↑ | [69] |
| RagA/B | Small GTPase, heterodimerizes with RagC/D, binds to Raptor, GTP loaded to be active | ↑ | [41,48] |
| RagC/D | Small GTPase, heterodimerizes with RagA/B, binds to Raptor, GDP loaded to be active | ↑ | [41,48] |
| Raptor | Regulatory component of mTORC1, involved in substrate recognition | ↑ | [73-74] |
| Rheb | Small GTPase, Rheb-GTP activates mTORC1 via binding | ↑ | [52] |
| SH3BP4 | Binds to the inactive Rag complex | ↓ | [83] |
| VPS34 | Lipid kinase, reported to promote mTORC1 activation | ↑ | [95] |
| VPS39 | Saccharomyces cerevisiae homolog Vam6 reported as Gtr1 GEF, VPS39 doesn’t appear to be RagA/B GEF | ↑ | [55, 57] |
| v-ATPase | Maintains PH homeostasis & function of lysosome, binds to Rags & Ragulator | ↑ | [62] |
| SLC7A5/SLC3A2 | Bidirectional transporter which imports leucine, involved in mTORC1 activation | ↑ | [45] |
| SLC1A5 | Regulates glutamine uptake, involved in mTORC1 activation | ↑ | [45] |
| T1R1/T1R3 | G-protein-coupled taste receptor, senses extracellular AAs to promote mTORC1 activation | ↑ | [96] |
| TTT-RUVBL1/2 | Binds to mTORC1, promotes mTORC1 activation | ↑ | [84] |
AMPK sensing of nutrients and energy
AMPK, a serine/threonine kinase, is a crucial cellular energy sensor found in all eukaryotes and is activated when there is an increase in cellular AMP or ADP. Under nutrient starvation conditions, AMPK activation conserves energy for the cell by phosphorylating numerous substrates to inhibit anabolic processes and promote catabolic processes. AMPK is a heterotrimeric complex composed of a catalytic (α) and two regulatory (β and γ) subunits. Myristoylation of the β subunit is required for membrane localization and activation[35]. Low energy status is directly sensed by AMPK; the γ subunit binds to AMP and ADP, enforcing a conformational change that blocks dephosphorylation of AMPK to keep it in an activated state. The phosphorylation of AMPK at threonine 172 in the activation loop is essential for its activation. Liver kinase B1 (LKB1; also known as STK11), a tumor suppressor gene mutated in Peutz-Jeghers syndrome, is the major upstream AMPK threonine 172 kinase. LKB1 is allosterically activated through the interaction with the pseudokinase STRAD and the adaptor protein MO25. Threonine 172 can also be phosphorylated by calcium/calmodulin-activated kinase, CAMKKβ (also known as CAMKK2). However, CAMKKβ mediated phosphorylation of AMPK occurs with an intracellular increase in calcium, and not necessarily changes in AMP and ADP levels[36].
Under nutrient starvation conditions AMPK acts as a so-called “metabolic checkpoint,” relaying messages to mTORC1 through direct phosphorylation of TSC2 and Raptor to inhibit cell growth and preserve energy (Figure 2). AMPK blocks mTORC1 activation and signaling by phosphorylating TSC2 and activating its GAP activity [37, 38]. An increase in the GAP activity of TSC2 decreases Rheb-GTP and mTORC1 activation. Phosphorylation of TSC2 by AMPK acts as a primer for the phosphorylation and activation of TSC2 function by glycogen synthase kinase 3 β (GSK3-β). Wnt signaling promotes mTOR activation and cell growth through the inhibition GSK3-β[37]. In addition to mTORC1 inhibition through TSC2, AMPK inhibits mTORC1 activation by direct phosphorylation of Raptor, inducing 14-3-3 and Raptor binding[38]. Hypoxia promotes AMPK-TSC activation and suppresses mTORC1 by reducing ATP production. Also, increased expression of the hypoxia-inducible regulated in development and DNA damage response 1 (REDD1) gene can inhibit mTORC1 in a TSC-dependent manner[39, 40]. Many well worked out nutrient sensing mechanisms by AMPK in controlling mTORC1 have been documented. It will be interesting what future findings will uncover, especially what additional crosstalk and cues regulate AMPK, which could impact mTORC1 and cell growth.
Figure 2. Energy sensing by AMPK and control of mTORC1.

AMP-activated protein kinase (AMPK) acts as a metabolic checkpoint under nutrient starvation conditions, translating signals to mTORC1 through direct phosphorylation of tuberous sclerosis complex 2 (TSC2) and Raptor and inhibiting cell growth. The phosphorylation of AMPK at threonine 172 by liver kinase B1 (LKB1) in the activation loop is essential for AMPK activation. LKB1 is in a complex with STRAD and MO25. AMPK phosphorylates and activates TSC1/2 by phosphorylating TSC2, which inhibits mTORC1. AMPK also phosphorylates regulatory-associated protein of mTOR (Raptor) in a parallel pathway on sites that induce Raptor-14-3-3 binding and inhibition of mTORC1. RTK denotes receptor tyrosine kinase. P represents phosphorylation.
Amino acid signaling to mTORC1
Compared to glucose and energy sensing by AMPK, how AAs are sensed and regulate mTORC1 is poorly understood. AAs are building blocks of proteins that promote cell growth, and not surprisingly AAs drive the mTORC1 pathway. Organisms tightly coordinate the balance between anabolic and catabolic events to best utilize not only energy but also AAs. In fact, AAs are essential for mTORC1 activation, as growth factors and other stimuli cannot efficiently activate mTOR when AAs are limiting[41-43]. Specific AAs required for the activation of mTORC1 are not completely understood, although withdrawal of the essential AAs leucine and arginine is as efficient as total AA removal in down-regulating mTOR signaling[44]. Additionally, glutamine is required for extracellular leucine to activate mTOR[45] and recently glutamine metabolism has been shown to control mTORC1 (Box 1) [46, 47]. Exactly how AAs control mTORC1 is currently a hot topic, resulting in many recent publications identifying new components aiming to decode the mechanisms involved in this signaling cascade (Table 1).
Box 1. Regulation of mTORC1 by glutamine metabolism.
Glutamine is the most abundant AA in the blood. The metabolism of glutamine occurs through a process of double deamination called glutaminolysis, to produce α-ketoglutarate (αKG) (Figure I). First, glutamine is de-aminated by glutaminase (GLS) to produce glutamate. Conversion of glutamate to αKG is performed by glutamate dehydrogenase (GDH). Leucine is an allosteric regulator of GDH, directly binding to GDH, aiding in the promotion of glutaminolysis by stimulating de-amination of glutamate to form αKG. Glutaminolysis has recently been implicated to stimulate Rag-mTORC1 signaling. αKG is thought to activate mTORC1 through a family of αKG-dependent dioxygenases, prolyl hydroxylases. Glutamine metabolism is important to sustain ATP levels, through αKG replenishing the TCA cycle, and cancer cells are thought to be addicted to glutamine. Glutamine in combination with leucine increases RagA/BGTP promoting mTORC1 activation (Figure 3). Chemical inhibition of glutaminolysis by 6-diazo-5-oxo-L-norleucine (DON) decreases RagA/BGTP formation, mTOR lysosomal localization, thus mTORC1 activation. Consistently, overexpression of the constitutively active Rag complex, RagA/BGTP-RagC/DGDP, reverses mTORC1 inhibition by DON. Furthermore, glutaminolysis was demonstrated to regulate autophagy and cell size through mTORC1 [46]. Consistently, elevation of glutamine synthetase (GS), the enzyme that catalyzes the opposite reaction to that of GLS, inhibits mTORC1 lysosomal translocation and activation, and promotes autophagy[88]. These findings demonstrate the crosstalk between metabolism and cell signaling and begin to build a model in how mTORC1 may sense the fluctuations of glutamine and leucine together. αKG production from glutamine activates mTORC1 and inhibits autophagy, whereas the synthesis of glutamine inhibits mTORC1 and promotes autophagy.
Figure I. mTORC1 activation controlled by glutamine.
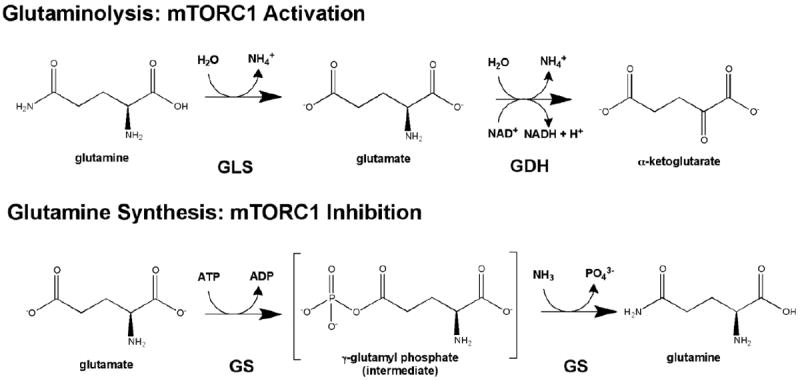
Glutamine metabolism controls mTORC1. Top- Glutaminolysis, the metabolism of glutamine to α-ketoglutarate (αKG), is a two step process which results in the actiavtion of mTORC1. Glutamine is first de-aminated by glutaminase (GLS) to produce glutamate, and then glutamate dehydrogenase (GDH) produces a-KG. Bottom- In contrast, the synthesis of glutamine inhibits mTORC1 activation. Glutamine is synthesized by glutamine synthetase from glutamate.
Rag GTPases
Center stage in the AA-dependent induction of mTORC1 is the Rag family of GTPases, possibly the strongest link to date between AA signaling and mTORC1 (Figure 3). Two independent groups identified this connection using different approaches. One study in mammalian cells identified the Rags as Raptor-interacting proteins through an immunoprecipitation-mass spectrometric analysis approach, and the other screened small GTPases using RNA interference (RNAi) in Drosophila cells[41, 48]. There are four Rag proteins in mammals: RagA and RagB (~98% sequence similarity) and RagC and RagD (~87% sequence similarity)[12]. RagA or RagB forms a heterodimer with RagC or RagD; thus, four different complexes are possible. Ras family GTPases have not been reported to form stable dimers, making this an exclusive feature of the Rags. In yeast, the Rag proteins are homologs to Gtr1p (RagA or RagB) and Gtr2p (RagC or RagD). The first connection between the Rags and TORC1 in yeast was the discovery of Gtr2p as a potential downstream effector of TORC1 [49]. Structurally, both Gtr1p and Gtr2p are composed of a N-terminal Ras-like GTPase domain important in guanine nucleotide binding and a C-terminal domain that is crucial for Gtr1p-Gtr2p interaction[50]. AAs promote the active conformation of the Rag complex, in which RagA/B is loaded with GTP (RagA/BGTP) and RagC/D is loaded with GDP (RagC/DGDP). Upon AA-rich conditions the RagA/BGTP-RagC/DGDP heterodimer physically interacts with Raptor to recruit mTORC1 from an undefined location within the cell to the lysosome. Immunofluorescent studies show withdrawal of AAs results in the dispersion of endogenous mTOR throughout the cell, whereas AA stimulation promotes the co-localization of mTOR and LAMP2 (lysosomal-associated membrane protein). The Rags are thought to shuttle mTORC1 to the lysosome, placing it in close proximity to Rheb, a potent activator of mTORC1. A mutated constitutively active Rag complex (RagA/BGTP-RagC/DGDP) can bind to and activate mTORC1 in the absence of AAs. Consistently, the inactive Rag Complex (RagA/BGDP-RagC/DGTP) does not interact with mTORC1 and suppresses mTORC1 activity even in the presence of AAs[51]. Therefore, the guanine nucleotide status of the Rag complex is crucial in modulating the activation of mTORC1. The identification of Rag GEFs and GAPs will be significant in understanding AA signaling to mTORC1 (Box 2).
Figure 3. Amino acid signaling to mTORC1.
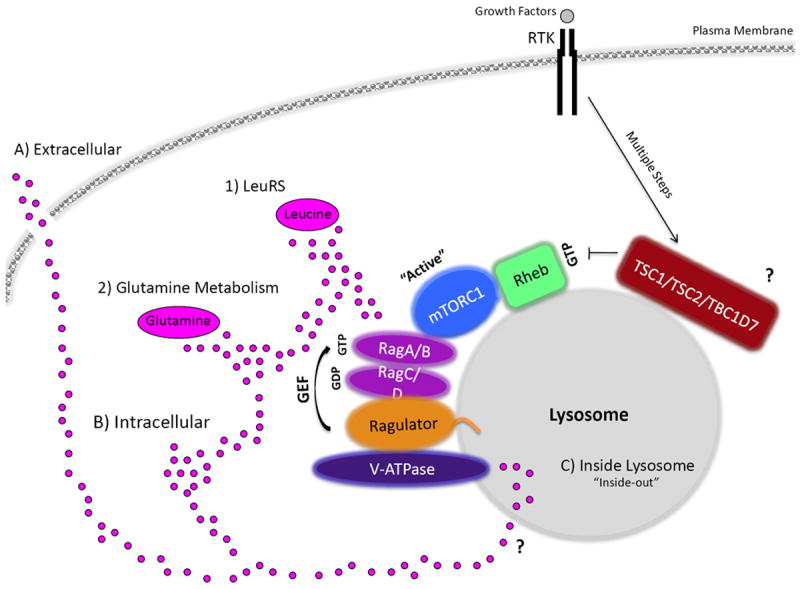
Amino acids (AAs), depicted as pink circles, may be sensed A) extracellularly, B) intracellularly, from within the C) lysosome, or through a combination. In addition, the specific AA sensed to promote mTORC1 activation is still not clear. For example, mTORC1 can sense 1) leucine through leucyl-tRNA synthetase (LeuRS) and 2) glutamine levels. Glutaminolysis results in mTORC1 activation, whereas glutamine synthesis seems to inhibit mTORC1 (Box 1). In response to AAs, an active RagA/BGTP-RagC/DGDP physically interacts with regulatory-associated protein of mTOR (Raptor) and recruits mTORC1 to the lysosome to promote its activation by Rheb, another GTPase. Recently, TSC2 has also been reported to localize at the lysosome (TSC1 and TBC1D7 have not yet been identified to reside at the lysosome, denoted by ?). The mTORC1 interaction with Rag A/BGTP-Rag C/DGDP is anchored to the lysosome by a complex called the Ragulator. The Ragulator also serves as a guanine exchange factor (GEF) for Rag A/BGTP, promoting mTORC1 activation. Moreover, the v-ATPase has been observed to be upstream of the Ragulator in AA sensing to mTORC1. The v-ATPase senses AA from within the lysosomal lumen, through an “inside-out” type of mechanism. How AAs accumulate inside the lysosome and what transporter(s) are involved in this are still unknown, denoted by ?. Growth factors, through the PI3K-AKT-TSC pathway, activate Rheb (RhebGTP) so that it can turn on the kinase activity of mTORC1, whereas AAs through the Rags localize mTORC1 in close proximately to Rheb at the lysosome. RTK denotes receptor tyrosine kinase.
Box 2. GEFs and GAPs involved in amino acid signaling to mTORC1.
An important piece in amino acid (AA) signaling to mTOR and cell growth is the identification of the Rag guanine nucleotide exchange factors (GEFs) and the opposing GTPase activating proteins (GAPs). GEFs usually activate GTPases by promoting the exchange of guanosine diphosphate (GDP) to guanosine triphosphate (GTP), whereas GAPs act antagonistically to deactivate GTPases by increasing their intrinsic rate of GTP hydrolysis[89]. RagA/BGTP must heterodimerize with RagC/DGDP in order to be active. Therefore, orchestration of the GEFs and GAPs for the Rags and other small GTPases that are involved in signaling to mTORC1 is key for mTORC1 activation. A new study, identifying the lysosomal localized RagA/B GEF as the Ragulator, has just emerged. The Ragulator is now classified a pentamer, with the identification of the two new components, C7orf59 and HBXIP (LAMTOR4 and LAMTOR5, respectively). All five components of the Ragulator complex appears to be required for GEF activity towards RagA/B[55]. Vam6 has been reported to function as a Gtr1p (RagA/B homolog) GEF in Saccharomyces cerevisiae[57]. However, the mammalian VPS39 homolog of Vam6 has no GEF activity towards RagA/B[55]. Two studies identified leucyl-tRNA synthetase (LeuRS) as a component playing a role in mTORC1 activation by AAs. Interestingly, LeuRS has been shown both in yeast and mammalian cells to be involved in AA signaling to mTORC1, although the proposed mechanisms were rather different between yeast and mammals. In yeast, LeuRS directly interacts with Gtr1pGTP, inhibiting an unknown GAP and keeping TORC1 active[72]. In mammals, LeuRS binds to and acts as a GAP for RagD (a Gtr2p homolog) in the cytoplasm. LeuRS facilitates an active RagD form by promoting RagDGDP[71]. Missing in the current model is RagA/B GAP and Rheb GEF [90]. Future studies identifying GEFs and GAPs and their regulation will provide critical new insights into AA sensing and cell growth control. Furthermore, in addition to the localization of additional GEFs/GAPs, which AAs control them and where they are originating from (extracellular, intracellular, or from within the lysosome) will be of great interest.
Rheb is required for AA-induced mTORC1 activation, as evidenced by the observation that AAs fail to activate mTOR in Rheb knockout cells[42, 52]. Notably, Rheb may not be directly involved in AA signaling to mTORC1. Fluorescent microscopy studies using overexpressed tagged Rheb suggest that it resides at the lysosome, but no efficient endogenous antibody is currently available to confirm this result. Recently, additional localization studies using an efficient endogenous TSC2 antibody showed TSC2 is also localized at the lysosome in further support of Rheb residing there[33]. As previously mentioned, growth factors such as insulin signal through the PI3K-AKT-TSC pathway to increase RhebGTP and promote mTORC1 activation, whereas AAs translocate mTORC1 to lysosome via the Rags. This is an attractive model because it explains how mTORC1 may combine multiple inputs, such as growth factors and AAs, in controlling cell growth. Further supporting this model, it has been reported that AAs promote Rheb-mTOR binding [52, 53]. Rheb overexpression can activate mTORC1 even under AA starvation. The overexpressed Rheb may be localized throughout the cell [51, 54]; therefore it is available to activate mTORC1 even when mTORC1 is not localized on lysosomes. Consistent with this model, artificially targeting Raptor to the lysosome surface activates mTORC1 regardless of the AA status [51].
Amino acid sensing at the lysosome
The Rag proteins lack membrane-targeting sequences, unlike other typical small GTPases such as Rheb. Thus, the Rag-mTORC1 complex is anchored to the lysosome surface by a complex called the “Ragulator.” The Ragulator was originally characterized as a trimeric complex composed of three small proteins: p18, p14, and MP1, which are encoded by the LAMTOR1, LAMTOR2, and LAMTOR3 genes, respectively. Recently, two additional members have been added to the Ragulator complex, C7orf59 and HBXIP (suggested to be renamed LAMTOR4 and LAMTOR5, respectively, for consistency), resulting in a pentameric Ragulator complex. Significantly, the pentameric Ragulator complex has guanine nucleotide exchange factors (GEF) activity, but not the individual subunits or the trimeric Ragulator towards RagA/B (Box 2)[55]. Rag binding to the Ragulator holds mTORC1 at the lysosome due to p18 N-terminal myristoylation and palmitoylation[51]. Components of the Ragulator complex have also been documented to act as an anchor element for MEK1 in the MEK/ERK pathway although the functional relationship to mTORC1 activation is unclear[56]. Depletion of the Ragulator components disrupts Rag localization to the lysosome and mTORC1 activation. It is currently unclear if the Rags are always lysosomally localized, or if they relocalize in response to particular conditions. In yeast, there are no obvious homologs of the Ragulator components; but the EGO complex that is also localized to the vacuole might similarly regulate Gtr1p-Gtr2p [49, 57]. Although the Ragulator components are not conserved, the high-resolution crystal structure of Ego3 is strikingly similar to that of MP1 and p14, suggesting an overall structural conservation[58]. Interestingly, the structures of MP1 and p14 contain a region called a roadblock domain, which is also predicted to be present in RagA/B and RagC/D[50, 59, 60]. Understanding the function and significance of this domain may be of great interest in better understanding AA signaling to mTORC1 and identifying additional components implicated in this pathway.
The precise location of where AAs originate from and are sensed is still unclear. Recent findings suggest that AA sensing may begin within the lysosome in an ‘inside-out’ type of signaling mechanism. Autophagy generates an AA supply for the cell through degradation of autophagosome contents within lysosomes under nutrient starvation conditions[12]. Inhibition of mTORC1 is required for the initiation of autophagy, but complete mTORC1 inhibition blocks termination of autophagy[61]. Moreover, mTORC1 is re-activated after prolonged starvation conditions in an autophagy-dependent manner, possibly due to the generation of AAs by autophagy. These observations suggest that some type of intricate relationship and balance between AAs, mTOR, and autophagy is needed to maintain activation of mTORC1 and cell growth[21, 61]. It will be intriguing to understand if the generation of AAs by autophagy controls mTORC1, and under what conditions. Sabatini and colleagues have reported a model in which AAs accumulate inside the lysosomal lumen and are eventually sensed by the large multisubunit vacuolar H+-adenosine triphosphate ATPase (v-ATPase), which in turn signals to the Ragulator-Rag complex via direct interaction, hence, the “inside-out” mechanism [62]. The v-ATPase, which consists of V1 and V0 domains, is essential for maintaining the low pH necessary for the lysosome to function properly. The V1 domain hydrolyses ATP, rotating the V0 membrane domain to pump protons across the plasma membrane and into the lysosomal lumen, acidifying it. Depletion of the v-ATPase subunits inhibits mTORC1 localization and activation. Supporting this, treatment of cells with two different v-ATPase inhibitors, concanamycin A and salicylihalamide A, halts mTOR localization to the lysosome and activation in response to AAs[62]. Although many components have been shown to be involved in AA mTORC1 activation at the lysosome, the precise sensor of AAs is still not known. Furthermore, the source of the build-up of AAs inside the lysosome is unclear. Whether this lysosomal pool of AAs is the end result of autophagy, shuttled in from outside of the lysosome (extracellularly or intracellularly), or a combination of the two, needs further investigation.
Many AA transporters have been identified at the lysosome and some have been documented to modulate mTORC1 activity[63-65]. In addition, a study demonstrated through the radiolabeling of AAs that extracellular AAs can accumulate within and be shuttled out of the lysosome [62]. One such transporter identified is the proton-assisted SLC36 AA transporters (PATs), which have a potent effect on mTORC1-mediated growth[65]. This mechanism may be conserved: Drosophila CG3424 and CG1139, two PAT-like transporters, control growth[66]; in mammals PAT1 is required for mTORC1 activation and cellular proliferation[65]. Instead of transporting AA into the lysosome, PAT1 exports AAs from the lysosomal lumen to the cytosol [63]. A recent study proposes a model where AAs promote the formation of a complex, termed the “nutrisome” or AA-sensing engine, comprising of PAT1, Rags, Ragulator, and the v-ATPase, that altogether are required to activate mTORC1. In this model, growth factor stimulated activation promotes the shuttling of PAT1 from the cell surface to the endosome/lysosome, where PAT1 is able to form the “nutrisome”. The v-ATPase pumps protons into the lysosome, cycling protons for PAT1 to transport the AAs out of the lysosome and somehow activating mTORC1 in the process[67]. Another group found that the overexpression of PAT1 completely suppressed AA-dependent mTORC1 activation by depleting the AA pool from within the lysosome. Inhibition was relieved by overexpressing RagA/BGTP, which is as efficient as the RagA/BGTP-RagC/DGDP heterodimer in rescuing mTORC1 inhibition under AA starvation conditions. This model implies that maintaining the AA pool inside the lysosome is important for the activation of mTOR, and that PAT1 decreases mTOR activation by facilitating the export of AAs out of the lysosome[62]. Also worth noting, PAT1 is specific for transporting alanine, glycine, and proline. This somewhat complicates the picture due to several studies implicating leucine, arginine, and glutamine, as the main activators of mTORC1[68].
Small GTPases associated with vesicle transport or lysosomes have been documented to play a role in AA mTORC1 activation. Besides Rheb and the Rags, RalA, Rab5, and Arf1 have been implicated in this pathway. Depletion of Ras-related protein RalA and its activator RalGDS antagonizes the ability of AAs and glucose, but not insulin, to signal to mTORC1[69]. AA availability regulates the amount of GTP-bound RalA and thus its activation; active RalA partially rescues mTORC1 activation in the absence of Rheb. Though this is a promising finding, the question of whether or not active RalA can rescue mTORC1 in AA depletion studies has yet to be tested. Rab and Arf, which are key mediators in vesicle transport, were identified as mTORC1 inhibitors through knockdown studies on small GTPases in Drosophila. Interestingly, activated Rab5 and Arf1 also block AA-induced mTORC1 signaling in mammalian cells, whereas glucose-stimulated mTORC1 signaling is unaffected. Furthermore, active Rab5 selectively inhibits mTOR through the Rags but not Rheb[70]. These data indicate that intracellular vesicle movement is important for AA-induced mTORC1 activation. Whether or not additional small GTPases play a role in AA signaling to mTOR has yet to be determined.
Other components implicated in amino acid signaling to mTORC1
Leucyl-tRNA synthetase
Two recent studies showed that leucyl-tRNA synthetase (LeuRS), the enzyme that charges leucine to its cognate tRNA, also functions as a leucine sensor in the activation of mTORC1[71, 72]. Leucine appears to be crucial for AA-dependent mTORC1 activation in most cells [44]. Interestingly, this mechanism was shown in both yeast and mammalian cells, although the details differ greatly. In yeast, LeuRS binds to Gtr1pGTP (the homolog in mammalian RagA/BGTP), preventing GTP hydrolysis and locking Gtr1p in its active form, which signals to TORC1[72]. In mammals, LeuRS directly interacts with and functions as a GAP for RagD, but not RagC, promoting its activation in a leucine dependent manner. Unexpectedly, LeuRS bound to the C-terminal domain of RagD, which has been shown to be crucial for Gtr1p-Gtr2p binding and thus TORC1 activation in yeast[71]. Notably, the arginine residue in human LeuRS that is essential for its GAP activity is not conserved in the Drosophila LeuRS homolog. This is rather surprising because activation of mTORC1 by AA is conserved in all eukaryotes, including Drosophila. LeuRS sensing occurs in the cytoplasm and not at the lysosome, possibly implicating multiple AA sensing pathways controlling mTORC1 (Figure 3). Further work is needed to elucidate the precise mechanism or role of LeuRS in translating AA levels into mTORC1 activation. Multiple other components have been reported to sense AAs not only at the lysosome, but also in the cytoplasm and at the plasma membrane and incorporating them all into a simplified model is going to be a very difficult task (Table 1).
mLST8 and IPMK: components of the nutrient sensitive mTOR-Raptor complex
Nutrients can regulate mTOR-Raptor binding. In nutrient deprivation, the Raptor-mTOR interaction is slightly “tighter,” which somehow inhibits the kinase activity of mTOR but is lost with the addition of AAs[73]. mLST8 (GβL) is required for both the nutrient- and rapamycin-dependent interaction between Raptor and mTOR, possibly implicating its involvement in AA sensing[74]. In addition to mLST8, inositol polyphosphate multikinase (IPMK) has been shown to regulate the nutrient sensitive mTOR-Raptor complex. IPMK controls the mTOR-Raptor interaction in response to AAs, a relationship that appears to be independent of IPMK catalytic activity. Disruption of the mTOR and Raptor interaction through IMPK depletion correlated with a significantly weakened association among mTOR and the active Rag complex, although Raptor-Rag binding remained unaffected. Like mLST8, nutrient deprivation enhanced Raptor, mTOR, and IPMK binding, which was reversed by the addition of AA. These results suggest that mTOR-Raptor binding may be nutrient sensitive, possibly through mLST8 and IPMK[75].
GCN2
General control non-repressed 2 (GCN2) is a protein kinase that monitors nutrient availability by binding to uncharged transfer RNA (tRNA). Increased GCN2 activity can increase the expression of genes involved in AA biosynthesis and transport through the transcription factor ATF4[76]. One possible pathway for the AA GCN2 mechanism is through the protein phosphatase 1 regulatory protein GADD34, which is upregulated by ATF4 and binds to TSC1/2, promoting the dephosphorylation of a crucial AKT site on TSC2. This causes an increase in TSC2 GAP activity, a decrease in RhebGTP, and inhibition of mTORC1[77, 78]. However, this transcription-dependent mechanism is at odds with the rapid activation of mTORC1 by AA, which causes S6K phosphorylation to increase within minutes. Interestingly, GCN2 knockout mice have impaired phosphorylation of mTORC1 substrates (4EBP1 and S6K1) and demonstrate an increased lethality when they are deprived of leucine[79, 80]. Understanding the role of GCN2 in AA signaling to mTORC1 will be interesting.
PLD
Another study showed that Rheb binds to phospholipase D 1 (PLD1), an upstream serum-activated positive regulator of mTORC1, in a Rheb bound GTP-dependent manner, and thus stimulates the activity of mTORC1 in vitro. AA starvation inhibited PLD activation in response to serum, linking PLD-Rheb to the AA induction of mTORC1. The class III PI3K, VPS34, which phosphorylates phosphatidylinositol at the 3’-OH to form phosphatidylinositol 3-phosphate (PI3P), has also been implicated as a bridge between AA signals and mTORC1. AA-induced VPS34 activation has been shown to promote PLD localization to the lysosome, in close proximately to Rheb, through production of PI3P. PI3P binds numerous proteins containing PI3P-targeting phox homology (PX) domains, including PLD [81, 82].
SH3BP4
A negative regulator of AA-induced mTORC1 activation is SH3 domain binding protein 4 (SH3BP4), which is often deleted in breast and renal cancers. It inhibits AA signaling to mTORC1 in leucine-deprived conditions by binding to the inactive Rag complex via its Src homology 3 (SH3) domain, preventing the formation of the active Rag complex. This results in decreased Raptor binding and shuttling of mTORC1 to the lysosome. Consistently, knockdown of SH3BP4 in leucine-stimulated conditions improved mTORC1 activation, increasing cell proliferation and size[83].
TTT-RUVBL1/2 Complex
The TTT-RUVBL1/2 complex was identified as a strong regulator of mTORC1 in TSC2-/- cells, indicating it regulates mTORC1 independently of growth factors[84]. TTT-RUVBL1/2 consists of the components Tel2, Tti1, and Ttil2, which were previously identified to regulate the assembly of PIKK-containing complexes such as mTOR[85-87]. Additionally, the TTT-RUVBL1/2 complex also contains the ATPases RUVBL1/2, and other components such as RPAP3, PIH1D1, and Hsp90. Interestingly, TTT-RUVBL1/2 complex mRNAs appear to be elevated in cancer tissues. Energy stress disassembled the ATP-dependent TTT-RUVBL1/2 complex, which in turn resulted in a decreased TTT-RUVBL1/2-mTORC1 interaction. Loss of TTT-RUVBL1/2 results in the mislocalization of mTOR to the lysosome, mTORC1 dimerization, and a decrease in mTORC1-Rag interaction[84].
Concluding remarks
The sensing of nutrients, growth factors, and stress all converge on the master regulator mTOR in order to appropriately control cell growth and physiology. Although many of the signaling cascades from growth factors and stress to mTORC1 are well characterized, the pathways that lead from nutrients to mTORC1 activation are less clear. Much progress has been made in understanding energy sensing by AMPK to mTORC1, however significant work is still needed in the AA sensing pathway. Numerous proteins have been identified and characterized to function in this signaling cascade and multiple pathways may be involved. The Rag GTPases have emerged as a key molecular switch downstream of AAs to modulate mTORC1 activity. Clarifying which AAs are critical and where they are originating from (extracellularly, intracellularly, or from within the lysosome) in mTORC1 activation would be beneficial in identifying sensors. Connecting all of the current known components to precise mechanisms, and to one another, is going to be a major challenge in completely understanding how mTOR senses AAs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Heitman J, et al. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 2.Kunz J, et al. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell. 1993;73:585–596. doi: 10.1016/0092-8674(93)90144-f. [DOI] [PubMed] [Google Scholar]
- 3.Sabatini DM, et al. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78:35–43. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 4.Sabers CJ, et al. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem. 1995;270:815–822. doi: 10.1074/jbc.270.2.815. [DOI] [PubMed] [Google Scholar]
- 5.Brown EJ, et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- 6.Populo H, et al. The mTOR Signalling Pathway in Human Cancer. Int J Mol Sci. 2012;13:1886–1918. doi: 10.3390/ijms13021886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loewith R, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 8.Sarbassov DD, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 9.Jacinto E, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 10.Phung TL, et al. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell. 2006;10:159–170. doi: 10.1016/j.ccr.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarbassov DD, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 12.Kim J, Guan KL. Amino acid signaling in TOR activation. Annu Rev Biochem. 2011;80:1001–1032. doi: 10.1146/annurev-biochem-062209-094414. [DOI] [PubMed] [Google Scholar]
- 13.Oh WJ, Jacinto E. mTOR complex 2 signaling and functions. Cell Cycle. 2011;10:2305–2316. doi: 10.4161/cc.10.14.16586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laplante M, Sabatini DM. mTOR Signaling in Growth Control and Disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao D, et al. mTOR drives its own activation via SCF(betaTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol Cell. 2011;44:290–303. doi: 10.1016/j.molcel.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y, et al. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(betaTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell. 2011;44:304–316. doi: 10.1016/j.molcel.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duan S, et al. mTOR generates an auto-amplification loop by triggering the betaTrCP- and CK1alpha-dependent degradation of DEPTOR. Mol Cell. 2011;44:317–324. doi: 10.1016/j.molcel.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 19.Thoreen CC, et al. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109–113. doi: 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsieh AC, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim J, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jung CH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ganley IG, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hosokawa N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benvenuto G, et al. The tuberous sclerosis-1 (TSC1) gene product hamartin suppresses cell growth and augments the expression of the TSC2 product tuberin by inhibiting its ubiquitination. Oncogene. 2000;19:6306–6316. doi: 10.1038/sj.onc.1204009. [DOI] [PubMed] [Google Scholar]
- 26.Chong-Kopera H, et al. TSC1 stabilizes TSC2 by inhibiting the interaction between TSC2 and the HERC1 ubiquitin ligase. J Biol Chem. 2006;281:8313–8316. doi: 10.1074/jbc.C500451200. [DOI] [PubMed] [Google Scholar]
- 27.Garami A, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11:1457–1466. doi: 10.1016/s1097-2765(03)00220-x. [DOI] [PubMed] [Google Scholar]
- 28.Inoki K, et al. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saucedo LJ, et al. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol. 2003;5:566–571. doi: 10.1038/ncb996. [DOI] [PubMed] [Google Scholar]
- 30.Stocker H, et al. Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol. 2003;5:559–565. doi: 10.1038/ncb995. [DOI] [PubMed] [Google Scholar]
- 31.Tee AR, et al. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci U S A. 2002;99:13571–13576. doi: 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, et al. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol. 2003;5:578–581. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
- 33.Dibble CC, et al. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell. 2012;47:535–546. doi: 10.1016/j.molcel.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zoncu R, et al. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hardie DG, et al. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inoki K, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 38.Gwinn DM, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brugarolas J, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–2904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reiling JH, Hafen E. The hypoxia-induced paralogs Scylla and Charybdis inhibit growth by down-regulating S6K activity upstream of TSC in Drosophila. Genes Dev. 2004;18:2879–2892. doi: 10.1101/gad.322704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sancak Y, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hara K, et al. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem. 1998;273:14484–14494. doi: 10.1074/jbc.273.23.14484. [DOI] [PubMed] [Google Scholar]
- 43.Wang X, et al. Amino acid availability regulates p70 S6 kinase and multiple translation factors. Biochem J. 1998;334(Pt 1):261–267. doi: 10.1042/bj3340261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Avruch J, et al. Amino acid regulation of TOR complex 1. Am J Physiol Endocrinol Metab. 2009;296:E592–602. doi: 10.1152/ajpendo.90645.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicklin P, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136:521–534. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duran RV, et al. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell. 2012;47:349–358. doi: 10.1016/j.molcel.2012.05.043. [DOI] [PubMed] [Google Scholar]
- 47.van der Vos KE, Coffer PJ. Glutamine metabolism links growth factor signaling to the regulation of autophagy. Autophagy. 2012;8:1862–1864. doi: 10.4161/auto.22152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim E, Guan KL. RAG GTPases in nutrient-mediated TOR signaling pathway. Cell Cycle. 2009;8:1014–1018. doi: 10.4161/cc.8.7.8124. [DOI] [PubMed] [Google Scholar]
- 49.Dubouloz F, et al. The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol Cell. 2005;19:15–26. doi: 10.1016/j.molcel.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 50.Gong R, et al. Crystal structure of the Gtr1p-Gtr2p complex reveals new insights into the amino acid-induced TORC1 activation. Genes Dev. 2011;25:1668–1673. doi: 10.1101/gad.16968011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sancak Y, et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Long X, et al. Rheb binding to mammalian target of rapamycin (mTOR) is regulated by amino acid sufficiency. J Biol Chem. 2005;280:23433–23436. doi: 10.1074/jbc.C500169200. [DOI] [PubMed] [Google Scholar]
- 53.Long X, et al. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15:702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 54.Buerger C, et al. Localization of Rheb to the endomembrane is critical for its signaling function. Biochem Biophys Res Commun. 2006;344:869–880. doi: 10.1016/j.bbrc.2006.03.220. [DOI] [PubMed] [Google Scholar]
- 55.Bar-Peled L, et al. Ragulator Is a GEF for the Rag GTPases that Signal Amino Acid Levels to mTORC1. Cell. 2012;150:1196–1208. doi: 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nada S, et al. The novel lipid raft adaptor p18 controls endosome dynamics by anchoring the MEK-ERK pathway to late endosomes. EMBO J. 2009;28:477–489. doi: 10.1038/emboj.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Binda M, et al. The Vam6 GEF controls TORC1 by activating the EGO complex. Mol Cell. 2009;35:563–573. doi: 10.1016/j.molcel.2009.06.033. [DOI] [PubMed] [Google Scholar]
- 58.Kogan K, et al. Structural conservation of components in the amino acid sensing branch of the TOR pathway in yeast and mammals. J Mol Biol. 2010;402:388–398. doi: 10.1016/j.jmb.2010.07.034. [DOI] [PubMed] [Google Scholar]
- 59.Lunin VV, et al. The structure of the MAPK scaffold, MP1, bound to its partner, p14. A complex with a critical role in endosomal map kinase signaling. J Biol Chem. 2004;279:23422–23430. doi: 10.1074/jbc.M401648200. [DOI] [PubMed] [Google Scholar]
- 60.Kurzbauer R, et al. Crystal structure of the p14/MP1 scaffolding complex: how a twin couple attaches mitogen-activated protein kinase signaling to late endosomes. Proc Natl Acad Sci U S A. 2004;101:10984–10989. doi: 10.1073/pnas.0403435101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–946. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zoncu R, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sagne C, et al. Identification and characterization of a lysosomal transporter for small neutral amino acids. Proc Natl Acad Sci U S A. 2001;98:7206–7211. doi: 10.1073/pnas.121183498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ruivo R, et al. Mechanism of proton/substrate coupling in the heptahelical lysosomal transporter cystinosin. Proc Natl Acad Sci U S A. 2012;109:E210–217. doi: 10.1073/pnas.1115581109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heublein S, et al. Proton-assisted amino-acid transporters are conserved regulators of proliferation and amino-acid-dependent mTORC1 activation. Oncogene. 2010;29:4068–4079. doi: 10.1038/onc.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goberdhan DC, et al. PAT-related amino acid transporters regulate growth via a novel mechanism that does not require bulk transport of amino acids. Development. 2005;132:2365–2375. doi: 10.1242/dev.01821. [DOI] [PubMed] [Google Scholar]
- 67.Ogmundsdottir MH, et al. Proton-Assisted Amino Acid Transporter PAT1 Complexes with Rag GTPases and Activates TORC1 on Late Endosomal and Lysosomal Membranes. PLoS One. 2012;7:e36616. doi: 10.1371/journal.pone.0036616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Metzner L, et al. Substrate specificity of the amino acid transporter PAT1. Amino Acids. 2006;31:111–117. doi: 10.1007/s00726-005-0314-6. [DOI] [PubMed] [Google Scholar]
- 69.Maehama T, et al. RalA functions as an indispensable signal mediator for the nutrient-sensing system. J Biol Chem. 2008;283:35053–35059. doi: 10.1074/jbc.M805822200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li L, et al. Regulation of mTORC1 by the Rab and Arf GTPases. J Biol Chem. 2010;285:19705–19709. doi: 10.1074/jbc.C110.102483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Han JM, et al. Leucyl-tRNA Synthetase Is an Intracellular Leucine Sensor for the mTORC1-Signaling Pathway. Cell. 2012 doi: 10.1016/j.cell.2012.02.044. [DOI] [PubMed] [Google Scholar]
- 72.Bonfils G, et al. Leucyl-tRNA Synthetase Controls TORC1 via the EGO Complex. Mol Cell. 2012 doi: 10.1016/j.molcel.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 73.Kim DH, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 74.Kim DH, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. 2003;11:895–904. doi: 10.1016/s1097-2765(03)00114-x. [DOI] [PubMed] [Google Scholar]
- 75.Kim S, et al. Amino acid signaling to mTOR mediated by inositol polyphosphate multikinase. Cell Metab. 2011;13:215–221. doi: 10.1016/j.cmet.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kilberg MS, et al. Nutritional control of gene expression: how mammalian cells respond to amino acid limitation. Annu Rev Nutr. 2005;25:59–85. doi: 10.1146/annurev.nutr.24.012003.132145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Novoa I, et al. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Watanabe R, et al. GADD34 inhibits mammalian target of rapamycin signaling via tuberous sclerosis complex and controls cell survival under bioenergetic stress. Int J Mol Med. 2007;19:475–483. [PubMed] [Google Scholar]
- 79.Zhang P, et al. The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol. 2002;22:6681–6688. doi: 10.1128/MCB.22.19.6681-6688.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Anthony TG, et al. Preservation of liver protein synthesis during dietary leucine deprivation occurs at the expense of skeletal muscle mass in mice deleted for eIF2 kinase GCN2. J Biol Chem. 2004;279:36553–36561. doi: 10.1074/jbc.M404559200. [DOI] [PubMed] [Google Scholar]
- 81.Sun Y, Chen J. mTOR signaling: PLD takes center stage. Cell Cycle. 2008;7:3118–3123. doi: 10.4161/cc.7.20.6881. [DOI] [PubMed] [Google Scholar]
- 82.Yoon MS, et al. Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J Cell Biol. 2011;195:435–447. doi: 10.1083/jcb.201107033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim YM, et al. SH3BP4 Is a Negative Regulator of Amino Acid-Rag GTPase-mTORC1 Signaling. Mol Cell. 2012 doi: 10.1016/j.molcel.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim SG, et al. Metabolic Stress Controls mTORC1 Lysosomal Localization and Dimerization by Regulating the TTT-RUVBL1/2 Complex. Mol Cell. 2012 doi: 10.1016/j.molcel.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Horejsi Z, et al. CK2 phospho-dependent binding of R2TP complex to TEL2 is essential for mTOR and SMG1 stability. Mol Cell. 2010;39:839–850. doi: 10.1016/j.molcel.2010.08.037. [DOI] [PubMed] [Google Scholar]
- 86.Kaizuka T, et al. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J Biol Chem. 2010;285:20109–20116. doi: 10.1074/jbc.M110.121699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Takai H, et al. Tel2 regulates the stability of PI3K-related protein kinases. Cell. 2007;131:1248–1259. doi: 10.1016/j.cell.2007.10.052. [DOI] [PubMed] [Google Scholar]
- 88.van der Vos KE, et al. Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat Cell Biol. 2012;14:829–837. doi: 10.1038/ncb2536. [DOI] [PubMed] [Google Scholar]
- 89.Bos JL, et al. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 90.Inoki K, et al. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 91.Findlay GM, et al. A MAP4 kinase related to Ste20 is a nutrient-sensitive regulator of mTOR signalling. Biochem J. 2007;403:13–20. doi: 10.1042/BJ20061881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yan L, et al. PP2A T61 epsilon is an inhibitor of MAP4K3 in nutrient signaling to mTOR. Mol Cell. 2010;37:633–642. doi: 10.1016/j.molcel.2010.01.031. [DOI] [PubMed] [Google Scholar]
- 93.Efeyan A, et al. Amino acids and mTORC1: from lysosomes to disease. Trends Mol Med. 2012;18:524–533. doi: 10.1016/j.molmed.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Duran A, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44:134–146. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gulati P, et al. Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab. 2008;7:456–465. doi: 10.1016/j.cmet.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wauson EM, et al. The G protein-coupled taste receptor T1R1/T1R3 regulates mTORC1 and autophagy. Mol Cell. 2012;47:851–862. doi: 10.1016/j.molcel.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]