

Abstract
OBJECTIVES:
The present study was performed to investigate 1) whether aerobic exercise training prior to myocardial infarction would prevent cardiac dysfunction and structural deterioration and 2) whether the potential cardiac benefits of aerobic exercise training would be associated with preserved morphological and contractile properties of cardiomyocytes in post-infarct remodeled myocardium.
METHODS:
Male Wistar rats underwent an aerobic exercise training protocol for eight weeks. The rats were then assigned to sham surgery (SHAM), sedentary lifestyle and myocardial infarction or exercise training and myocardial infarction groups and were evaluated 15 days after the surgery. Left ventricular tissue was analyzed histologically, and the contractile function of isolated myocytes was measured. Student's t-test was used to analyze infarct size and ventricular wall thickness, and the other parameters were analyzed by the Kruskal-Wallis test followed by Dunn's test or a one-way analysis of variance followed by Tukey's test (p<0.05).
RESULTS:
Myocardial infarctions in exercise-trained animals resulted in a smaller myocardial infarction extension, a thicker infarcted wall and less collagen accumulation as compared to myocardial infarctions in sedentary animals. Myocardial infarction-induced left ventricular dilation and cardiac dysfunction, as evaluated by +dP/dt and -dP/dt, were both prevented by previous aerobic exercise training. Moreover, aerobic exercise training preserved cardiac myocyte shortening, improved the maximum shortening and relengthening velocities in infarcted hearts and enhanced responsiveness to calcium.
CONCLUSION:
Previous aerobic exercise training attenuated the cardiac dysfunction and structural deterioration promoted by myocardial infarction, and such benefits were associated with preserved cardiomyocyte morphological and contractile properties.
Keywords: Cardiac Function; Cardiac Myocyte; Cardiac Remodeling; Physical Activity, Myocardial Infarction; Cardioprotection
INTRODUCTION
A sedentary lifestyle is known to be a major cardiovascular risk factor predisposing individuals to cardiac events such as myocardial infarction (MI) (1), which is the most common etiology of heart failure (HF). In recent years, studies have provided strong evidence for the benefits of aerobic exercise training (AET) in cardiac rehabilitation, which highlights its use as an adjuvant therapy for a variety of cardiovascular diseases (2-4). Despite the well-known benefits of AET in terms of the clinical outcomes of cardiac patients, the potential protective effect of AET performed prior to cardiac insult remains incompletely understood and is poorly addressed in the literature.
A small number of studies have evaluated how increasing physical activity prior to MI affects hemodynamic parameters and cardiac function and structure. However, despite distinct experimental designs and exercise regimens, results from different studies seem to be fairly convergent and suggest that exercise training protects against the maladaptive remodeling and global function induced by MI in rodents (5-8). Nevertheless, the mechanisms underlying these morphological and functional benefits of prior AET in post-remodeled myocardium are poorly understood and may be related to the morphological and contractile properties of cardiomyocytes.
The role played by cardiomyocyte contractile function in reducing blood pumping capacity after MI is controversial. Some authors have reported cardiac dysfunction paralleled by impaired cardiomyocyte contractility (9-12), while others have observed preserved cardiomyocyte contractility in infarcted ventricles (13-15). Based on these findings, left ventricular (LV) remodeling (e.g., increased collagen content, pathological cardiac hypertrophy and LV dilation) has been proposed as the main determinant of cardiac dysfunction after MI; however, further studies are needed to obtain a more thorough understanding of such a relationship.
As AET improves cardiomyocyte fractional shortening as well as maximal shortening and relengthening velocities (16-18), it is possible that improving cardiomyocyte contractile function by AET could provide protection against MI-induced cardiac dysfunction. Therefore, the present study was undertaken to investigate 1) whether AET prior to MI prevents cardiac dysfunction and structural deterioration and 2) whether the potential cardiac benefits of AET would be associated with preserved morphological and contractile properties of cardiomyocytes in post-infarct remodeled myocardium.
MATERIALS AND METHODS
Study population
Experiments were performed according to the principles of laboratory animal care (NIH publication No. 86-23, revised 1985) and the Brazilian College of Animal Experimentation and were approved by the Institutional Animal Care Committee (protocol 55/2009). Male Wistar rats (100-120 g) provided by the animal facilities at the Federal University of Viçosa, Brazil were randomly assigned to the following groups: control (SHAM; n = 15); sedentary lifestyle and MI (SED-MI; n = 20); and exercise training and MI (ET-MI; n = 20). The large initial number of animals was chosen to provide at least six rats per group for each of the measurements, as a high mortality rate is expected for rats subjected to MI (30-40%). The animals were kept under a 12:12 hour light-dark cycle in a temperature-controlled (22°C) room with free access to standard laboratory chow (Nuvital, Colombo, Paraná, Brazil) and tap water.
Aerobic Exercise Training and test protocols
The ET-MI group underwent AET on a motor-driven treadmill (Insight Scientific Equipments, Ribeirão Preto, São Paulo, Brazil) five days per week for eight weeks prior to MI surgery. The running speed, treadmill inclination and session duration were progressively increased throughout the protocol in such a way that by the 6th week, the rats were running continuously for 60 min at 18 m.min-1 and a 10° grade, which was maintained until the end of the protocol. The running speed of 18 m.min-1 was chosen because a pilot study in our lab failed to verify cardiomyocyte adaptations after AET at lower intensities. To verify the relative intensity of the training protocol, a subset of five sedentary rats underwent graded treadmill running tests at a 10° grade until exhaustion to measure the maximum running workload. Treadmill running tests performed until exhaustion in a separate batch of age-matched rats revealed that 18 m.min-1 corresponded to 55-70% of the maximal running speed in all animals; therefore, the AET protocol was performed at a moderate intensity. SHAM and SED-MI groups were placed on the treadmill twice a week and walked for 10 min at 20% of the speed of the exercise-trained group to mimic the running stress associated with the experimental protocol and maintenance of exercise skills. Forty-eight hours after the last training session (ET-MI) or adaptation (SHAM and SED-MI), all animals underwent a graded treadmill exercise test until exhaustion, as described by Koch and Britton (19). Briefly, the initial treadmill speed was 10 m.min-1, which was increased by 1 m.min-1 every 2 min until the rats were no longer able to run. The tests were carried out by experienced observers (LHMB and MFS) over the course of three days.
Myocardial infarction
Coronary artery occlusion was performed in the MI groups 48 hours after the last graded treadmill running test. Rats were deeply anesthetized with ketamine (50 mg/kg, ip) and xylazine (10 mg/kg, ip), and a left thoracotomy was then performed. The mediastinum was accessed by incising the intercostal muscles between the 3rd and 4th ribs, the heart was carefully exteriorized, and then the left anterior descending (LAD) coronary artery was occluded with a 6-0 thread. After LAD ligation, the thorax was closed, and lung collapse was prevented by rapid withdrawal of air from the pleural cavity using a syringe. Sham-operated animals underwent a similar left thoracotomy and cardiac exteriorization but did not undergo LAD ligation. The peri-operative mortality was 30% in sedentary and 15% in exercise-trained animals. After MI induction, the animals were kept sedentary, and all measurements were performed 15 days after the surgical procedures. A similar number of animals from each of the three groups was allocated for either (1) the in vivo assessment of hemodynamic parameters and cardiac structure or (2) isolated cardiomyocyte structure and function evaluation.
In vivo hemodynamic and cardiac function measurements
Fifteen days after the surgical procedures, the animals were deeply anesthetized with ketamine (70 mg.kg-1, ip) and xylazine (10 mg.kg-1, ip) for LV catheterization. The right common carotid artery was separated from its connective tissue and catheterized with a fluid-filled polyethylene catheter (P50). The catheter was connected to a pressure transducer (TRI 21, Letica Scientific Instruments, Hospitalet, Barcelona, Spain) and to a digitalizing unit (Powerlab/4SP ML750, ADInstrument, Sidney, New South Wales, Australia) for data recording. After acquiring arterial systolic (SBP) and diastolic blood pressure (DBP) values, the catheter was moved to the left ventricle to obtain the following parameters: heart rate (HR); left ventricular systolic (LVSP) and end-diastolic pressures (LVEDP); and the maximum rates of pressure rise (+dP/dt) and fall (-dP/dt).
In situ LV pressure-volume relationship
After hemodynamic measurements were collected, each heart was arrested with 3 M KCl (0.2 mL, iv), and a double lumen catheter (P50 inserted into P200) was inserted into the left ventricle through the aorta to determine the in situ left ventricle diastolic pressure-volume relationship, as previously described (20). Briefly, the atrio-ventricular groove was occluded, and a small incision was made in the right ventricular free wall to hinder any compression effect. Then, 0.9% NaCl was infused with an infusion pump (Insight Scientific Equipments, Ribeirão Preto, São Paulo, Brazil) at a constant rate of 0.68 mL.min-1 into the P200, and pressure was continuously monitored through the P50. The pressure followed a linear pattern during volume infusion from 0 to 5 mmHg, indicating the passive distention of ventricular wall; therefore, the slope was used to estimate LV dilatation because no stiffness was observed up to 5 mmHg (20).
Histological evaluation
After in vivo hemodynamic measurements, the heart was removed, mounted for routine histological procedures, transversely sectioned in 5 μm-thick sections and stained with picrosirius red for analysis of infarct size and collagen content. Endocardial and epicardial circumferences of the infarcted tissue and left ventricle were determined using Image J software (NIH, Bethesda, Maryland, USA). Infarct size was calculated as (endocardial + epicardial circumference of infarcted tissue)/(endocardial + epicardial circumference of the left ventricle) and was expressed as a percentage. The infarcted wall thickness was measured at the medial point of the scar area. The stereological parameter of collagen volume density (Vv) was estimated from the remote region by point counting for collagen according to the following formula: Vv[collagen] = PP[collagen]/72; where PP is the number of points that hit the structure and 72 is the total number of test points. A points test system was used for analysis in a standard test area of 0.15 mm2 (21).
Cardiomyocyte isolation
Cardiac myocytes from a region adjacent to the MI were enzymatically isolated as previously described (22). Briefly, after euthanasia, the heart was removed rapidly, and extraneous tissue was dissected away. The heat was then flushed with a modified Hepes-Tyrode solution at room temperature containing the following ingredients: 130 mM Na+, 5.4 mM K+, 1.4 mM Mg2+, 140 mM Cl-, 0.75 mM Ca2+, 5 mM Hepes, 10 mM glucose, 20 mM taurine and 10 mM creatine, pH 7.3. The heart was then blotted dry and weighed before being mounted on a custom-designed Langendorff apparatus. After perfusion for 3 min with a 750 μM CaCl2 solution, the heart was perfused for 3-5 min with a Ca2+-free solution containing EGTA (0.1 mM). Afterwards, the heart was perfused for 15-20 min with a solution containing 1 mg.mL-1 collagenase type II (Worthington, Lakewood, New Jersey, USA). After tissue digestion, fragments of the MI-adjacent region were obtained, and single cells were isolated by mechanical dispersion and stored at 5°C until use.
Cardiomyocyte contractile function and morphology
Cellular contractility was measured as previously described (22). Briefly, isolated cardiomyocytes were placed in an experimental chamber with a glass coverslip base mounted on the stage of an inverted microscope. The chamber was perfused with HEPES Tyrode's solution containing 0.6, 1 or 5 mM CaCl2 and field-stimulated at a frequency of 3 Hz (20 V, 5 ms-long square pulses) at room temperature (25 - 30°C). Cells were imaged using a digital video camera in partial scanning mode, and these images were used to measure cell shortening (our index of contractility) in response to electrical stimulation using a video-edge detection system (Milton, Massachusetts, Ionoptix, USA). Cell images were sampled at a frame rate of 240 Hz. Cell shortening (expressed as a percentage of resting cell length), shortening velocity and relengthening velocity were calculated as previously described (22). Only Ca2+-tolerant, quiescent and rod-shaped myocytes showing clear cross striations were studied. The same images were also used to determine cell length and width, which were used to estimate myocyte volume (23) (130-180 cells per experimental group).
Statistics
The data are presented as the mean ± SEM. The Student's t-test was used to analyze infarct size and ventricular wall thickness. The other parameters were analyzed using the Kruskal-Wallis test followed by Dunn's test (nonparametric data) or a one-way analysis of variance (ANOVA) followed by Tukey's test (parametric data). Statistical significance was defined as a p value below 0.05.
RESULTS
Running performance
To verify the AET efficacy, all animals underwent graded treadmill running tests until exhaustion at the end of the training period prior to MI induction. As expected, the time until exhaustion, i.e., the running capacity, was substantially increased in exercise-trained rats (Table 1). Importantly, prior to the surgical procedure, the running capacity of the SHAM and SED-MI groups was similar, demonstrating homogeneity between these two groups at that time point.
Table 1.
Physiological parameters.
| SHAM | SED-MI | ET-MI | |
| BW, g | 377±6 | 360±12 | 345±12 |
| Running time, min | 8.7±0.8 | 9.0±1.3 | 19.4±0.6*† |
| HW, mg | 1.9±0.1 | 2.4±0.1* | 2.5±0.1* |
| HW/BW, mg/g | 5.0±0.2 | 6.6±0.4* | 7.5±0.4* |
| Lung wet/dry ratio | 4.9±0.09 | 5.2±0.05* | 5.2±0.07* |
| HR, bpm | 246±7 | 269±8 | 251±7 |
| SBP, mmHg | 116 ±2 | 108±3 | 110±3 |
| DBP, mmHg | 73±2 | 74±3 | 75±2 |
Data are presented as the mean±SEM. SHAM, control. SED-MI, sedentary and given a MI. ET-MI, exercise-trained and given a MI. HW: heart weight. HW/BW: heart weight to body weight ratio. HR: heart rate. SBP: systolic blood pressure. DBP: diastolic blood pressure. *p<0.05 vs. SHAM; †p<0.05 vs. SED-MI (ANOVA followed by Tukey's test).
Physiological parameters
Fifteen days after infarction, significant LV remodeling had occurred. MI-induced cardiac macroscopic alterations were verified by increased heart weights (HW) and heart weight to body weight ratios (HW/BW) in both MI groups as compared to the SHAM animals (Table 1). No significant effect of prior AET was found for HW or the HW/BW ratio (Table 1). Pulmonary congestion, estimated according to the lung wet-to-dry ratio, was detected in both MI groups, without a significant difference between the SED-MI and ET-MI groups (Table 1). Histological analysis revealed that MI extension was greater in SED-MI than ET-MI animals (Fig. 1A and B), and infarcted wall thickness was also greater in ET-MI as compared to SED-MI animals (Fig. 1A and C). MI also increased cardiac collagen content in both groups, although this occurred to a lesser extent in rats that had undergone AET (Fig. 1D).
Figure 1.
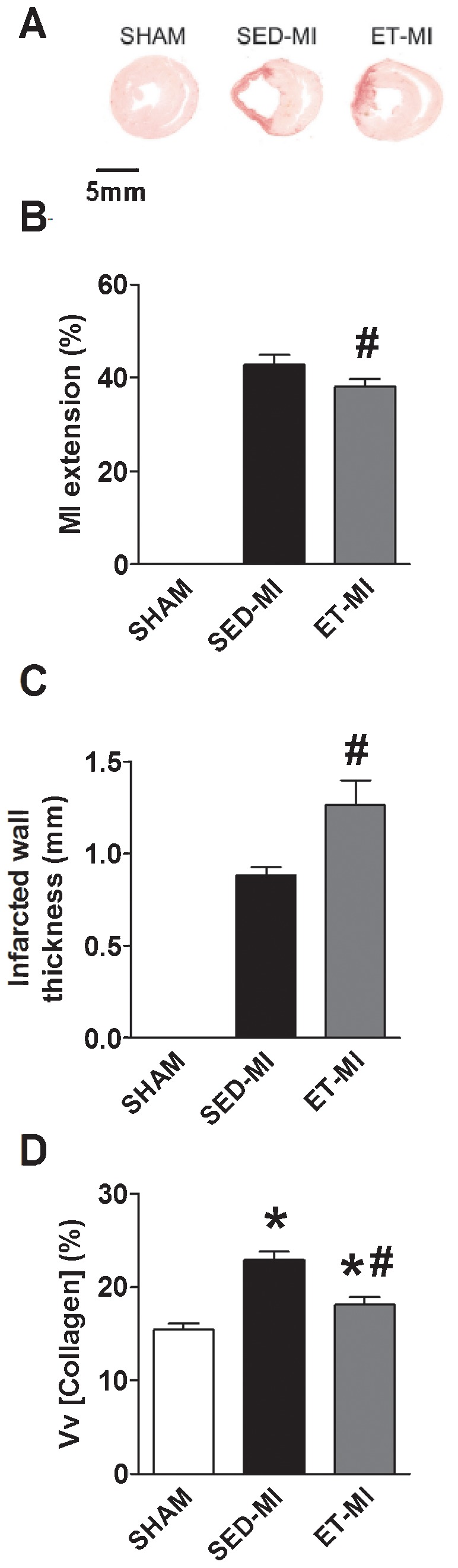
A Representative photomicrography of cardiac tissue. B Myocardial infarct extension. C Infarcted wall thickness. D Cardiac collagen content. SHAM, control. SED-MI, sedentary and given a MI. ET-MI, exercise trained and given a MI. Vv, volume density. Data are presented as the mean±SEM. *p<0.05 vs. SHAM; #p<0.05 vs. SED-MI (ANOVA followed by Tukey's test).
Hemodynamic parameters
HR, SBP and DBP under anesthesia were similar among groups (Table 1). MI decreased LVSP in the SED-MI group, which was not observed in the ET-MI group (Fig. 2A). However, AET did not prevent the elevation of LVEDP by MI (Fig. 2B). Inotropic function, evaluated according to +dP/dt, was reduced in SED-MI animals, and prior AET significantly attenuated inotropic dysfunction in MI rats (Fig. 2C). Accordingly, prior AET partially restrained the effects of MI on lusitropic function (-dP/dt) (Fig. 2D).
Figure 2.

A Left ventricular systolic pressure (LVSP). B Left ventricular end-diastolic pressure (LVEDP). C Inotropic function (+ dP/dt). D Lusitropic function (- dP/dt). SHAM, control. SED-MI, sedentary and given a MI. ET-MI, exercise trained and given a MI. *p<0.05 vs. SHAM; #p<0.05 vs. SED-MI (ANOVA followed by Tukey's test).
In situ LV pressure-volume relationship
The linear rise in LV pressure up to 5 mmHg in response to fluid infusion indicates passive chamber filling. Thus, the LV volume at 5 mmHg was compared among groups and provided an estimate of LV dilation (20). As shown in Figure 3, SED-MI animals displayed LV dilation as compared to SHAM animals, which was prevented by prior AET.
Figure 3.

In situ LV pressure-volume relationship. SHAM, control. SED-MI, sedentary and given a MI. ET-MI, exercise trained and given a MI. Data are presented as the mean±SEM. *p<0.05 vs. SHAM; #p<0.05 vs. SED-MI (ANOVA followed by Tukey's test).
Cardiomyocyte morphology and contractile function
As illustrated in Figure 4, increased myocyte length without any change in cell width was observed in the SED-MI group. In contrast, the ET-MI group displayed myocyte length and width values greater than those observed in the SED-MI and SHAM groups (Fig. 4A and B). Estimated cardiomyocyte volume was increased only in ET-MI animals (Fig. 4C). Interestingly, increased cardiomyocyte dimensions in the ET-MI group were paralleled by preserved cell shortening at 1 mM and 5 mM concentrations of extracellular Ca2+ ([Ca2+]e), whereas SED-MI animals demonstrated reduced cell shortening. No difference in cell shortening was observed among groups at 0.6 mM of [Ca2+]e (Fig. 5A), and MI did not impair maximal cell shortening and relengthening velocities at any [Ca2+]e. In contrast, prior AET significantly increased both parameters in MI rats (Fig. 5B and C).
Figure 4.
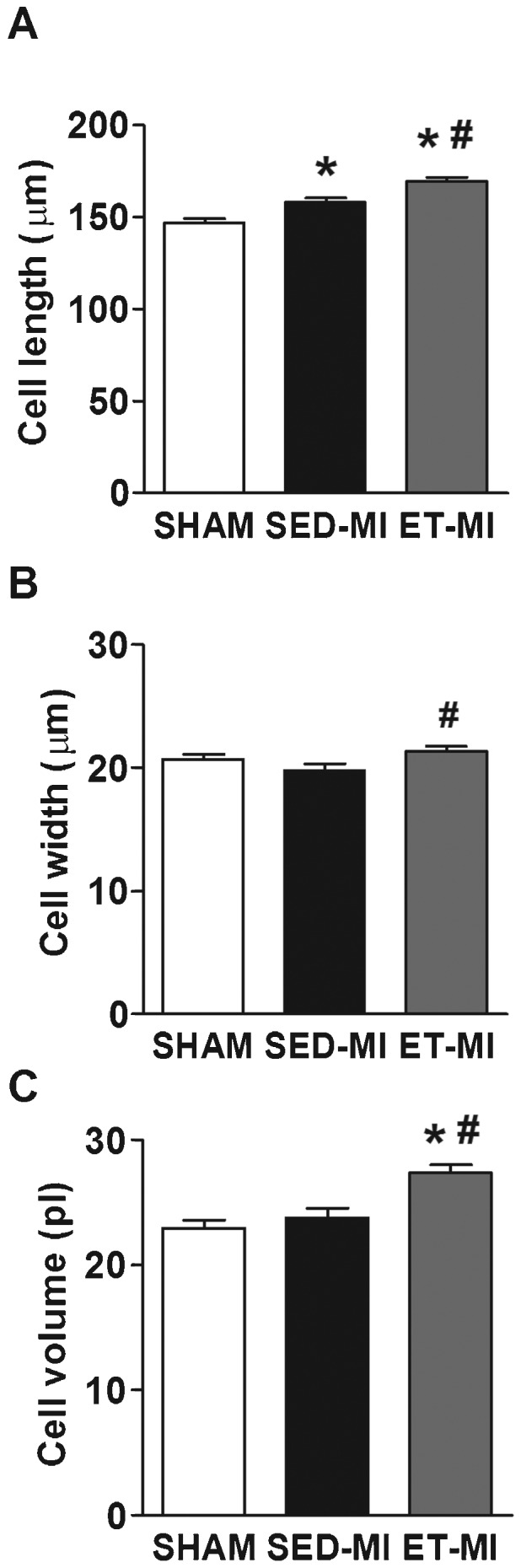
Cardiomyocyte morphology. A Cell length. B Cell width. C Cell volume. SHAM, control. SED-MI, sedentary and given a MI. ET-MI, exercise trained and given a MI. Data are presented as the mean±SEM. *p<0.05 vs. SHAM; #p<0.05 vs. SED-MI (Kruskal-Wallis followed by Dunn's test).
Figure 5.

Cardiomyocyte contractile function. A Cell shortening (% of resting cell length). B Shortening velocity. C Relengthening velocity. SHAM, control. SED-MI, sedentary and given MI. ET-MI, exercise trained and given MI. Data are presented as the mean±SEM. *p<0.05 vs. SHAM; #p<0.05 vs. SED-MI. +p<0.05 vs. lower [Ca+2] (Kruskal-Wallis followed by Dunn's test).
DISCUSSION
AET is used as an adjuvant therapy in preventive cardiology. In addition, cardiovascular risk factors are known to arise in rats artificially selected for inborn low aerobic capacity, suggesting that there are cardioprotective effects of increased aerobic capacity (24). The present study provides strong evidence for the attenuation of MI-induced cardiac dysfunction by prior moderate-intensity AET, and this effect was accompanied by diminished deleterious remodeling and improved cardiomyocyte morphological and contractile properties. The cardioprotective effect of prior AET was also confirmed by the reduced peri-operative mortality observed in exercise-trained animals.
The efficacy of AET is typically demonstrated by increased exercise performance and resting bradycardia (25,26). In this context, the AET employed in the present study was efficient in improving the running performance of MI rats in a graded treadmill test. Although HR was unchanged between the treatment groups, it must be considered that it was assessed under deep anesthesia.
We demonstrated that rats given AET had a smaller infarct extension compared to SED-MI animals, and this may reflect exercise-associated improvements in cardiac vascularity or activation of cardioprotective signaling pathways. Improved myocardial capillarization, intracellular redox balance, increased levels of anti-apoptotic proteins and downregulation of the MI-induced renin angiotensin system and sympathetic nervous activity by prior AET may also have accounted for this observation. Such parameters are directly involved in MI expansion and the progression of HF (27-32) and are improved by AET (32-37). Additionally, cardiomyocyte sliding occurs after MI and the thinning and elongation of the infarcted area, which accelerates MI expansion and leads to LV dilation (38). Therefore, the thicker infarcted areas observed in ET-MI animals as compared to SED-MI animals may also have contributed to the smaller MI extension and prevented LV dilation in rats receiving both exercise training and MI. In fact, a reduction in infarct area thickness is directly associated with MI expansion (38). Moreover, the attenuation of LV collagen accumulation in the ET-MI group confirms the reduced deleterious effects of cardiac remodeling promoted by AET (39).
Attenuated cardiac deterioration by AET was paralleled by reduced cardiac dysfunction in the ET-MI group when compared to SED-MI group. Moreover, blunting of inotropic (+dP/dt) and lusitropic (-dP/dt) dysfunction by prior AET can be explained, at least in part, by the lesser degree of cardiac tissue deterioration during remodeling observed in the ET-MI group.
Although other studies corroborate our hypothesis (5,6), Veiga et al. (40) did not observe any cardioprotective effects following the administration of a different AET protocol. These divergent findings might be explained by the fact that animals in this previous study underwent swimming training in the absence of an external load, which may not have provided enough AET intensity to promote cardioprotection. In fact, it has been shown that exercise-induced cardioprotection is intensity-dependent, such that low-intensity exercise does not provide significant protection against cardiac damage (41). Moreover, further differences in experimental conditions, such as resting time after MI and animal gender, may also be responsible for the distinct findings between our study and that of Veiga et al. (40).
LV remodeling following MI involves structural rearrangement of the remaining cardiomyocytes in an attempt to evenly distribute the increased wall stress (42,43). In fact, we observed increased cardiomyocyte length in the SED-MI group as compared to the ET-MI group. It could be argued that AET aggravated MI-induced myocyte remodeling; however, several lines of evidence have demonstrated that physiological cardiomyocyte enlargement associated with a gain of function occurs after AET (16,18,44). Despite structural similarities, studies have confirmed that distinct molecular pathways separate pathological from physiological myocyte hypertrophy. While activation of the calcineurin-NFAT-GATA4 pathway resulting from MI normally results in cardiomyocyte dysfunction (45,46), activation of the cardiac PI3k-Akt-mTOR cascade by AET promotes cellular hypertrophy associated with functional gains (47,48). Therefore, despite not measuring hypertrophy markers, the notion of physiological hypertrophy associated with a gain of function is supported by our data showing preserved cell shortening in the ET-MI group, whereas SED-MI cell shortening was depressed under physiological (1 mM [Ca2+]e) and stimulated (5 mM [Ca2+]e) conditions. Extending our findings on cellular function, we also evaluated cardiomyocyte shortening and relengthening velocities in response to increasing inotropic stimuli (i.e., [Ca2+]e). Of interest, AET prior to MI not only improved both parameters but also promoted enhanced responsiveness to increasing [Ca2+]e.
Studies reporting impaired Ca2+ handling and decreased myofilament sensitivity to Ca2+ in post-infarction HF in rats (10-12) support our findings of reduced responsiveness to increasing [Ca2+]e in the SED-MI group.
For the first time, we demonstrated that AET performed prior to MI preserved cardiomyocyte shortening, as well as the shortening and relengthening velocities. The mechanisms underlying such findings could be explained, at least partly, by preserved or increased myofilament Ca+2 sensitivity (17,49). The reduction in myofilament Ca+2 sensitivity in infarcted hearts may result from increased reactive oxidative species (ROS) production, which upregulates kinases involved in the phosphorylation of troponin T and I, and culminates in disrupted actin–myosin interactions (50). In this regard, it has also been shown that AET improves antioxidant defenses as well as, and also reduces ROS release by the heart's major source of ROS, the mitochondria (51). Therefore, it is reasonable to suggest that previous AET improved the mitochondrial profile after MI, which could have reduced ROS release and thereby preserved myofilament Ca+2 sensitivity. In fact, studies in healthy and post-MI rats have demonstrated that AET improves myofilament Ca+2 sensitivity, which was also associated with improved cardiomyocyte function (17,49). It is also possible that prior AET promotes a shift in myosin heavy chain composition (i.e., towards the fast V1 isoform) in the cardiac muscle (52), which may explain the changes in the contraction and relaxation velocities. Furthermore, increases in transient intracellular Ca+2 through increased SERCA2a expression and activity, as well as NCX activity, have been shown to be modulated by AET in healthy animals [16-18,26,53]. Altogether, such exercise-induced cardiac adaptations may have provided protection against MI-induced cardiomyocyte dysfunction.
In conclusion, AET performed prior to MI effectively attenuated LV dysfunction and cardiac remodeling, which were associated with preserved cardiomyocyte fractional shortening, improved shortening and relengthening velocities and physiological cellular hypertrophy. Taken together, our data support the notion that AET provides considerable cardioprotection against the deleterious effects of MI, which reinforces the importance of maintaining a physically active lifestyle.
Study limitations
We cannot exclude the possibility of variations in the ischemic region between SED-MI and ET-MI animals. To minimize this potential effect, the surgeries were performed by an experienced surgeon (MPB) who was blinded to the animal groupings. Correlations between in vivo global cardiac function and cardiomyocyte parameters from the same rats were not possible because such experiments were performed in distinct subsets of animals; however, our findings of improved cardiac function and cardiomyocyte contractility in trained animals suggest an association between these variables. Although the absence of an exercise-trained SHAM group did not allow us to directly confirm that exacerbated cellular hypertrophy in the ET-MI group was due to physiological cardiac remodeling promoted by AET, this hypothesis is supported by several previous publications (16,18,44) as well as our data on cardiac and myocyte function.
ACKNOWLEDGMENTS
This study received financial support from the Fundo de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG – PRONEX) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). LHMB held a master degree scholarship from CAPES/REUNI-Federal University of Viçosa (# 164584). AJN is a CNPq fellow.
Footnotes
No potential conflict of interest was reported.
REFERENCES
- 1.Booth FW, Roberts CK. Linking performance and chronic disease risk: indices of physical performance are surrogates for health. Br J Sports Med. Circulation. 2008;42(12):950–2. doi: 10.1136/bjsm.2008.052589. [DOI] [PubMed] [Google Scholar]
- 2.Wisloff U, Stoylen A, Loennechen JP, Bruvold M, Rognmo O, Haram PM, et al. Superior cardiovascular effect of aerobic interval training versus moderate continuous training in heart failure patients: a randomized study. Circulation. 2007;115(24):3086–94. doi: 10.1161/CIRCULATIONAHA.106.675041. [DOI] [PubMed] [Google Scholar]
- 3.Fraga R, Franco FG, Roveda F, de Matos LN, Braga AM, Rondon MU, et al. Exercise training reduces sympathetic nerve activity in heart failure patients treated with carvedilol. Eur J Heart Fail. 2007;9(6-7):630–6. doi: 10.1016/j.ejheart.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Rognmo O, Hetland E, Helgerud J, Hoff J, Slordahl SA. High intensity aerobic interval exercise is superior to moderate intensity exercise for increasing aerobic capacity in patients with coronary artery disease. Eur J Cardiovasc Prev Rehabil. 2004;11(3):216–22. doi: 10.1097/01.hjr.0000131677.96762.0c. [DOI] [PubMed] [Google Scholar]
- 5.Dayan A, Feinberg MS, Holbova R, Deshet N, Scheinowitz M. Swimming exercise training prior to acute myocardial infarction attenuates left ventricular remodeling and improves left ventricular function in rats. Annals of clinical and laboratory science. 2005;35(1):73–8. [PubMed] [Google Scholar]
- 6.Freimann S, Scheinowitz M, Yekutieli D, Feinberg MS, Eldar M, Kessler-Icekson G. Prior exercise training improves the outcome of acute myocardial infarction in the rat. Heart structure, function, and gene expression. J Am Coll Cardiol. 2005;45(6):931–8. doi: 10.1016/j.jacc.2004.11.052. [DOI] [PubMed] [Google Scholar]
- 7.de Waard MC, Duncker DJ. Prior exercise improves survival, infarct healing, and left ventricular function after myocardial infarction. J Appl Physiol. 2009;107(3):928–36. doi: 10.1152/japplphysiol.91281.2008. [DOI] [PubMed] [Google Scholar]
- 8.Galvao TF, Matos KC, Brum PC, Negrao CE, Luz PL, Chagas AC. Cardioprotection conferred by exercise training is blunted by blockade of the opioid system. Clinics (Sao Paulo) 2011;66(1):151–7. doi: 10.1590/S1807-59322011000100026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li P, Park C, Micheletti R, Li B, Cheng W, Sonnenblick EH, et al. Myocyte performance during evolution of myocardial infarction in rats: effects of propionyl-L-carnitine. Am J Physiol. 1995;268(4 Pt 2):H1702–13. doi: 10.1152/ajpheart.1995.268.4.H1702. [DOI] [PubMed] [Google Scholar]
- 10.Holt E, Tonnessen T, Lunde PK, Semb SO, Wasserstrom JA, Sejersted OM, et al. Mechanisms of cardiomyocyte dysfunction in heart failure following myocardial infarction in rats. J Mol Cell Cardiol. 1998;30(8):1581–93. doi: 10.1006/jmcc.1998.0724. [DOI] [PubMed] [Google Scholar]
- 11.Loennechen JP, Wisloff U, Falck G, Ellingsen O. Cardiomyocyte contractility and calcium handling partially recover after early deterioration during post-infarction failure in rat. Acta Physiol Scand. 2002;176(1):17–26. doi: 10.1046/j.1365-201X.2002.01011.x. [DOI] [PubMed] [Google Scholar]
- 12.Kim YK, Kim SJ, Kramer CM, Yatani A, Takagi G, Mankad S, et al. Altered excitation-contraction coupling in myocytes from remodeled myocardium after chronic myocardial infarction. J Mol Cell Cardiol. 2002;34(1):63–73. doi: 10.1006/jmcc.2001.1490. [DOI] [PubMed] [Google Scholar]
- 13.Gupta S, Prahash AJ, Anand IS. Myocyte contractile function is intact in the post-infarct remodeled rat heart despite molecular alterations. Cardiovasc Res. 2000;48(1):77–88. doi: 10.1016/s0008-6363(00)00160-7. [DOI] [PubMed] [Google Scholar]
- 14.Song J, Zhang XQ, Wang J, Carl LL, Ahlers BA, Rothblum LI, et al. Sprint training improves contractility in postinfarction rat myocytes: role of Na+/Ca2+ exchange. J Appl Physiol. 2004;97(2):484–90. doi: 10.1152/japplphysiol.00061.2004. [DOI] [PubMed] [Google Scholar]
- 15.Ahlers BA, Song J, Wang J, Zhang XQ, Carl LL, Tadros GM, et al. Effects of sarcoplasmic reticulum Ca2+-ATPase overexpression in postinfarction rat myocytes. J Appl Physiol. 2005;98(6):2169–76. doi: 10.1152/japplphysiol.00013.2005. [DOI] [PubMed] [Google Scholar]
- 16.Natali AJ, Wilson LA, Peckham M, Turner DL, Harrison SM, White E. Different regional effects of voluntary exercise on the mechanical and electrical properties of rat ventricular myocytes. J Physiol. 2002;541(Pt 3):863–75. doi: 10.1113/jphysiol.2001.013415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wisloff U, Loennechen JP, Falck G, Beisvag V, Currie S, Smith G, et al. Increased contractility and calcium sensitivity in cardiac myocytes isolated from endurance trained rats. Cardiovasc Res. 2001;50(3):495–508. doi: 10.1016/s0008-6363(01)00210-3. [DOI] [PubMed] [Google Scholar]
- 18.Kemi OJ, Ellingsen O, Ceci M, Grimaldi S, Smith GL, Condorelli G, et al. Aerobic interval training enhances cardiomyocyte contractility and Ca2+ cycling by phosphorylation of CaMKII and Thr-17 of phospholamban. J Mol Cell Cardiol. 2007;43(3):354–61. doi: 10.1016/j.yjmcc.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koch LG, Britton SL. Artificial selection for intrinsic aerobic endurance running capacity in rats. Physiological genomics. 2001;5(1):45–52. doi: 10.1152/physiolgenomics.2001.5.1.45. [DOI] [PubMed] [Google Scholar]
- 20.Baldo MP, Zaniqueli D, Forechi L, Machado RC, Rodrigues SL, Mill JG. Effects of spironolactone in spontaneously hypertensive adult rats subjected to high salt intake. Clinics. 2011;66(3):477–82. doi: 10.1590/S1807-59322011000300020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bezerra DG, Lacerda Andrade LM, Pinto da Cruz FO, Mandarim-de-Lacerda CA. Atorvastatin attenuates cardiomyocyte loss in adult rats from protein-restricted dams. J Card Fail. 2008;14(2):151–60. doi: 10.1016/j.cardfail.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 22.Roman-Campos D, Duarte HL, Sales PA, Jr, Natali AJ, Ropert C, Gazzinelli RT, et al. Changes in cellular contractility and cytokines profile during Trypanosoma cruzi infection in mice. Basic Res Cardiol. 2009;104(3):238–46. doi: 10.1007/s00395-009-0776-x. [DOI] [PubMed] [Google Scholar]
- 23.Satoh H, Delbridge LM, Blatter LA, Bers DM. Surface: volume relationship in cardiac myocytes studied with confocal microscopy and membrane capacitance measurements: species-dependence and developmental effects. Biophys J. 1996;70(3):1494–504. doi: 10.1016/S0006-3495(96)79711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wisloff U, Najjar SM, Ellingsen O, Haram PM, Swoap S, Al-Share Q, et al. Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science. 2005;307(5708):418–20. doi: 10.1126/science.1108177. [DOI] [PubMed] [Google Scholar]
- 25.Rolim NP, Medeiros A, Rosa KT, Mattos KC, Irigoyen MC, Krieger EM, et al. Exercise training improves the net balance of cardiac Ca2+ handling protein expression in heart failure. Physiol Genomics. 2007;29(3):246–52. doi: 10.1152/physiolgenomics.00188.2006. [DOI] [PubMed] [Google Scholar]
- 26.Medeiros A, Rolim NP, Oliveira RS, Rosa KT, Mattos KC, Casarini DE, et al. Exercise training delays cardiac dysfunction and prevents calcium handling abnormalities in sympathetic hyperactivity-induced heart failure mice. J Appl Physiol. 2008;104(1):103–9. doi: 10.1152/japplphysiol.00493.2007. [DOI] [PubMed] [Google Scholar]
- 27.Di Filippo C, Cuzzocrea S, Rossi F, Marfella R, D'Amico M. Oxidative stress as the leading cause of acute myocardial infarction in diabetics. Cardiovasc Drug Rev. 2006;24(2):77–87. doi: 10.1111/j.1527-3466.2006.00077.x. [DOI] [PubMed] [Google Scholar]
- 28.Hori M, Nishida K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc Res. 2009;81(3):457–64. doi: 10.1093/cvr/cvn335. [DOI] [PubMed] [Google Scholar]
- 29.Lepore DA, Knight KR, Anderson RL, Morrison WA. Role of priming stresses and Hsp70 in protection from ischemia-reperfusion injury in cardiac and skeletal muscle. Cell Stress Chaperones. 2001;6(2):93–6. doi: 10.1379/1466-1268(2001)006<0093:ropsah>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen EP, Bittner HB, Davis RD, Folz RJ, Van Trigt P. Extracellular superoxide dismutase transgene overexpression preserves postischemic myocardial function in isolated murine hearts. Circulation. 1996;94(9 Suppl):II412–7. [PubMed] [Google Scholar]
- 31.Hayakawa K, Takemura G, Kanoh M, Li Y, Koda M, Kawase Y, et al. Inhibition of granulation tissue cell apoptosis during the subacute stage of myocardial infarction improves cardiac remodeling and dysfunction at the chronic stage. Circulation. 2003;108(1):104–9. doi: 10.1161/01.CIR.0000074225.62168.68. [DOI] [PubMed] [Google Scholar]
- 32.Liu JL, Irvine S, Reid IA, Patel KP, Zucker IH. Chronic exercise reduces sympathetic nerve activity in rabbits with pacing-induced heart failure: A role for angiotensin II. Circulation. 2000;102(15):1854–62. doi: 10.1161/01.cir.102.15.1854. [DOI] [PubMed] [Google Scholar]
- 33.Melling CW, Thorp DB, Milne KJ, Krause MP, Noble EG. Exercise-mediated regulation of Hsp70 expression following aerobic exercise training. Am J Physiol Heart Circ Physiol. 2007;293(6):H3692–8. doi: 10.1152/ajpheart.00827.2007. [DOI] [PubMed] [Google Scholar]
- 34.Gao L, Wang W, Liu D, Zucker IH. Exercise training normalizes sympathetic outflow by central antioxidant mechanisms in rabbits with pacing-induced chronic heart failure. Circulation. 2007;115(24):3095–102. doi: 10.1161/CIRCULATIONAHA.106.677989. [DOI] [PubMed] [Google Scholar]
- 35.Xu X, Zhao W, Wan W, Ji LL, Powers AS, Erikson JM, et al. Exercise training combined with angiotensin II receptor blockade reduces oxidative stress after myocardial infarction in rats. Exp Physiol. 2010;95(10):1008–15. doi: 10.1113/expphysiol.2010.054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soufi FG, Farajnia S, Aslanabadi N, Ahmadiasl N, Alipour M, Doustar Y, et al. Long-term exercise training affects age-induced changes in HSP70 and apoptosis in rat heart. Gen Physiol Biophys. 2008;27(4):263–70. [PubMed] [Google Scholar]
- 37.Ziada AM, Hassan MO, Tahlilkar KI, Inuwa IM. Long-term exercise training and angiotensin-converting enzyme inhibition differentially enhance myocardial capillarization in the spontaneously hypertensive rat. J Hypertens. 2005;23(6):1233–40. doi: 10.1097/01.hjh.0000170387.61579.ab. [DOI] [PubMed] [Google Scholar]
- 38.Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81(4):1161–72. doi: 10.1161/01.cir.81.4.1161. [DOI] [PubMed] [Google Scholar]
- 39.Oliveira RS, Ferreira JC, Gomes ER, Paixao NA, Rolim NP, Medeiros A, et al. Cardiac anti-remodelling effect of aerobic training is associated with a reduction in the calcineurin/NFAT signalling pathway in heart failure mice. J Physiol. 2009;587(Pt 15):3899–910. doi: 10.1113/jphysiol.2009.173948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Veiga EC, Antonio EL, Bocalini DS, Murad N, Abreu LC, Tucci PJ, et al. Prior exercise training does not prevent acute cardiac alterations after myocardial infarction in female rats. Clinics. 2011;66(5):889–93. doi: 10.1590/S1807-59322011000500028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Starnes JW, Taylor RP, Ciccolo JT. Habitual low-intensity exercise does not protect against myocardial dysfunction after ischemia in rats. Eur J Cardiovasc Prev Rehabil. 2005;12(2):169–74. doi: 10.1097/01.hjr.0000159319.62466.95. [DOI] [PubMed] [Google Scholar]
- 42.Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101(25):2981–8. doi: 10.1161/01.cir.101.25.2981. [DOI] [PubMed] [Google Scholar]
- 43.Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999;79(1):215–62. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- 44.Kemi OJ, Haram PM, Wisloff U, Ellingsen O. Aerobic fitness is associated with cardiomyocyte contractile capacity and endothelial function in exercise training and detraining. Circulation. 2004;109(23):2897–904. doi: 10.1161/01.CIR.0000129308.04757.72. [DOI] [PubMed] [Google Scholar]
- 45.Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, et al. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94(1):110–8. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 46.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7(8):589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 47.DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, et al. Akt1 is required for physiological cardiac growth. Circulation. 2006;113(17):2097–104. doi: 10.1161/CIRCULATIONAHA.105.595231. [DOI] [PubMed] [Google Scholar]
- 48.Kemi OJ, Ceci M, Wisloff U, Grimaldi S, Gallo P, Smith GL, et al. Activation or inactivation of cardiac Akt/mTOR signaling diverges physiological from pathological hypertrophy. Journal of cellular physiology. 2008;214(2):316–21. doi: 10.1002/jcp.21197. [DOI] [PubMed] [Google Scholar]
- 49.Wisloff U, Loennechen JP, Currie S, Smith GL, Ellingsen O. Aerobic exercise reduces cardiomyocyte hypertrophy and increases contractility, Ca2+ sensitivity and SERCA-2 in rat after myocardial infarction. Cardiovasc Res. 2002;54(1):162–74. doi: 10.1016/s0008-6363(01)00565-x. [DOI] [PubMed] [Google Scholar]
- 50.He X, Liu Y, Sharma V, Dirksen RT, Waugh R, Sheu SS, et al. ASK1 associates with troponin T and induces troponin T phosphorylation and contractile dysfunction in cardiomyocytes. The American journal of pathology. 2003;163(1):243–51. doi: 10.1016/S0002-9440(10)63647-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee Y, Min K, Talbert EE, Kavazis AN, Smuder AJ, Willis WT, et al. Exercise protects cardiac mitochondria against ischemia-reperfusion injury. Med Sci Sports Exerc. 2012;44(3):397–405. doi: 10.1249/MSS.0b013e318231c037. [DOI] [PubMed] [Google Scholar]
- 52.Orenstein TL, Parker TG, Butany JW, Goodman JM, Dawood F, Wen WH, et al. Favorable left ventricular remodeling following large myocardial infarction by exercise training. Effect on ventricular morphology and gene expression. The Journal of clinical investigation. 1995;96(2):858–66. doi: 10.1172/JCI118132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diffee GM, Nagle DF. Regional differences in effects of exercise training on contractile and biochemical properties of rat cardiac myocytes. J Appl Physiol. 2003;95(1):35–42. doi: 10.1152/japplphysiol.00951.2002. [DOI] [PubMed] [Google Scholar]