

Abstract
Minor histocompatibility (H) antigens are targets of graft-versus-host disease and graft-versus-tumor responses after human leukocyte antigen (HLA) matched allogeneic hematopoietic stem cell transplantation. Recently, we reported a strategy for genetic mapping of linkage disequilibrium (LD) blocks that encoded novel minor H antigens using the large data set from the International HapMap Project combined with conventional immunologic assays to assess recognition of HapMap B lymphoid cell line (B-LCL) by minor H antigen-specific T cells. In this study, we have constructed and provide an online interactive program and demonstrate its utility for searching for single nucleotide polymorphisms (SNPs) responsible for minor H antigen generation. The website is available as “HapMap SNP Scanner”, and can incorporate T cell recognition and other data with genotyping data sets from CEU, JPT, CHB and YRI to provide a list of candidate SNPs that correlate with observed phenotypes. This method should substantially facilitate discovery of novel SNPs responsible for minor H antigens and be applicable for assaying of other specific cell phenotypes (e.g. drug sensitivity) to identify individuals who may benefit from SNP-based customized therapies.
Keywords: genome-wide association study, HapMap, minor histocompatibility antigen, online software, single nucleotide polymorphism
Introduction
Variations in the DNA sequences of humans can determine susceptibility to a variety of diseases and responses to drugs, vaccines, and other agents, and will therefore contribute to the development of personalized medicines (1). Around only 0.1% of DNA sequences in one individual differs from that of another, and of the 0.1% difference, single nucleotide polymorphisms (SNPs) account for over 80% (2). In HLA-identical allogeneic hematopoietic stem cell transplantation (HSCT), most minor histocompatibility (H) antigens recognized by cytotoxic T lymphocytes (CTL) or helper T lymphocytes of the donor result from nonsynonymous SNPs that differ between the HSCT donor and recipient and result in a novel peptide sequence presented by major histocompatibility complex (MHC) molecules on recipient cells (3, 4). Disparities in minor H antigens result in graft-versus-host disease (GVHD), graft rejection, and graft-versus-tumor (GVT) effect following allo-HSCT, but despite the efforts of several laboratories worldwide, only a small number of determinants have been molecularly characterized. Several methods have been developed to identify minor H antigens, including peptide elution from MHC molecules (5, 6), cDNA expression cloning (7, 8), linkage analysis (9–11), and whole genome association scanning using SNP array (12).
Recently, we and others (13, 14) reported a powerful approach for rapidly identifying minor H antigen loci using a large genotyping data set and B-lymphoid cell lines (B-LCLs) from the International HapMap Project for genome wide association analysis (15–17). Haplotypes are generally shared between populations, but their frequency can differ widely. Until recently, four populations were selected for inclusion in the HapMap: 30 adult-and-both-parents trios from (i.e., 90 individuals) Ibadan, Nigeria (YRI), 30 trios of U.S. residents of northern and western European ancestry (CEU), 44 unrelated individuals from Tokyo, Japan (JPT) and 45 unrelated Han Chinese individuals from Beijing, China (CHB) (15–17). By assigning HapMap B-LCLs into antigen positive and antigen negative groups based on T cell recognition, one can then determine the likely locations and haplotypes (i.e. a set of tag SNPs) from the HapMap data that are involved in the antigen positive phenotype.
Although the approach is straightforward and powerful, the computational calculations required genetic analysis capabilities that are currently unavailable to most conventional laboratories. In this report, we describe an online program, HapMap SNP Scanner, that is freely available on the web, and can be conveniently used by research groups engaged in minor H antigen discovery to search for candidate SNP(s) responsible for destructive and beneficial alloreactivity after HSCT. This program could also be applied to evaluate a potential genetic basis for differences in other cell phenotypes such as sensitivities to drugs (e.g. etoposide (18)), cytokines and other agents.
Materials and Methods
Online Programs
The HapMap SNP Scanner consists of a set of cooperating programs. Dynamic HTML pages for user query and results demonstrating a list of the candidate SNPs are generated by PHP scripts and are linked with the main program written in C++ that finds candidate SNPs associating the cell phenotype of interest running on a Unix clone (13). Computation of statistics was performed on a Macintosh equipped with 2 × quadcore 3.2 GHz Zeon processors (Apple, Cupertino, CA, USA). The exact algorithm used to identify the candidate minor histocompatibility antigen locus was described previously (13). In brief, the relevant loci were explored through association analysis, in which χ2 test statistics was calculated for all the Phase III HapMap SNPs according to the dominant model, which is expected for the phenotype (i.e., the susceptibility to lysis in CTL assays or interferon-γ production by ELISA). The software and HLA typing database for HapMap individuals (see below) are available via a web interface at http://hapmap-scanner.fujita-hu.ac.jp/. [The site is currently password protected. Please use “hapmap” as ID and Password for viewing]. An example demonstrating the utility of this program for discovering a novel minor H antigen-associated SNP is described in the Results.
HLA typing of the HapMap B-LCLs
HapMap B-LCLs were obtained from the Coriell Cell Repositories (Coriell Institute for Medical Research, Camden, NJ, USA). DNA of all samples from individual B-LCL (1–2 × 106 cells) was isolated using the QIAamp DNA Mini Kit (QIAGEN Inc, Valencia, CA, USA) according to the recommendations of the manufacturer. DNA samples were then genotyped for HLA-A, B, C and DRB1 loci using Luminex200 (Luminex Technology, Austin, TX, USA) at the HLA laboratory (Kyoto, Kyoto, Japan).
CTL clone and immunological typing
All samples were obtained after written informed consent based on a protocol approved by the Institutional Review Board Committee at Fred Hutchinson Cancer Research Center and Aichi Cancer Center according to the Declaration of Helsinki. Minor H antigen-specific CD8+ CTL clones were generated from a donor’s peripheral blood by stimulation with dendritic cells prepared from his HLA-identical sibling (patient) followed by standard limiting dilution as described (11). An HLA-B*07:02-restricted CTL clone, K9.3 that exhibited sufficient lytic activity and growth, was chosen for analysis using the HapMap Scanner.
HLA-B*07:02-transduced CEU HapMap LCL panels were phenotyped for the presence or absence of minor H antigen by CTL-K9.3 using an enzyme-linked immunosorbent assay (ELISA). In brief, ten thousand B-LCLs were plated in each well of 96-well flat-bottomed plates with ten thousand K9.3-CTL overnight at 37°C, then 50 µL of supernatant was collected and IFN-γ production was measured by ELISA. All assays were performed at least in duplicate, then, the minor H antigen status (phenotype) of each HapMap B-LCL was defined as positive (+), negative (−), or undetermined.
Confirmatory genotyping, epitope mapping and reconstitution assay
Confirmatory genotyping was carried out by direct sequencing of genomic DNA for the candidate SNPs. DNA was extracted from B-LCLs, and PCR was performed with primers corresponding to the genes of interest. Amplified DNA samples were sequenced using BigDye Terminator 3.1 (Applied Biosystems, Foster City, CA, USA).
Peptides spanning the polymorphic amino acid and predicted to bind to the corresponding HLA molecule by BIMAS (http://bimas.dcrt.nih.gov/molbio/hla_bind/) (19) and/or SYFPEITHI algorithms (http://www.syfpeithi.de)(20) were synthesized by standard methods. Chromium-labeled minor H antigen-negative B-LCL from the HSCT donor was incubated for 30 min in medium containing 10-fold serial dilutions of the peptides and then used as target cells in a standard cytotoxic assay.
Real time PCR assay
First-strand cDNA from poly (A) RNA from normal human tissues (Human Multiple Tissue cDNA panels I and II; Clontech) was analyzed for expression of CD45 and TMEM214 by polymerase chain reaction (PCR) of duplicate samples. Real-time PCR analysis was performed using the TaqMan assay as described previously (11). Data were normalized to expression of porphobilinogen deaminase (PBGD). Amplifications were performed on an ABI Prism 7900 (Applied Biosystems) with a 50-µL reaction mixture consisting of 2.5 ng of cDNA, 25 µL of TaqMan Gene Expression Master Mix (Applied Biosystems), 300nM gene-specific forward and reverse primers, and 250nM gene-specific TaqMan probe. All results were normalized with respect to the internal control, and are expressed relative to the levels found in recipient B-LCL which were susceptible to lysis by the minor H antigen specific CTL-K9.3.
Results and Discussion
HLA typing database of individual HapMap B-LCL
Minor H antigen peptides are presented to alloreactive T cells in the context of specific HLA alleles, therefore it is essential that the B-LCL to be used as target cells express the HLA restricting allele to appropriately assign an antigen positive or antigen negative phenotype. If necessary, the B-LCL can be transduced with a cDNA encoding the HLA restricting allele before immunological phenotyping by T cell clones (21). However, to avoid unnecessary gene transduction, we performed HLA genotyping on individual B-LCLs so that this information is available to select B-LCL for analysis (Figure 1, and complete data except YRI individuals are available in the supplementary Table 1, 2 and 3). In the case of CTL clone K9.3 that recognizes a minor H antigen presented by HLA-B*07:02, it was evident that of 60 CEU parents (devoid of their child), 18 (30%) individuals possessed the HLA-B*07:02 allele, thus transduction of these LCL prior to cytotoxicity assay was unnecessary. If available, links tagged to the individual B-LCLs on our website are directed to the related information available at the Coriell Institute for Medical Research (http://ccr.coriell.org/), and the HLA genotyping data that is provided may serve as standard quality controls for clinical and research laboratories.
Figure 1.

HLA genotyping of HapMap CEU B-LCL cell lines. Genomic DNA was extracted from individual B-LCLs and genotyped with Luminex200 technology. The HLA genotypes are expressed as 4-digit nomenclature. The first column indicates the catalogue IDs which are directly linked to Coriell databases available on-line. The whole HLA typing data for CEU, JPT and CHB B-LCLs are provided in the Supplementary Tables 1, 2 and 3.
HapMap SNP Scanner-based genome wide search for minor H antigen loci and estimation of linkage disequilibrium (LD) block(s)
We next performed interferon-γ production assays with CTL-K9.3 against a B-LCL panel that either endogenously expressed, or were transduced to express, HLA-B*07:02. The minor H antigen phenotype was assigned for each B-LCL (33, positive; 25, negative; and 3, undetermined, supplemental Figure 1) and entered by selecting “Pos” for minor H antigen+, “Neg” for minor H antigen− or “NA” for typing not available, in a pull-down menu for individual HapMap B-LCLs shown in the entry page of the HapMap SNP Scanner for CEU (Figure 2A). The number of permutations can be entered before submission. From this dataset, a list of candidate SNPs will be derived and shown automatically in a new page according to their χ2 values (Figure 2B). Each SNP is linked to the International HapMap Project web site showing the search result for the chosen SNP (http://hapmap.ncbi.nlm.nih.gov/cgi-perl/gbrowse/hapmap27_B36/#search). By choosing an appropriate magnification such as 100kb from the Scroll and Zoom pull-down menu with LD plot checkbox activated, it is possible to obtain the LD block of interest region as an intrinsic feature of the International HapMap Project browser (Figure 3, 100kb width).
Figure 2.
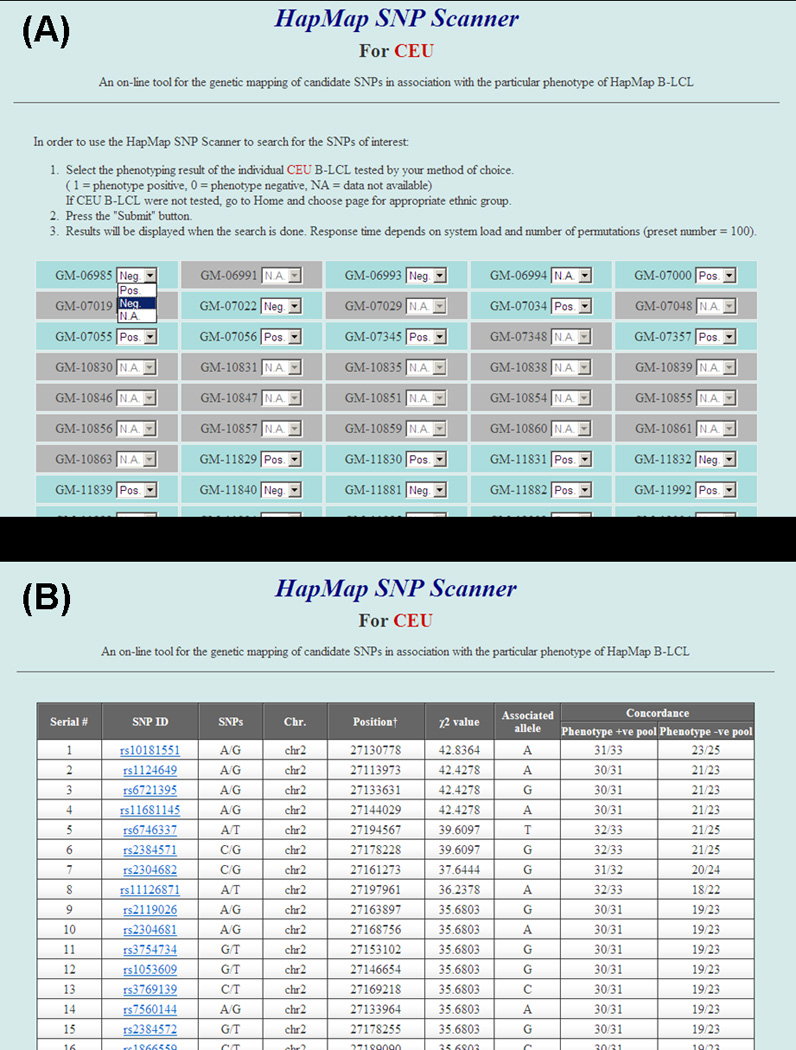
SNP search using HapMap SNP Scanner. (A) Phenotyping data entry page for CEU. ID numbers shown in gray correspond to the offspring of the individual trios that are not included in our association study currently. The phenotyping results obtained by appropriate biological assays can be entered by selecting “Pos” for phenotype+, “Neg” for phenotype− or “NA” for typing not available, in a pull-down menu for individual HapMap B-LCLs. The number of permutations can be entered (out of the range of the figure because it is located in the bottom of the web page). After submitting the query, a new page with results will automatically appear. (B) Result page. A list of candidate SNPs aligned as a descending manner according to their χ2 values. Each SNP (underlined) is linked to the International HapMap Project web site showing its search result for the chosen SNP.
Figure 3.

LD structures analysis around the SNP showing peak statistical value. LD plot around rs1124649 was drawn by the International HapMap project webpage by clicking the candidate SNP unique number appeared in the Result page. The size of the region containing a putative minor H antigen locus is adjusted according to the condition of linkage disequilibrium. The position of the SNPs showing the highest χ2 value of 42.8346 (rs1124649, rs10181551, rs6721395 and rs11681145) are indicated as follows: ▼, □, ○, ◊, respectively. The candidate genes, TMEM214 and AGBL5 are also shown.
Analysis of the data generated for CTL clone K9.3 revealed that the top 4 SNPs with the highest χ2 value of 42.8346 (Figure 2B) were located either in TMEM214 (rs1124649) or AGBL5 (rs10181551, rs6721395 and rs11681145). Direct sequencing of amplified products spanning individual SNPs by genomic PCR of DNA from donor and recipient cells revealed that only the SNP rs1124649 genotype was disparate in the GVHD/GVT direction, which excluded rs10181551, rs6721395 and rs11681145 as candidates (data not shown). The donor was G/G homozygous and the recipient A/G heterozygous at rs1124649, resulting in a predicted amino acid substitution from valine (GTG) to methionine (ATG). Analysis of the predicted amino acid sequences of peptides that spanned the SNP rs1124649 for binding motifs for HLA-B*07:02 using SYFPEITHI software (20) identified YPRLKVLAF and YPRLKMLAF as candidate sequences from donor and recipient alleles, respectively that would bind to HLA- B*07:02. These peptides were synthesized and pulsed onto donor cells in epitope reconstitution assays performed. As shown in Figure 4, the peptide with the recipient amino acid sequence (YPRLKMLAF) was recognized by the K 9.3-CTL with concentration at half maximal cytolysis of 10 nM, but the donor sequence (YPRLKVLAF) was not recognized. Thus, the genetic and functional data indicate that the minor H antigen recognized by K 9.3 CTL is YPRLKMLAF, resulting from the SNP at rs1124649. We performed TaqMan qPCR for TMEM214 on a panel of cDNAs derived from non-hematopoietic tissues and found that the gene is not hematopoietic specific (data not shown), consistent with the data in the BioGPS database (http://biogps.org/).
Figure 4.
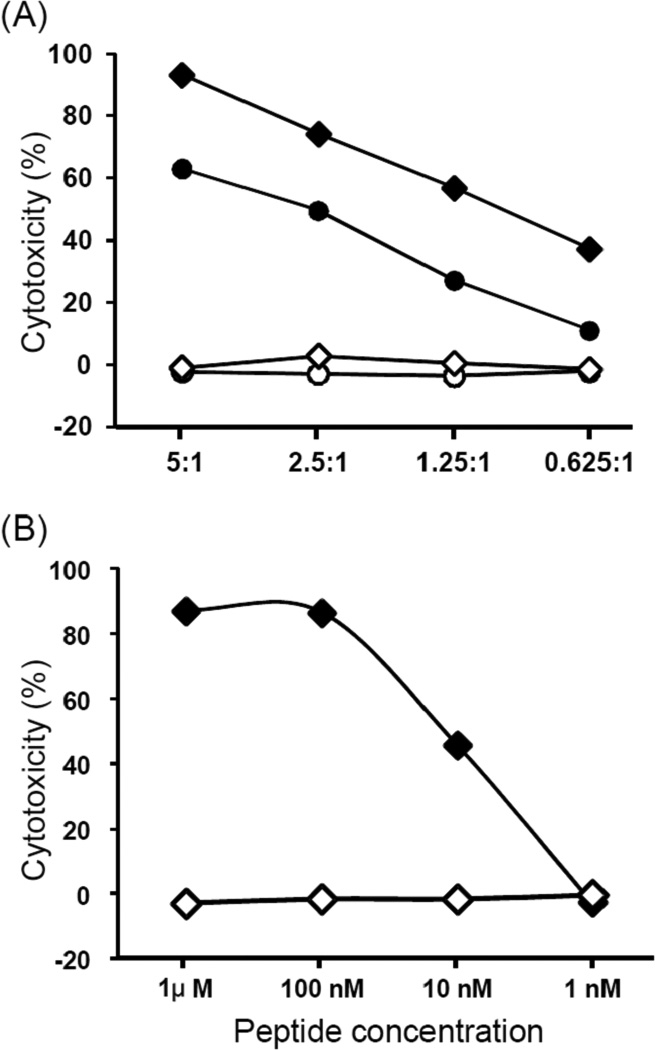
(A) Lysis of recipient B-LCL (●), donor B-LCL (○), donor B-LCL pulsed with 1 µM of peptide, YPRLKMLAF (♦), and donor B-LCL pulsed with 1 µM of peptide YPRLKVLAF (◊) by the CTL clone K 9.3. (B) Lysis of donor B-LCL pulsed with various concentrations of YPRLKMLAF (♦) or YPRLKVLAF (◊) peptides. Data is shown at an E/T of 20:1.
The International HapMap Project has had a profound impact on our ability to interrogate human genome sequence polymorphisms and identify genes that affect human health and disease. The publicly available HapMap resources enable gene-wide scanning, and we and others have recently shown that these resources are amenable to high-throughput identification of minor H antigen genes without highly specialized equipment or expensive microarrays (13, 14, 22). Although many labs are focused on discovering clinically relevant minor H antigens, an obstacle that remains for widespread use of the HapMap resources is the lack of a readily available analysis platform for integrating the results of cytotoxicity or other assays of T cell recognition with the complex genetic data of the HapMap LCL lines used in the assays. In this study, we have developed and provide an online program with a user-friendly interface where any phenotyping results of HapMap individuals can be entered by simply choosing a pull-down menu, and the analyzed results will be shown in a new table in ascending order according to χ2 values of candidate SNPs.
Another potential use of HapMap data is to identify SNPs that are associated with significant biological effects in response to drugs. A small percentage of people given a drug might either have no response or have a life-threatening response or death. The current limitation is that all available biological samples for the International HapMap Projects are only B-LCL, immortalized B cells with Epstein Barr virus, and extracted DNA. However, since B-LCLs have been reprogrammed to Epstein Barr virus-free pluripotent stem cells using an episomal vector system and differentiated into other cell types (23), the online program we have developed may also be useful for exploring other genetic traits or molecular targets (e.g., differential responses to drug of interest), if they can be discriminated accurately through appropriate biological assays.
Supplementary Material
Acknowledgements
We thank Dr. W. Ho for critically reading the manuscript; Ms. Keiko Nishida, Ms. Miyoko Tanimoto, Ms. Yasuko Watarai, and Ms. Hiromi Tamaki for their technical expertise. This study was supported in part by Grant-in-Aid for Scientific Research (C)(21591256), from the Ministry of Education, Culture, Science, Sports, and Technology, Japan; Grants for Research on the Human Genome, Tissue Engineering Food Biotechnology and the Second and Third Team Comprehensive 10-year Strategy for Cancer Control, from the Ministry of Health, Labour, and Welfare, Japan; a Grant-in-Aid from Core Research for Evolutional Science and Technology (CREST) of Japan. The National Cancer Institute CA18029 (SRR) and K23CA154532 (MB) also provided support. Dr. Marie Bleakley is the Damon Runyon-Richard Lumsden Foundation Clinical Investigator supported by the Damon Runyon Cancer Research Foundation (CI-57-11). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Footnotes
Conflict of interest:
The authors declare no conflict of interest.
References
- 1.Nebert DW, Zhang G, Vesell ES. From human genetics and genomics to pharmacogenetics and pharmacogenomics: past lessons, future directions. Drug metabolism reviews. 2008;40:187–224. doi: 10.1080/03602530801952864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weiner MP, Hudson TJ. Introduction to SNPs: discovery of markers for disease. BioTechniques. 2002;10(Suppl:4–7):2–3. [PubMed] [Google Scholar]
- 3.Bleakley M, Riddell SR. Molecules and mechanisms of the graft-versus-leukaemia effect. Nature reviews. 2004;4:371–380. doi: 10.1038/nrc1365. [DOI] [PubMed] [Google Scholar]
- 4.Spierings E, Goulmy E. Expanding the immunotherapeutic potential of minor histocompatibility antigens. The Journal of clinical investigation. 2005;115:3397–400. doi: 10.1172/JCI27094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brickner AG, Warren EH, Caldwell JA, et al. The immunogenicity of a new human minor histocompatibility antigen results from differential antigen processing. The Journal of experimental medicine. 2001;193:195–206. doi: 10.1084/jem.193.2.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.den Haan JM, Meadows LM, Wang W, et al. The minor histocompatibility antigen HA-1: a diallelic gene with a single amino acid polymorphism. Science (New York, NY. 1998;279:1054–1057. doi: 10.1126/science.279.5353.1054. [DOI] [PubMed] [Google Scholar]
- 7.Dolstra H, Fredrix H, Maas F, et al. A human minor histocompatibility antigen specific for B cell acute lymphoblastic leukemia. The Journal of experimental medicine. 1999;189:301–308. doi: 10.1084/jem.189.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawase T, Akatsuka Y, Torikai H, et al. Alternative splicing due to an intronic SNP in HMSD generates a novel minor histocompatibility antigen. Blood. 2007;110:1055–1063. doi: 10.1182/blood-2007-02-075911. [DOI] [PubMed] [Google Scholar]
- 9.de Rijke B, van Horssen-Zoetbrood A, Beekman JM, et al. A frameshift polymorphism in P2X5 elicits an allogeneic cytotoxic T lymphocyte response associated with remission of chronic myeloid leukemia. The Journal of clinical investigation. 2005;115:3506–3516. doi: 10.1172/JCI24832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akatsuka Y, Nishida T, Kondo E, et al. Identification of a polymorphic gene, BCL2A1, encoding two novel hematopoietic lineage-specific minor histocompatibility antigens. The Journal of experimental medicine. 2003;197:1489–1500. doi: 10.1084/jem.20021925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bleakley M, Otterud BE, Richardt JL, et al. Leukemia-associated minor histocompatibility antigen discovery using T-cell clones isolated by in vitro stimulation of naive CD8+ T cells. Blood. 2011;115:4923–4933. doi: 10.1182/blood-2009-12-260539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawase T, Nannya Y, Torikai H, et al. Identification of human minor histocompatibility antigens based on genetic association with highly parallel genotyping of pooled DNA. Blood. 2008;111:3286–3294. doi: 10.1182/blood-2007-10-118950. [DOI] [PubMed] [Google Scholar]
- 13.Kamei M, Nannya Y, Torikai H, et al. HapMap scanning of novel human minor histocompatibility antigens. Blood. 2009;113:5041–5048. doi: 10.1182/blood-2008-07-171678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spaapen RM, Lokhorst HM, van den Oudenalder K, et al. Toward targeting B cell cancers with CD4+ CTLs: identification of a CD19-encoded minor histocompatibility antigen using a novel genome-wide analysis. The Journal of experimental medicine. 2008;205:2863–2872. doi: 10.1084/jem.20080713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 16.Frazer KA, Ballinger DG, Cox DR, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang RS, Duan S, Bleibel WK, et al. A genome-wide approach to identify genetic variants that contribute to etoposide-induced cytotoxicity. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:9758–9763. doi: 10.1073/pnas.0703736104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 20.Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol. 1994;152:163–175. [PubMed] [Google Scholar]
- 21.Akatsuka Y, Goldberg TA, Kondo E, et al. Efficient cloning and expression of HLA class I cDNA in human B-lymphoblastoid cell lines. Tissue antigens. 2002;59:502–511. doi: 10.1034/j.1399-0039.2002.590607.x. [DOI] [PubMed] [Google Scholar]
- 22.Spaapen RM, de Kort RA, van den Oudenalder K, et al. Rapid identification of clinical relevant minor histocompatibility antigens via genome-wide zygosity-genotype correlation analysis. Clin Cancer Res. 2009;15:7137–7143. doi: 10.1158/1078-0432.CCR-09-1914. [DOI] [PubMed] [Google Scholar]
- 23.Rajesh D, Dickerson SJ, Yu J, Brown ME, Thomson JA, Seay NJ. Human lymphoblastoid B-cell lines reprogrammed to EBV-free induced pluripotent stem cells. Blood. 118:1797–1800. doi: 10.1182/blood-2011-01-332064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.