

Abstract
Cryptosporidium parvum and related species are zoonotic intracellular parasites of the intestine. Cryptosporidium is a leading cause of diarrhea in small children around the world. Infection can cause severe pathology in children and immunocompromised patients. This waterborne parasite is resistant to common methods of water treatment and therefore a prominent threat to drinking and recreation water even in countries with strong water safety systems. The drugs currently used to combat these organisms are ineffective. Genomic analysis revealed that the parasite relies solely on inosine-5′-monophosphate dehydrogenase (IMPDH) for the biosynthesis of guanine nucleotides. Herein, we report a selective urea-based inhibitor of C. parvum IMPDH (CpIMPDH) identified by high throughput screening. We performed a SAR study of these inhibitors with some analogues exhibiting high potency (IC50 < 2 nM) against CpIMPDH, excellent selectivity > 1000-fold versus human IMPDH type 2 and good stability in mouse liver microsomes. A subset of inhibitors also displayed potent antiparasitic activity in a Toxoplasma gondii model.
Introduction
Cryptosporidium parasites such as C. parvum and C. hominis are major causes of the ‘vicious cycle of diarrhea and malnutrition’ that afflicts the developing world1. This infection is self-limiting in healthy adult individuals, but can be chronic and fatal in immunocompromised patients and children under 5 years of age2. Parasite oocysts are resistant to commonly used methods of water treatment, and contaminated drinking and recreational water are major sources of host to host transmission3. While cryptosporidiosis is more prevalent in developing countries4, the developed world also has a significant disease burden. Fifteen documented Cryptosporidium waterborne outbreaks were reported during 2000 in the USA, resulting in enormous medical expenses5. Since the oocysts can be obtained with relative ease and the water supply is readily accessed, C. parvum has the potential to be used for bio-terrorism. Consequently, it is considered a class B bio-warfare agent6. Neither vaccines nor the approved drug nitazoxanide are effective treatments for cryptosporidiosis in immunocompromised patients, so there is an urgent need for new chemotherapeutic agents.
Genomic analysis revealed that C. parvum and C. hominis rely on the enzyme inosine 5′-monophosphate dehydrogenase (IMPDH) for the production of guanine nucleotides2b, 7 (Scheme 1; CpIMPDH will be used to denote the parasite enzyme, which is identical in C. parvum and C. hominis) 8. Remarkably, these parasites appear to have obtained their IMPDH gene from an ε-proteobacterium by lateral gene transfer9. The bacterial origin of CpIMPDH and the different kinetic properties of these enzymes compared with their human counterparts suggested CpIMPDH-selective inhibitors could be obtained10. A high throughput screen for selective inhibitors yielded an urea-based inhibitor with an IC50 value of 340 nM against CpIMPDH (Compound 1, Table 1). Herein, we report a SAR study of 1 with respect to CpIMPDH inhibition and antiparasitic activity and optimization of mouse microsomal stability.
Scheme 1.
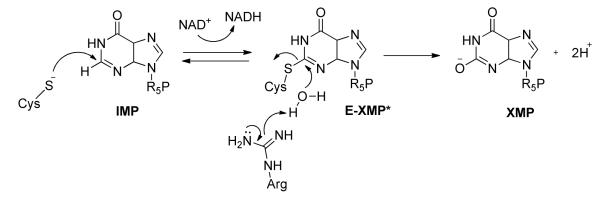
IMPDH catalyzed conversion of IMP to XMP. R5P = ribose-5′-phosphate
Table 1.
SAR of the anilide. Assays as described in methods. No significant inhibition of human IMPDH type 2 was observed (IC50 ≥ 5 μM).
| IC50 (nM) |
|||
|---|---|---|---|
| ID | R | (−) BSA | (+) BSA |
| 5a | Ph | 250 ± 20 | 410 ± 20 |
| 5b | 4-Cl-Ph | 20 ± 7 | 37 ± 7 |
| 5c | 4-Br-Ph | 10 ± 4 | 20 ± 10 |
| 5d | 3-Cl-Ph | 70 ± 10 | 330 ± 30 |
| 5e | 2-Cl-Ph | >5000 a | n.d. |
| 5f | 4-OMe-Ph | 9 ± 1 | 21 ± 7 |
| 5g | 4-tBu-Ph | >5000 a | n.d. |
| 5h | 3,4-di-ClPh | 6 ± 1 | 50 ± 20 |
| 5i | 3-CF3-4-Cl-Ph | 4 ± 1 | 20 ± 6 |
| 5j | 3-OMe,4-Cl-Ph | 1.3 ± 0.2 | 5 ± 1 |
| 5k | 3-CONH2,4-Cl-Ph | 2.3 ± 0.8 | 3 ± 1 |
| 5l | 3-CONHCH3,4-Cl-Ph | 7 ± 2 | 14 ± 3 |
| 5m | 3-CON(Me)2,4-Cl-Ph | 8 ± 1 | 11.7 ± 0.5 |
| 5n | 3,4-(OCH2CH2O)-Ph | 7.2 ± 0.6 | 110 ± 30 |
| 5o | 2-Naphthyl | 2.1 ± 0.8 | 40 ± 20 |
| 5p | 1-Naphthyl | >5000 a | n.d. |
| 5q | 6-Quinolyl | 1.8 ± 0.4 | 4 ± 1 |
| 5r | 7-Quinolyl | 0.8 ± 0.1 | 6 ± 2 |
| 5s | 3-Quinoline | 70 ± 10 | 308 ± 8 |
| 5t | 2-Quinoline | 250 ± 20 | 1100 ± 300 |
One determination. n.d., not determined.
Results and Discussion
High throughput screen
Following our previously described assay protocol 10, we performed a high throughput screen of 85,000 compounds to identify inhibitors of CpIMPDH. This screen targeted the NAD site, which is the most highly diverged region of the active site relative to the human enzymes. This screen yielded 50 compounds that inhibited CpIMPDH but not human IMPDH type 2 (IC50 > 5 μM). Six of these compounds were urea-based, of which the most potent was compound 1 (Figure 1) with IC50 of 340 nM. Interestingly, two urea-based inhibitors of human IMPDHs, merimepodib and AVN944, have entered clinical trials11, which further encouraged development of this series
Figure 1.
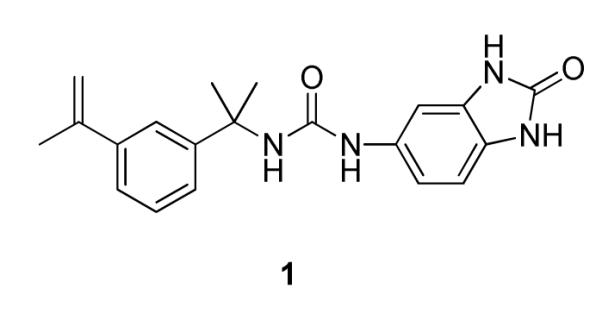
CpIMPDH inhibitor identified by HTS.
Chemistry
Various derivatives of 1 were prepared following the synthetic method outlined in Scheme 2. Ozonolysis followed by reductive work-up with dimethyl sulfide transformed 3-isopropenyl-α, α-dimethyl benzyl isocyanate 2 into 3-acetyl-α, α-dimethyl benzyl isocyanate 3 in moderate yield. Upon treating 2 and 3 with substituted anilines 4, the corresponding ureas 5 and 6 were obtained. The acetyl group on 6 was converted to the corresponding oxime 7 and methyl oxime 8 in good yields by heating with a slight excess of hydroxylamine hydrochloride and methoxyamine hydrochloride, respectively, in pyridine.12 Oxime 7 was alkylated in the presence of sodium hydride with 2-chloroethylamine hydrochloride to give amine derivative 9. Lithium aluminum hydride (LAH) reduction of acetyl derivative 6 gave alcohol 10. Hydrogenation of isoprenyl derivative 5 with 10% palladium on carbon yielded the saturated urea derivative 11 in good yield.
Scheme 2.

General procedure for preparation of urea derivatives.*
*Reagents and conditions: (a) (i) O3, DCM, -78 °C; (ii) Me2S, 10 h, 58%; (b) DCM, R-PhNH2 (4), 2-4 h, 70-85%; (c) THF, R-PhNH2 (4), 6 h, 70 0C, 70-75%; (d) NH OH•HCl, pyridine, 2 h, 90 °2 C, 80-85%; (e) NH2OMe•HCl, pyridine, 2 h, 90 oC, 60-85 %; (f) (i) NaH, THF, 0 oC, 30 min; (ii) Cl(CH ) o2 2NH2•HCl, rt, 10 h, 53%; (g) LAH, THF, 1 h, 0 C, 74%; (h) H2/Pd-C, MeOH, 1 h, 90%.
Scheme 3 outlines the reaction sequence used for the synthesis of a mono-methyl urea derivative. 3-(1-Cyanoethyl)benzoic acid, 12, was converted into acetyl derivative 13 using a step wise process that involved the conversion of the acid to the corresponding acid chloride followed by treatment with Meldrum’s acid in the presence of base and then hydrolytic decarboxylation13. Subsequent hydrolysis of nitrile derivative 13 gave substituted hydratropic acid 14, which was treated with thionyl chloride followed by sodium azide in water to generate acyl azide 15 that was used without isolation. Acyl azide 15 was heated to reflux in benzene under a nitrogen atmosphere to generate isocyanate 1614. Finally, the isocyanate was treated with 4-chloroaniline to give urea derivative 17.
Scheme 3.
Preparation of mono-methyl derivative 17.*
*Reagents and conditions (a) (i) SOCl , reflux, 2 h; (ii) Meldrum’s acid, pyridine, DCM, 0-5 ° 2 C, 2 h; iii) AcOH, H2O, reflux, 4 h, 28%; (b) Conc. HCl, 1,4-dioxane, reflux, 2 h, 78%; (c) (i) SOCl2, reflux; (ii) NaN3, DCM; (d) benzene, reflux, 1.5 h, 92%; (e) 4-Cl-PhNH2, THF, reflux, 6 h, 70%.
An analogue of 1 that lacks both methyl groups on the benzylic position was prepared according to the procedure outlined in Scheme 4. 3-Acetylbenzonitrile, 18, was treated with ethylene glycol15 to give the ketal protected benzonitrile derivative 19. LAH reduction generated benzylamine derivative 20, which was subsequently converted to urea 22 in two steps via the intermediate p-nitrophenyl carbamate 2116. Removal of the ketal was carried out under acidic conditions to give 23.
Scheme 4.
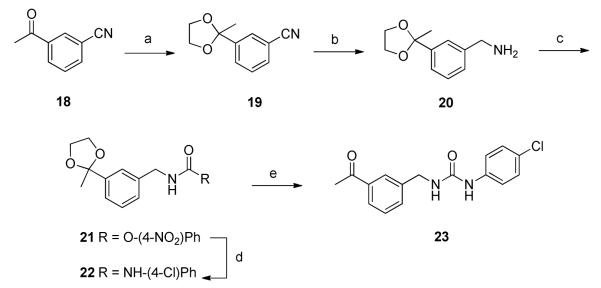
Preparation of benzyl derivative 23.*
*Reagents and conditions (a) (CH2OH)2, p-TSA, benzene, 4 h, 110 °C, Dean-Stark, 76%; (b) LAH, ether, 0 °C, 1.5 h, 60%; (c) DIPEA, 4-NO2-PhOCOCl, DCM/THF (1:1), rt, 24 h, 82%; (d) 4-Cl-PhNH2, DCM, rt, 10 h, 71%; (e) 2N HCl, THF, rt, 2 h, 91%.
A series of carbamate and amide derivatives were prepared using the methods outlined in Scheme 5. Isocyanate 3 was refluxed with 8N HCl to give benzylamine derivative 24 in good yield. This material was converted to carbamate 25 by treatment with 4-chlorophenyl chloroformate in the presence of base. Amine 24 was also converted to amide 26 in good yield. In addition, 24 was treated with chlorosulfonic acid, followed by exposure to aqueous potassium nitrite and subsequent decomposition with sulfate buffer (pH = 2.8) to give the substituted benzyl alcohol 2717. Upon treatment with 4-chlorophenyl isocyanate, this material was converted to carbamate 28.
Scheme 5.

Preparation of carbamate and amide derivatives.*
*Reagents and conditions (a) 8N HCl, 70 °C, 3 h, 55%; (b) 4-ClPhOCOCl, DIPEA, DCM/THF, rt, 24 h, 81% ; (c) 4-Cl-PhCH COCl, DCM, 0 °C to rt, 80%; (d) (i) ClSO o2 3H, CHCl3, 0 C (ii) KNO2 (iii) Water, pH = 2.8, 30%; (e) TEA, benzene, 4-Cl-Ph-NCO, 70 °C, 3 h, 62%.
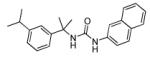
Several biphenyl derivatives were prepared using the methods outlined in Scheme 6. 3-Phenylcyanobenzene, 29, was allowed to react with 3 equivalents of methyl magnesium bromide in the presence of the Lewis acid catalyst titanium (IV) isopropoxide to give tert-alkyamine 3018. Biphenylcyclopropylamine 31 was similarly prepared from 29 by treatment with 2 equivalents of ethyl magnesium bromide and titanium (IV) isopropoxide19. Amines 30, 31 and 32 were converted to corresponding urea derivatives 36, 37 and 38, respectively, using a two-step procedure via intermediates 33, 34 and 35.
Scheme 6.

Preparation of biphenyl derivatives 36, 37 and unsubstituted benzylderivative 38.*
*Reagents and conditions (a) (i) 3 equiv. CH3MgBr, ether, rt, 30 min; (ii) Ti(i-PrO)4, 10 h, 57%; (b) (i) 2 equiv. EtMgBr, Ti(i-PrO)4, -78 °C to rt, 1 h; (ii) 2 equiv. BF3•OEt2, 1 h, 52%; (c) 4-NO2PhOCOCl, DIPEA, DCM/THF, 10 h; (d) 4-Cl-PhNH2, TEA, DCM, rt, 10 h, 71%.
Evaluation of enzyme inhibition
IMPDH activity was assayed by monitoring the production of NADH. The IC50 values reported in the tables are the average of three independent experiments unless otherwise noted. Nonspecific protein binding was assessed by measuring IC50 values in presence of 0.05% fatty acid free bovine serum albumin (BSA)20. Our previous experience indicates that the IC50 in the presence of BSA is the better predictor of antiparasitic activity21.
For the SAR study, initial attempts were made to optimize the anilide substituent. Removal of the cyclic urea moiety in 1 yielded compound 5a, which demonstrated a slight increase in CpIMPDH inhibitory activity with an IC50 of 250 nM (Table 1). Replacing the imidazolone with a 4-ClPh or 4-BrPh increased potency by 16-fold (5b) and 32-fold (5c), respectively. A similar trend has also been observed with other CpIMPDH inhibitor series22. However, translocation of the chlorine atom to the 3-position (5d) resulted in a 3-fold loss of activity, while the 2-chloro derivative (5e) was completely inactive. The electronic nature of the substituent in the 4-position had little effect on inhibitory activity. For example, an electron donating methoxy (5f) retained activity comparable to the bromide (5c) with IC50 of 9 nM. However, sterics did have an effect as illustrated by the 4-tert+butyl (5g) derivative lacking activity. Introduction of substituents in the 3,4-positions increased activity. For example, 3,4-di-chloro (5h), 4-chloro-3-trifluoromethyl (5i) and a variety of 3-amido-4-chloro (5k, 5l, and 5m) substituents increased potency by about 2.5 to 10-fold as compared to 5b. Again the electronic nature of the 3,4-substitents had little effect as exemplified by 4-chloro-3-methoxy (5j) and the 3,4-methylenedioxy (5n) analogues, although the latter compound demonstrated significantly weaker activity in the presence of BSA. Given these results, several fused carbo- and heteroaromatic derivatives were examined. The 2-naphthyl derivative (5o) retained potent active, while the regioisomer 1-naphthyl was devoid of activity. However, the inhibitory activity of 5o decreased in presence of BSA. Replacement of the 2-naphthyl with 6- and 7-quinolyl (5q and 5r) resulted in highly potent compounds that were only modestly reduced in the presence of BSA. The 2- and 3-quinolyl analogues (5t and 5s) were less potent.
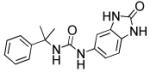
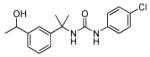
Next, the isoprenyl group was examined (Table 2). Saturation of the alkene to the resulting isopropyl (11) or removal (38) resulted in complete loss of inhibitory activity. Quite surprisingly, replacement of the alkene with an alcohol (10) resulted in moderate activity suggesting that polar substituents could be tolerated at this position. Incorporation of the alkene into a phenyl (36a) retained potent activity. However, transposing the phenyl (36b) to the para-position resulted in complete loss of inhibitory activity. Given the relative importance of the sp2-hybridized substituent at the 3-position and moderate tolerance of a hydrophilic alcohol in 10, replacement of the alkene with a ketone was explored. In three cases examined (6a-c) good to moderate inhibitory activity was observed.
Table 2.
SAR of the isopropenyl substituent.
| Compound | Structure | IC50 (nM) |
|
|---|---|---|---|
| (−) BSA | (+) BSA | ||
| 38 |
|
>5000 | n.d. |
| 11 |
|
>5000 | n.d. |
| 10 |
|
80 ± 20 | 120 ± 10 |
| 36a |
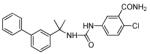
|
4 ± 4 | 14 ± 3 |
| 36b |
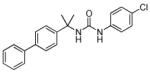
|
>5000a | n.d. |
| 6a |
|
39 ± 10b | 64 ± 22b |
| 6b |
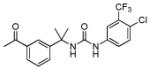
|
9 ± 5 | 20 ± 2 |
| 6c |
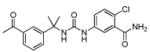
|
54 ± 7 | 52 ± 4 |
One determination,
Two determinations, n.d., not determined.
The SAR of the central portion of 1 was interrogated next (Table 3). First, the importance of the gem-dimethyl was confirmed. For example, the mono-methyl derivative 17 was less active compared to 6a and elimination of the methyl groups (23) resulted in complete loss of activity. Incorporation of the two methyl groups into a cyclopropane (37) was also detrimental. Several changes to the urea were examined. For example, N-methylation (5u) of the aniline portion of the urea and replacement of the nitrogen with oxygen (25) or carbon (26) were deleterious. Replacement of the benzylamine nitrogen with oxygen (28) was better tolerated, demonstrating only a 2-fold reduction in activity.
Table 3.
SAR of the urea moiety.
| Compound | Structure | IC50 (nM) |
|
|---|---|---|---|
| (−) BSA | (+) BSA | ||
| 17 |
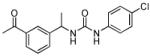
|
70 ± 20 | 110 ± 40 |
| 23 |
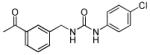
|
~5000a | n.d. |
| 37 |
|
40 ± 10 | 90 ± 30 |
| 5u |
|
900 ± 400 | 5000 |
| 25 |
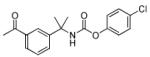
|
>5000 | n.d. |
| 26 |
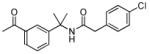
|
>5000 | n.d. |
| 28 |
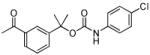
|
90 ± 20 | 120 ± 10 |
One determination,
n.d., not determined.
Finally, based on the encouraging results obtained with 6a the ketone functional group was transformed into hydrophilic oximes and O-methyloximes (Table 4). Gratifyingly, the oximes (7a, 7b, 7c, 7d and 7e) demonstrated excellent CpIMPDH inhibitory potencies, with IC50 ≤1 nM. Likewise, the O-methyloximes 8a, 8b and 8c proved to be quite active with IC50 ≤5 nM. O-Ethanolamine 9 also showed promising activity with an IC50 value of 20 nM.
Table 4.
SAR of oxime and methyloxime analogs
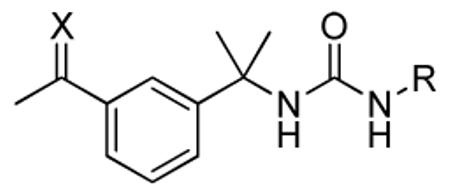
| ID | X | R | IC50 (nM) | |
|---|---|---|---|---|
| (−) BSA | (+) BSA | |||
| 7a | N-OH | 3-CF3,4-Cl-Ph | 1.0 ± 0.1 | 4 ± 2 |
| 7b | N-OH | 3-NO2,4-Cl-Ph | 0.66 ± 0.08 | 2.0 ± 0.4 |
| 7c | N-OH | 3-CONH2, 4-Cl-Ph |
5 ± 1 | 6 ± 2 |
| 7d | N-OH | 2-Naphthyl | 1.0 ± 0.2 | 2.3 ± 0.6 |
| 7e | N-OH | 7-Quinolyl | 0.9 ± 0.2 | 1.2 ± 0.3 |
| 8a | N-OMe | 3-CF3, 4-Cl-Ph | 5 ± 1 | 18 ± 3 |
| 8b | N-OMe | 3-CONH2, 4-Cl-Ph |
5 ± 2 | 5 ± 1 |
| 8c | N-OMe | 2-Naphthyl | 1.6 ± 0.1 | 13 ± 4 |
| 9 | N-O(CH2)2NH2 | 3-NO2,4-Cl-Ph | 20 ± 3.4 | 24 ± 17 |
The introduction of hydrophilic groups in place of the isoprenyl also resulted in improvements in mouse liver microsomal stability, which was a potential liability for 5k and 5r (Table 5). The ketone derivative 6c was very resistant to oxidative degradation in microsomes. The oximes demonstrated a range of stabilities, with 7c having a t½ of 190 min. While the O-methyloxime 8a was less stable, the aminoethyleneoxime derivative 9 showed excellent mouse liver microsomal stability with t1/2 > 2 hours. All of the compounds examined demonstrated excellent mouse plasma stability.
Table 5.
Mouse liver microsome and plasma stability: n.d., not determined.
| Compound | Mouse Microsomal Stability (t1/2, min) | Mouse Plasma Stability (t1/2, min) |
|---|---|---|
| 5k | 25 | >120 |
| 5r | 5.9 | >120 |
| 6c | >700 | >120 |
| 7a | 88 | n.d. |
| 7b | 11 | n.d. |
| 7c | 190 | >120 |
| 7e | 45 | >120 |
| 8a | 33 | n.d. |
| 9 | 121 | n.d |
Toxoplasma gondii is an intracellular parasite closely related to Cryptosporidium that, unlike C. parvum, can be continuously cultured in hTERT immortalized human foreskin fibroblasts. We engineered a T. gondii strain (Toxo/CpIMPDH) that relies on CpIMPDH, and is sensitive to CpIMPDH inhibitors21. In contrast, the wild type T. gondii strain RH (Toxo/WT) contains a typical eukaryotic IMPDH and is resistant to CpIMPDH inhibitors. Therefore, Toxo/WT serves as a proxy for host cell toxicity as well as provides important target validation.
Approximately thirty compounds with good inhibitory activity against CpIMPDH were tested in the Toxoplasma model. Eleven compounds had EC50 values ≤ 200 nM with selectivity ≥ 100-fold over Toxo/WT. Gratifyingly, 7b, our best inhibitor in the enzyme assay, also displayed the best antiparasitic activity with EC50 of 6 nM and 670-fold selectivity over Toxo/WT. Compounds 7a and 7e are also very promising candidates, with EC50 values of 10 nM and 58 nM, respectively, and selectivity ≥ 230.
Conclusion
In this study, the SAR of the CpIMPDH inhibitor 1 was investigated. Initially, the anilide portion of the molecule was explored. Several derivatives containing 3,4-di-substitution or 3,4-ring fusion were found to significantly increase inhibitory potency. The aniline nitrogen atom of the urea was necessary for potent activity, while the other nitrogen could be replace with oxygen. The gem-dimethyl group was also found to be best for CpIMPDH inhibition. The 3-isopropenyl substituent could be replaced with a number of hydrophobic or hydrophilic groups, but those with sp2-hybridization (e.g. phenyl, acetyl, oximes, or O-methyloximes) were best. Eleven compounds displayed potent and selective in vitro antiparastic activity, including two that also demonstrated good in vitro metabolic stability. These compounds appear to possess the necessary properties for evaluation in an animal model of cryptosporidiosis in order to determine the optimal pharmacological profile necessary for in vivo efficacy.
Experimental Section
Chemistry Materials and Methods
Unless otherwise noted, all reagents and solvents were purchased from commercial sources and used without further purification. All reactions were performed under nitrogen atmosphere unless otherwise noted. The NMR spectra were obtained using a 400 MHz spectrometer. All 1H NMR spectra are reported in δ units ppm and are reference to tetramethylsilane (TMS) if conducted in CDCl3 or to the central line of the quintet at 2.49 ppm for samples in DMSO-d6. All chemical shift values are also reported with multiplicity, coupling constants and proton count. All 13C NMR spectra are reported in δ units ppm and are reference to the central line of the triplet at 77.23 ppm if conducted in CDCl3 or to the central line of the septet at 39.5 ppm for samples in DMSO-d6. Coupling constants (J values) are reported in hertz. Column chromatography was carried out on SILICYCLE SiliaFlash silica gel F60 (40-63 Vm, mesh 230-400). High-resolution mass spectra were obtained using a Q-Tof Ultima mass spectrometer (University of Illinois Urbana-Champaign, Urbana, IL 61801). All melting points were taken in glass capillary tubes and are uncorrected. All test compounds reported in this manuscript had a purity ≥ 95% as determined by high performance liquid chromatography (HPLC) analyses using an instrument equipped with a quaternary pump and a Zorbaxρ SB-C8 column (30 × 4.6 mm, 3.5 μm). UV absorption was monitored at λ = 254 nm. The injection volume was 5 VL. HPLC gradient went from 5 % acetonitrile and 95 % water to 95 % acetonitrile and 5 % water (both solvents contain 0.1% trifluoroacetic acid) over 1.9 min with a total run time of 2.5 min and a flow rate of 3.0 mL/min.
Synthesis of 3-acetyl-α, α-dimethylbenzyl isocyanate (3)
A solution of 3-isopropenyl-α, α-dimethyl benzyl isocyanate (2.04 g, 10.14 mmol) in dichloromethane (40 mL) was cooled to -78 oC and then treated with dry ozone in oxygen until a blue color persist. Excess ozone was flushed off with oxygen. Dimethyl sulfide (0.74 mL, 10.14 mmol) was added to the reaction mixture, which was then stirred overnight at room temperature. Excess Me2S was removed by evaporated on a water bath placed inside a fume hood. Water (30 mL) was added to the reaction mixture, which was then extracted with dichloromethane. The combined organic layers were washed with brine (30 mL) and dried over anhydrous MgSO4. The mixture was filtered and the filtrate concentrated. The residue was purified by silica gel column chromatography using ethylacetate/ hexane (1: 10) as an eluent to furnish 5b (1.19 g, 58%). 1H NMR (CDCl3, 400 MHz) δ 1.76 (s, 6H), 2.63 (s, 3H), 7.47 (t, J = 8 Hz, 1H), 7.67 (dd, J1 = 7.6 Hz, J2 = 2 Hz, 1H), 7.86 (dt, J1 = 7.6 Hz, J2 = 1.2 Hz, 1H), 8.04 (t, J = 2 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 26.9, 33.2, 60.9, 124.2, 127.6, 129.0, 129.4, 137.4, 146.6, 198.1.
General procedure for the preparation of urea derivatives 5 : Exemplified for the preparation of 1-(4-chlorophenyl)-3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)urea (5b)
To a solution of 3-isopropenyl α, α-dimethylbenzyl isocyanate 2 (473 mg, 2.35 mmol) in dichloromethane (6 mL) at 0 °C was added 4-chloroaniline (300 mg, 2.35 mmol) in dichloromethane (3 mL). The reaction was stirred until complete consumption of starting materials. The precipitated product was collected by filtration and washed with dichloromethane to give 5b (852 mg, 80%). mp 234-236 °C Yield 80 %; 1H NMR (DMSO+d6, 400 MHz) δ 1.58 (s, 6H), 2.08 (s, 3H), 5.06 (s, 1H), 5.36 (s, 1H), 6.64 (s, 1H), 7.20 (d, J = 6.4 Hz, 2H), 7.23-7.32 (m, 5H), 7.47 (s, 1H), 8.55 (s, 1H); 13C NMR (DMSO+d6, 100 MHz) δ 22.24, 30.30, 55.0, 113.06, 119.50, 122.35, 123.69, 124.86, 124.95, 128.65, 129.09, 140.11, 140.73, 143.60, 148.95, 154.53; ESI-HRMS for C19H22N2OCl (M+H)+ calcd. 329.1421, found 329.1418.
1-Phenyl-3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)urea (5a)
mp 188-190 °C; Yield 81%; 1H NMR (DMSO-d6, 400 MHz) δ 1.58 (s, 6H), 2.08 (s, 3H), 5.06 (s, 1H), 5.36 (s, 1H), 6.59 (s, 1H), 6.83 (t, J =7.2 Hz, 1H), 7.15 (t, J = 8 Hz, 2H), 7.24-7.31 (m, 5H), 7.47 (s, 1H), 8.39 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 22.25, 30.39, 54.97, 113.05, 118.00, 121.55, 122.36, 123.65, 124.87, 128.64, 129.28, 140.70, 141.12, 143.62, 149.11, 154.71; ESI-HRMS for C19H23N2O (M+H)+ calcd. 295.1810, found 389.1815.
1-(4-Bromophenyl)-3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)urea (5c)
Yield 78 %; 1H NMR (DMSO-d6, 400 MHz) δ 1.61 (s, 6H), 2.10 (s, 3H), 5.08 (s, 1H), 5.38 (s, 1H), 6.66 (s, 1H), 7.28-7.35 (m, 5H), 7.49 (s, 1H), 8.57 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 22.23, 30.29, 55.07, 112.76, 113.06, 119.96, 122.35, 123.70, 124.85, 128.65, 131.97, 140.53, 140.74, 143.62, 148.94, 154.50; ESI-HRMS for C19H22N2OBr (M+H)+ calcd. 373.0915, found 373.0915.
1-(3-Chlorophenyl)-3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)urea (5d)
Yield 83 %; 1H NMR (DMSO-d6, 400 MHz) δ 1.59 (s, 6H), 2.09 (s, 3H), 5.07 (s, 1H), 5.37 (s, 1H), 6.70 (s, 1H), 6.89 (d, J = 7.6 Hz, 1H), 7.05 (d, J = 7.6 Hz 1H), 7.19 (t, J = 8 Hz ,1H), 7.28-7.30 (m, 3H), 7.48 (s, 1H), 7.60 (s, 1H), 8.63 (s,1H) ; 13C NMR (DMSO-d6, 100 MHz) δ 22.23, 30.26, 55.08, 113.08, 116.41, 117.32, 121.15, 122.32, 123.71, 124.83, 128.67, 130.88, 133.78, 140.74, 142.63, 143.60, 148.88, 154.44; ESI-HRMS for C19H22N2OCl (M+H)+ calcd. 329.1421, found 329.1430.
1-(2-Chlorophenyl)-3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)urea (5e)
Yield 65 %; 1H NMR (DMSO-d6, 400 MHz) δ 1.62 (s, 6H), 2.1 (s, 3H), 5.08 (s, 1H), 5.38 (s, 1H), 6.91 (t, J = 7.2 Hz, 1H), 7.16 (t, J = 8.8 Hz, 1H), 7.30-7.32 (m, 3H), 7.37 (dd, 1H, J = 7.2 Hz, J2 = 3.2 Hz), 7.49-7.52 (m, 2H), 8.02 (dd, J1= 7.8 Hz, J = 2 Hz), 8.09 (d, J = 2 Hz, 1H); 13 2 C NMR (DMSO-d6, 100 MHz) δ 22.22, 30.29, 55.17, 113.06, 121.08, 121.50, 122.32, 122.86, 123.69, 124.86, 127.99, 128.68, 129.67, 137.38, 140.71, 143.59, 148.92, 154.22; ESI-HRMS for C19H22N2OCl (M+H)+ calcd. 329.1421, found 329.1430.
1-(4-Methoxyphenyl)-3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)urea (5f)
Yield 85%; 1H NMR (DMSO-d6, 400 MHz) δ 1.59 (s, 6H), 2.10 (s, 3H), 3.66 (s, 3H), 5.08 (s, 1H), 5.37 (s, 1H), 6.50 (s, 1H), 6.77 (d, J = 8.4 Hz, 2H), 7.20 (d, J = 8.4 Hz, 2H), 7.29-7.31 (m, 3H), 7.49 (s, 1H), 8.22 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 22.23, 30.46, 54.92, 55.74, 113.01, 114.47, 119.64, 122.36, 123.60, 124.88, 128.61, 134.28, 140.67, 143.63, 149.25, 154.40, 154.95; ESI-HRMS for C20H25N2O2 (M+H)+ calcd. 325.1916, found 325.1919.
1-(4-(tert-Butyl)phenyl)-3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)urea-(5g)
Yield 87%; 1H NMR (DMSO-d6, 400 MHz) δ 1.20 (s, 9H), 1.58 (s, 6H), 2.09 (s, 3H), 5.06 (s, 1H), 5.36 (s, 1H), 6.54 (s, 1H), 7.15-7.19 (m, 4H), 7.27-7.30 (m, 3H), 7.48 (s, 1H), 8.31 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 22.25, 30.45, 31.93, 34.42, 54.95, 113.02, 117.88, 122.37, 123.62, 124.87, 125.84, 128.62, 138.52, 140.70, 143.63, 143.79, 149.19, 154.82; ESI-HRMS for C23H31N2O (M+H)+ calcd. 351.2436, found 3512433.
1-(3,4-Dichlorophenyl)-3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)urea (5h)
mp 198-200 °C Yield 80 %; 1H NMR (DMSO-d6, 400 MHz) δ 1.61 (s, 6H), 2.10 (s, 3H), 5.09 (s, 1H), 5.38 (s, 1H), 6.79 (s, 1H), 7.13 (t, J = 4.8 Hz, 1H), 7.31 - 7.34 (m, 3H), 7.42 (d, J =7.6 Hz ,1H), 7.49 (s, 1H), 7.79 (s, 1H), 8.79 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 22.23, 30.21, 55.14, 113.03, 118.13, 118.95, 122.31, 123.75, 124.82, 128.69, 131.07, 131.58, 140.76, 141.31, 141.34, 143.58, 148.79, 154.33; ESI-HRMS for C19H21N2OCl2 (M+H)+ calcd. 363.1031, found 363.1029.
1-(4-Chloro-3-(trifluoromethyl)phenyl)-3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)urea (5i)
Yield 82%; 1H NMR (DMSO-d6, 400 MHz) δ 1.61 (s, 6H), 2.10 (s, 3H), 5.09 (s, 1H), 5.38 (s, 1H), 6.81 (s, 1H), 7.30-7.33 (m, 3H), 7.42-7.44 (m, 1H), 7.49-7.53 (m, 2H), 8.01 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 22.22, 30.22, 55.20, 113.11, 116.35, 116.40, 121.90, 122.12, 122.30, 122.78, 123.77, 124.81, 128.70, 132.50, 140.66, 140.78, 143.58, 148.73, 154.36; ESI-HRMS for C20H21N2OClF3 (M+H)+ calcd. 397.1295, found 397.1296.
1-(4-Chloro-3-methoxyphenyl)-3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)urea (5j)
Yield 89%; 1H NMR (DMSO-d6, 400 MHz) δ 1.59 (s, 6H), 2.09 (s, 3H), 3.73 (s, 3H), 5.07 (s, 1H), 5.37 (s, 1H), 6.66 (s, 1H), 6.72 (d, J = 8.8 Hz, 1H), 7.18 (d, J = 8.8 Hz,1H), 7.28-7.32 (m, 4H), 7.48 (s, 1H), 8.59 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 22.23, 30.34, 55.03, 56.28, 102.50, 110.59, 112.95, 113.10, 122.33, 123.72, 124.85, 128.70, 130.16, 140.75, 141.47, 143.59, 148.97, 154.53, 155.10; ESI-HRMS for C20H24N2O2Cl (M+H)+ calcd. 359.1529, found 359.1532.
2-Chloro-5-(3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)ureido)benzamide (5k)
Yield 72 %; 1H NMR (DMSO-d6, 400 MHz) δ 1.57 (s, 6H), 2.06 (s, 3H), 5.04 (s, 1H), 5.34 (s, 1H), 6.65 (s, 1H), 7.22 - 7.27 (m, 5H), 7.44-7.48 (m, 3H), 7.75 (s, 1H), 8.61 (s,1H) ; 13C NMR (DMSO-d6, 100 MHz) δ 22.23, 30.27, 55.08, 113.08, 117.61, 119.67, 121.36, 122.31, 123.71, 124.82, 128.66, 130.33, 137.89, 139.95, 140.73, 143.59, 148.89, 154.45, 168.85; ESI-HRMS for C20H23N3O2Cl (M+H)+ calcd. 372.1479, found 372.1485.
2-Chloro-N-methyl-5-(3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)ureido)benzamide (5l)
Yield 72%; 1H NMR (CDCl3, 400 MHz) δ 1.64 (s, 6H), 2.17 (s, 3H), 2.89 (d, J = 4.8 Hz, 3H), 5.05 (s, 1H), 5.32 (s, 1H), 5.97 (s, 1H,), 6.61 (d, J = 4.4 Hz, 1H), 7.10-7.13 (m, 2H), 7.27-7.31 (m, 2H), 7.49 −7.51 (m, 3H), 7.81 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 22.25, 26.56, 30.26, 55.08, 113.09, 117.76, 119.75, 121.57, 122.31, 123.71, 124.82, 128.66, 130.32, 137.80, 140.01, 140.74, 143.59, 148.90, 167.41, 189.22; ESI-HRMS for C21H25N3O2Cl (M+H)+ cacld. 386.1635, found 386.1639.
2-Chloro-N,N-dimethyl-5-(3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)ureido)benzamide (5m)
Yield 72%; 1H NMR (CDCl3, 400 MHz) δ 1.66 (s, 6H), 2.13 (s, 3H), 2.86 (s, 3H), 3.07 (s, 3H), 5.05 (s, 1H), 5.33 (s, 1H), 6.02 (s, 1H,), 6.88 (d, J = 2.4 Hz, 1H), 7.11 (d, J = 8.8Hz, 2H), 7.25-7.35 (m, 4H), 7.50 (s, 1H), 8.01 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 22.17, 29.89, 30.16, 34.94, 38.49, 55.13, 112.51, 116.93, 120.92, 121.29, 122.18, 123.81, 124.24, 128.33, 129.97, 135.15, 139.83, 141.27, 143.81, 147.93, 154.48, 169.80; ESI-HRMS for C22H27N3O2Cl (M+H)+ calcd. 400.1792, found 400.1787.
1-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl) 3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)urea (5n)
Yield 75%; 1H NMR (DMSO-d6, 400 MHz) δ 1.59 (s, 6H), 2.10 (s, 3H), 4.15 (d, J = 7.6 Hz, 4H), 5.08 (s, 1H), 5.37 (s, 1H), 6.50 (s, 1H), 6.67 (d, J = 8.4 Hz, 2H), 6.97 (s, 1H), 7.29-7.31 (m, 3H), 7.49 (s, 1H), 8.22 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 22.22, 30.42, 54.93, 64.44, 64.82, 107.22, 111.36, 113.0, 117.31, 122.35, 123.60, 124.85, 128.60, 134.89, 138.31, 140.68, 143.62, 143.65, 149.17, 154.81; ESI-HRMS for C21H25N2O3 (M+H)+ calcd. 353.1865, found 353.186.
1-(Naphthalen-2-yl)-3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)urea (5o)
mp 201-203 °C; Yield 75%; 1H NMR (DMSO-d6, 400 MHz) δ 1.64 (s, 6H), 2.11 (s, 3H), 5.08 (s, 1H), 5.39 (s, 1H), 6.72 (s, 1H), 7.27-7.39 (m, 6H), 7.53 (d, J = 1.2 Hz, 1H), 7.68 (d, J = 8 Hz, 1H), 7.75 (d, J = 8.4 Hz, 2H), 7.98 (s, 1H), 8.64 (s, 1H) ; 13C NMR (DMSO-d6, 100 MHz) δ 22.23, 30.35, 55.04, 112.83, 113.06, 119.92, 122.37,123.69, 124.12, 124.90, 126.85, 127.38, 128.01, 128.67, 128.89, 129.29, 134.49, 138.73, 140.75, 143.63, 149.09, 154.78; ESI-HRMS for C23H25N2O (M+H)+ calcd. 345.1967, found 345.1964.
1-(Naphthalen-1-yl)-3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)urea (5p)
mp 206-208 °C; Yield 75%; 1H NMR (DMSO-d6, 400 MHz) δ 1.66 (s, 6H), 2.10 (s, 3H), 5.08 (s, 1H), 5.39 (s, 1H), 7.13 (s, 1H), 7.29-7.38 (m, 4H), 7.50-7.55 (m, 4H), 7.90 (d, J = 7.6 Hz, 1H), 7.87 (d, J = 8 Hz, 1H), 8.10 (d, J = 8.4 Hz, 1H), 8.54 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 22.24, 30.42, 55.16, 113.05, 116.51, 121.79, 122.40, 123.68, 124.92, 125.80, 126.01, 126.36, 126.52, 128.68, 129.06, 134.33, 135.77, 140.74, 143.61, 149.15, 155.02; ESI-HRMS for C23H25N2O (M+H)+ calcd. 345.1967, found 345.1976.
1-(2-(3-(Prop-1-en-2-yl)phenyl)propan-2-yl)-3-(quinolin-6-yl)urea (5q)
Yield 81%; 1H NMR (DMSO-d6, 400 MHz) δ 1.65 (s, 6H), 2.11 (s, 3H), 5.08 (s, 1H), 5.39 (s, 1H), 7.10 (s, 1H), 7.29-7.37 (m, 2H), 7.54 (s, 1H), 7.67-7.74 (m, 2H), 8.04 (d, J = 9.2 Hz, 1H), 8.27 (d, J = 2 Hz, 1H), 8.33 (s, 1H), 8.57 (d, J = 8.4 Hz, 1H), 8.87 (d, J = 4 Hz, 1H), 9.39 (s, 1H) ; 13C NMR (DMSO-d6, 100 MHz) δ 22.25, 30.31, 55.14, 112.04, 113.07, 122.37, 122.45, 123.68, 124.87, 125.70, 125.75, 128.65, 130.06, 138.77, 138.79, 140.72, 140.75, 143.58, 145.04, 148.96, 154.68; ESI-HRMS for C22H24N3O (M+H)+ calcd. 346.1919, found 346.1925.
1-(2-(3-(Prop-1-en-2-yl)phenyl)propan-2-yl)-3-(quinolin-7-yl)urea (5r)
Yield 72%; 1H NMR (DMSO-d6, 400 MHz) δ 1.64 (s, 6H), 2.11 (s, 3H), 5.08 (s, 1H), 5.39 (s, 1H), 6.79 (s, 1H), 7.28-7.37 (m, 4H), 7.47-7.53 (m, 2H), 7.80 (d, J = 8.8 Hz, 1H), 8.03 (d, J = 2 Hz, 1H), 8.18 (d, J = 8.4 Hz, 1H), 8.75 (dd, J1= 4.4 Hz, J2=1.6 Hz, 1H), 8.85 (s, 1H) ; 13C NMR (DMSO-d6, 100 MHz) δ 22.23, 30.30, 55.16, 113.09, 113.66, 119.69, 120.23, 122.37, 123.73, 124.88, 128.69, 128.98, 136.03, 140.77, 142.06, 143.61, 148.92, 149.48, 151.27, 154.55; ESI-HRMS for C22H24N3O (M+H)+ calcd. 346.1919, found 346.1915.
1-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)-3-(quinolin-3-yl)urea (5s)
Yield 69%; 1H NMR (DMSO-d6, 400 MHz) δ 1.65 (s, 6H), 2.11 (s, 3H), 5.09 (s, 1H), 5.39 (s, 1H), 6.88 (s, 1H), 7.31-7.38 (m, 3H), 7.49-7.55 (m, 3H), 7.79 (d, J = 7.2 Hz, 1H), 7.88 (d, J = 7.4 Hz, 1H), 8.41 (s, 1H), 8.66 (d, J = 2.4 Hz, 1H), 8.91 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 22.24, 30.25, 55.17, 113.11, 119.44, 122.35, 123.75, 124.89, 127.27, 127.49, 127.81, 128.71, 128.90, 129.10, 134.88, 140.77, 143.60, 143.82, 144.47, 148.88, 154.72; ESI-HRMS for C22H24N3O (M+H)+ calcd. 346.1919, found 346.1924.
1-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)-3-(quinolin-2-yl)urea (5t)
mp 126-128 °C; Yield 75%; 1H NMR (CDCl3, 400 MHz) δ 1.90 (s, 6H), 2.13 (s, 3H), 5.05 (s, 1H), 5.37 (s, 1H), 6.54 (d, J = 8.8 Hz, 1H), 7.25-7.38 (m, 3H), 7.48 (d, J = 7.6 Hz, 1H), 7.57-7.67 (m, 4H), 7.83 (d, J = 8.8 Hz, 1H), 9.69 (s, 1H), 10.92 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 22.20, 30.11, 55.62, 113.19, 114.22, 122.47, 123.95, 124.71, 124.84, 124.89, 126.57, 128.42, 128.82, 130.71, 139.03, 140.94, 143.57, 145.55, 148.65, 153.54, 154.10; ESI-HRMS for C22H24N3O (M+H)+ calcd. 346.1919, found 346.1917.
1-(4-Chlorophenyl)-1-methyl-3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)urea (5u)
Yield 56%; 1H NMR (DMSO-d6, 400 MHz) δ 1.50 (s, 6H), 2.06 (s, 3H), 3.11 (s, 3H), 5.04 (s, 1H), 5.32 (s, 1H), 6.11 (s, 1H), 7.21-7.25 (m, 5H), 7.34-7.40 (m, 3H); 13C NMR (DMSO-d6, 100 MHz) δ 22.25, 30.28, 37.49, 55.72, 112.90, 122.26, 123.40, 124.81, 127.92, 128.49, 129.50, 129.58, 140.54, 143.64, 144.18, 149.37, 156.06; ESI-HRMS for C20H24N2OCl (M+H)+ calcd. 343.1577, found 343.1570.
General procedure for the preparation of urea derivatives 6: Exemplified for the preparation of 5-(3-(2-(3-acetylphenyl)propan-2-yl)ureido)-2-chlorobenzamide (6c)
To a solution of 3-acetyl α, α dimethyl isocyanate 3 (118 mg, 0.584 mmol) in THF at room temperature was added 5-amino-2-chlorobenzamide (100 mg, 0.584 mmol). The reaction was heated to 70 °C for 6 h. Volatiles were removed under reduced pressure and the residue was purified by column chromatography using methanol/chloroform as eluent to obtain urea derivative 6c (156 mg, 72%). 1H NMR (DMSO-d6, 400 MHz) δ 1.61 (s, 6H), 2.58 (s, 3H), 6.80 (s, 1H), 7.26 (s, 2H), 7.45-7.52 (m, 3H), 7.66 (d, J = 7.2 Hz, 1H), 7.79-7.83 (m, 2H), 7.94 (s, 1H), 8.68 (s, 1H) ; 13C NMR (DMSO-d6, 100 MHz) δ 27.44, 30.19, 54.89, 117.63, 119.72, 121.43, 124.57, 127.12, 129.11, 130.42, 137.24, 137.89, 139.87, 149.54, 154.40, 168.87, 198.76; ESI-HRMS for C25H16N3O (M+H)+ calcd. 374.1293, found 374.1283.
1-(2-(3-acetylphenyl)propan-2-yl)-3-(4-chlorophenyl)urea (6a)
Yield 85 %; 1H NMR (DMSO-d6, 400 MHz) δ 1.57 (s, 6H), 2.52 (s, 3H), 6.71 (s, 1H), 7.17 (d, J = 9.2 Hz, 2H), 7.29 (d, J = 8.8 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 7.62 (d, J = 7.6 Hz, 1H), 7.77 (d, J = 7.6 Hz, 1H), 7.91 (s, 1H), 8.54 (s, 1H) ; 13C NMR (DMSO-d6, 100 MHz) δ 29.8, 32.6, 57.2, 121.9, 127.0, 127.4, 129.4, 131.5, 131.55, 132.8, 139.7, 142.4, 152.0, 156.9, 201.1; ESI-HRMS for C18H19ClN2O2 (M+H)+ : calcd. 331.1213, found 331.1214..
1-(2-(3-Acetylphenyl)propan-2-yl)-3-(4-chloro-3 (trifluoromethyl)phenyl)urea (6b)
Yield 69 %; 1H NMR (DMSO-d6, 400 MHz) δ 1.62 (s, 6H), 2.51 (s, 3H), 6.91 (s, 1H), 7.42 - 7.53 (m, 3H), 7.68 (d, J = 7.2 Hz, 1H), 7.84 (d, J = 7.2 Hz, 1H), 7.95 (s, 1H), 8.01 (s, 1H), 8.95 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 27.45, 30.14, 54.99, 116.36, 116.41, 116.47, 121.96, 122.83, 124.56, 127.17, 129.15, 130.42, 132.50, 137.28, 140.58, 149.37, 154.31, 198.71; ESI-HRMS for C19H19N2O2ClF3 (M+H)+ calcd. 399.1087, found 399.1089.
General procedure for the preparation of oxime derivatives 7: Exemplified for the preparation of (E) 2 chloro 5-(3-(2-(3-(1-(hydroxyimino)ethyl)phenyl)propan-2-yl)ureido)benzamide (7c)
Hydroxylamine hydrochloride (25 mg, 0.362 mmol) was added to a solution of 56 (100 mg, 0.302 mmol) in 3 mL of pyridine. The reaction solution was heated to 90 °C for 2 hours. The reaction was allowed to cool to room temperature and then the pyridine was removed by evaporation under reduced pressure. The resulting residue was dissolved in methanol and purified by column chromatography eluting with methanol/chloroform to obtained 7c (88 mg, 85%). 1H NMR (DMSO-d6, 400 MHz) δ 1.54 (s, 6H), 2.11 (s, 3H), 6.67 (s, 1H), 7.20 - 7.49 (m, 7H), 7.67 (s, 1H), 7.76 (s, 1H), 8.62 (s, 1H), 11.13 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 12.42, 30.27, 54.98, 117.59, 119.67, 121.38, 122.44, 124.21, 125.87, 128.76, 130.35, 137.33, 137.90, 139.96, 148.95, 153.79, 154.39, 168.90; ESI-HRMS for C19H22N4O3Cl (M+H)+ calcd. 389.1380, found 389.1384.
(E)-1-(4-Chloro-3-(trifluoromethyl)phenyl)-3-(2-(3-(1-(hydroxyimino)ethyl)phenyl)propan-2-yl)urea (7a)
mp 194-196 °C; Yield 71%; 1H NMR (DMSO-d6, 400 MHz) δ 1.61 (s, 6H), 2.15 (s, 3H), 6.82 (s, 1H), 7.34 (d, J = 8 Hz, 1H), 7.39 – 7.46 (m, 3H), 7.52 (d, J = 9.2 Hz, 1H), 7.70 (s, 1H), 8.01 (s, 1H), 8.91(s, 1H), 11.17 (bs, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 12.39, 30.19, 55.07, 116.31, 116.37, 116.42, 121.91, 122.44, 122.76, 124.25, 125.84, 128.78, 132.50, 137.36, 140.64, 149.77, 153.74, 154.29; ESI-HRMS for C19H20N3O2ClF3 (M+H)+ calcd. 414.1196, found 414.1191.
(E)-1-(4-Chloro-3-nitrophenyl)-3-(2-(3-(1-(hydroxyimino)ethyl)phenyl)propan-2-yl)urea (7b)
mp 183-185 °C; Yield 80%; 1H NMR (DMSO-d6, 400 MHz) δ 1.62 (s, 6H), 2.16 (s, 3H), 6.91 (s, 1H), 7.35 (t, J = 8.0 Hz, 1H), 7.35 (t, J = 8.0 Hz, 1H), 7.41-7.47 (m, 3H), 7.58 (d, J = 8.8 Hz, 1H), 7.71 (s, 1H), 8.22 (d, J = 2.4 Hz, 1H), 9.04 (s, 1H), 11.18 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 12.4, 30.13, 55.12, 113.84, 116.26, 122.42, 122.86, 124.27, 125.84, 128.79, 132.28, 137.36, 141.11, 148.14, 148.67, 153.74, 154.12; ESI-HRMS for C18H20N4O4Cl (M+H)+ calcd. 391.1173, found 391.1172.
(E)-1-(2-(3-(1-(Hydroxyimino)ethyl)phenyl)propan-2-yl) 3 (naphthalen-2-yl)urea-(7d)
Yield 85 %; 1H NMR (DMSO-d6, 400 MHz) δ 1.69 (s, 6H), 2.21 (s, 3H), 6.81 (s, 1H), 7.35-7.50 (m, 6H), 7.72 - 7.80 (m, 4H), 8.05 (s, 1H), 8.70 (s, 1H), 11.22 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 12.40, 30.32, 54.90, 112.75, 119.86, 122.49, 124.11, 124.16, 125.93, 126.85, 127.37, 128.00, 128.74, 128.90, 129.26, 134.48, 137.32, 138.70, 149.12, 153.77, 154.66; ESI-HRMS for C22H24N3O2 (M+H)+ calcd. 362.1869, found 362.1872.
(E)-1-(2-(3-(1-(Hydroxyimino)ethyl)phenyl)propan-2-yl)-3 (quinolin-7-yl)urea (7e)
mp 164-166 °C; Yield 85 %; 1H NMR (pyridine-d5, 400 MHz) δ 1.68 (s, 6H), 2.28 (s, 3H), 6.99-7.02 (m, 2H), 7.25 (t, J = 8 Hz, 1H), 7.53 -7.62 (m, 3H), 7.88 (t, J = 9.6 Hz, 2H), 8.16 (s, 1H), 8.63 (s, 1H), 8.77 (d, J = 1.6 Hz, 1H), 9.54 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 12.41, 30.29, 55.03, 113.61, 119.70, 120.20, 122.50, 123.78, 124.22, 125.93, 128.77, 129.01, 136.04, 137.35, 142.06, 148.97, 149.4, 151.29, 153.77, 154.47; ESI-HRMS for C21H23N4O2 (M+H)+ calcd. 363.1821, found 363.1825.
General procedure for the preparation of alkoxy oxime derivatives 8: Exemplified for the preparation of (E)-2-chloro-5-(3-(2-(3-(1-(methoxyimino)ethyl)phenyl)propan-2-yl)ureido)benzamide (8b)
Methoxyamine hydrochloride (30 mg, 0.362 mmol) was added to a solution of 6b (100 mg, 0.302 mmol) in 3 mL of pyridine. The reaction solution was heated to 90 °C for 2 h and then the pyridine was removed by evaporation under reduced pressure. The residue was dissolved in methanol and purified by column chromatography eluting with methanol/chloroform to obtained 8b (99 mg, 82%). 1H NMR (CDCl3, 400 MHz) δ 1.55 (s, 6H), 2.12 (s, 3H), 3.91 (s, 3H), 6.18 (s, 1H), 6.59 (s, 1H), 6.86 (s, 1H), 7.06 (d, J = 8.8 Hz, 1H), 7.15 (s, 1H), 7.25 (d, J = 6.8 Hz, 1H), 7.33 (d, J = 7.6 Hz, 1H), 7.37 (d, J = 7.6 Hz, 2H), 7.67 (1H, s), 7.97 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 13.29, 30.23, 54.97, 62.17, 117.68, 119.75, 121.42, 122.77, 124.49, 126.46, 128.83, 130.33, 136.30, 137.89, 139.93, 149.10, 154.43, 155.037, 168.88; ESI-HRMS for C20H24N4O3Cl (M+H)+ calcd. 403.1537, found 403.1532.
(E)-1-(4-Chloro-3-(trifluoromethyl)phenyl)-3-(2-(3-(1-(methoxyimino)ethyl)phenyl)propan-2-yl)urea (8a)
1H NMR (DMSO-d6, 400 MHz) δ 1.62 (s, 6H), 2.1 (s, 3H), 3.90 (s. 3H), 6.84 (s, 1H), 7.36 (dd, J1 = 7.6 Hz, J2 = 1.6 Hz, 1H), 7.43-7.48 (m, 3H), 7.52 (dd, J1 = 8.8 Hz, J2 = 1.6 Hz), 7.67 (s, 1H), 8.01 (s. 1H), 8.93 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 12.80, 29.86, 55.30, 62.04, 117.77, 117.82, 117.88, 122.29, 122.87, 124.88, 124.93, 125.63, 128.82, 131.77, 136.96, 137.96, 147.31, 154.63, 154.91; ESI-HRMS for C20H22N3O2F3Cl (M+H)+ calcd. 428.1353, found 428.1353.
(E)-1-(2-(3-(1-(Methoxyimino)ethyl)phenyl)propan-2-yl)-3 (naphthalen-2-yl)urea (8c)
Yield 58%; 1H NMR (DMSO-d6, 400 MHz) δ 1.65 (s, 6H) 2.19 (s, 3H), 3.90 (s, 3H), 6.78 (s, 1H), 7.30-7.42 (4H, m), 7.48 (d, J = 7.2 Hz, 2H), 7.68-7.72 (m, 2H), 7.76 ( d, J = 8.4 Hz, 2H), 8.00 (s, 1H), 8.67 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 13.30, 30.30, 54.91, 62.16, 112.8, 119.9, 122.8, 124.1, 124.4, 126.5, 126.8, 127.3, 128.0, 128.83, 128.89, 129.28, 134.47, 136.29, 138.68, 149.28, 154.71, 155.06; ESI-HRMS for C23H26N3O2 (M+H)+ calcd. 376.2025, found. 376.2026.
Preparation of (E)-1-(2-(3-(1-((2-aminoethoxy)imino)ethyl)phenyl)propan-2-yl)-3 (4-chloro-3-nitrophenyl)urea (9)
(E)-1-(4-Chloro-3-nitrophenyl)-3-(2-(3-(1-(hydroxyimino)ethyl)phenyl)propan-2-yl)urea (7b, 100 mg, 0.25 mmol) in 3 mL dry DMF was added drop wise at 0 °C to sodium hydride dispersion in mineral oil (15.3 mg, 0.50 mmol). The resulting mixture was stirred at 0 °C for 30 min. 2-Chloroethylamine hydrochloride (29.6 mg, 0.25 mmol) in 2 mL DMF was added and the reaction mixture was stirred at room temperature for 2 h and then the volatiles were removed under reduced pressure. The residue was dissolved in a minimal amount of methanol and purified by column chromatography using methanol/chloroform to obtained 9 (49 mg, 46 % ) 1H NMR (DMSO-d6, 400 MHz) δ 1.61 (s, 6H), 2.19 (s, 3H), 2.79 (t, J = 5.6 Hz, 2H), 4.05 (t, J = 5.6 Hz, 2H), 6.95 (s, 1H), 7.34 (t, J = 7.6 Hz, 1H), 7.42-7.47 (m, 3H), 7.56 (d, J = 8.8 Hz, 1H), 7.65 (s, 1H), 8.20 (d, J = 2 Hz, 1H), 9 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 13.3, 30.1, 41.6, 55.4, 76.6, 113.8, 116.2, 122.7, 122.9, 124.5, 126.3, 128.8, 132.2, 136.5, 141.1, 148.1, 148.8, 154.2, 154.9; ESI-HRMS for C20H24Cl N5O4 (M+H)+ calcd. 434.1595, found. 434.1595.
1-(4-chlorophenyl)-3-(2-(3-(1-hydroxyethyl)phenyl)propan-2-yl)urea (10)
A solution of 1-(2-(3-acetylphenyl)propan-2-yl)-3-(4-chlorophenyl)urea (25 mg, 0.07 mmol) in THF was cooled in an ice bath to 0 °C. Lithium aluminum hydride solution (2M in THF, 0.8 equiv) was added drop wise over 5 min and then the reaction was continued for approximately 1 h at 0 °C until starting material disappeared. The reaction was carefully quenched with a solution of sodium sulfate. The reaction mixture was filtered through a sintered funnel and the supernatant washed with dichloromethane. Combined organic fractions were concentrated under reduced pressure. The residue was purified by column chromatography using chloroform-methanol as a eluent to give 10 (19 mg, 74%). 1H NMR (DMSO-d6, 400 MHz) δ 1.25 (d, J = 6 Hz, 3H), 1.54 (s, 6H), 4.64 (pent, J = 4.4 Hz, 1H), 5.09 (d, J = 4 Hz, 1H), 6.56 (s, 1H), 7.11-7.19 (m, 5H), 7.28 (s, 1H), 7.31 (d, J = 9.2 Hz, 1H), 8.51 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 26.78, 30.33, 30.44, 55.06, 68.94, 119.47, 122.42, 123.70, 124.88, 128.28, 129.10, 140.18, 147.68, 148.65, 154.48; ESI-HRMS for C18H22N2O2Cl (M+H)+ calcd. 333.1370, found 333.1378.
1-(2-(3-Isopropylphenyl)propan-2-yl)-3-(naphthalen-2-yl)urea (11)
A solution of 1-(naphthalen-2-yl)-3-(2-(3-(prop-1-en-2-yl)phenyl)propan-2-yl)urea (5o, 25 mg, 0.072 mmol) in methanol (3 mL) containing a catalytic amount of 10% Pd/C was placed under an atmosphere of hydrogen. After 1h the reaction mixture was filtered through a short silica gel column and concentrated to give 11 (24 mg, 98%).1H NMR (DMSO-d6, 400 MHz) δ 1.17 (d, J = 4.4 Hz, 6H), 1.60 (s, 6H), 2.84 (hept, J = 6.8 Hz, 1H), 6.66 (s, 1H), 7.03-7.36 (m, 7H), 7.66 (d, J = 8 Hz, 1H), 7.75 (d, J = 8 Hz, 2H); 13C NMR (DMSO-d6, 100 MHz) δ 24.69, 30.42, 34.26, 55.09, 122.75, 118.22, 119.91, 123.01, 123.58, 124.08, 124.31, 126.84, 127.36, 128.01, 128.59, 128.87, 129.25, 134.49, 138.80, 149.00, 154.78; ESI-HRMS for C23H27N2O (M+H)+ calcd. 347.2123, found 347.2126.
2-(3-Acetylphenyl)propanenitrile (13)
Thionyl chloride (10 mL) was added at 0 °C to 3-(1-cyanoethyl)benzoic acid (12, 500 mg, 2.85 mmol). The reaction mixture was heated at 80 °C for 2 h. The excess thionyl chloride was removed to give 3-(1-cyanoethyl)benzoyl chloride, which was used without further purification.
Next, to a solution of Meldrum’s acid (408 mg, 2.84 mmol) in dichloromethane (15 mL) at 0 °C was added pyridine (0.457 mL, 5.68 mmol). The resulting mixture was stirred for 15 min and then 3-(1-cyanoethyl)benzoyl chloride (482.5 mg, 2.5 mmol) was added. The reaction mixture was stirred at 0 °C for 30 min and then for 1h at room temperature. The reaction mixture was diluted with dichloromethane and washed with 1N HCl. The organic layer was dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure. The crude product was dissolved in AcOH-H2O (1:2) and heated at reflux for 4 h. The reaction mixture was diluted with water and extracted with ethyl acetate (3 × 30 mL). The combined organic extracts were washed with NaHCO3 solution and brine, then dried over anhydrous magnesium sulfate, filtered and concentrated. The residue was purified by column chromatography using hexane-ethyl acetate (9:1) as eluent to give 13 (138 mg, 28%).1H NMR (CDCl3, 400 MHz) δ 1.68 (d, J = 7.2 Hz 3H), 2.63 (s, 3H), 3.99 (q, J = 7.2 Hz, 1H), 6.60 (d, J = 7.6 Hz, 1H), 7.53 (d, J = 2.8 Hz, 1H), 7.60 (d, J = 7.2 Hz, 1H), 7.91 (t, J = 2 Hz, 1H), 7.94 (s, 1H).
2-(3-Acetylphenyl) propanoic acid (14)
To a solution of 13 (100 mg, 0.57 mmol) in 2 mL of 1, 4-dioxane was added conc. HCl (1.5 mL) and then the resulting mixture was refluxed for 5 h. After the mixture was allowed to cool to room temperature, the volatiles were removed under reduced pressure. The residue was diluted with water (10 mL) and extracted with dichloromethane (3 × 10 mL). The organic extracts were combined, washed with brine (2 × 10 mL), dried over anhydrous magnesium sulfate, filtered and concentrated to give 14 as a white solid (86 mg, 78% yield). 1H NMR (CDCl3, 400 MHz) δ 1.57 (d, J = 7.2 Hz, 3H), 2.38 (s, 3H), 3.83 (q, J = 7.2 Hz, 1H), 7.42-7.50 (m, 2H), 7.61 (d, J = 7.6 Hz, 1H), 7.75 (s, 1H).
2-(3-Acetylphenyl)propanoyl azide (15)
Thionyl chloride (2 mL) was added to 14 (86 mg, 0.40 mmol) at 0 °C. The mixture was then heated at 80 °C for 2 h. The reaction mixture was concentrated under reduced pressure. The resulting acid chloride was used without further purification.
A solution of the acid chloride (84 mg, 0.40 mmol) in 3 mL dry acetone was added to a solution of sodium azide (325 mg, 5 mmol) in 2 mL of water at 0 °C over 10 min. The reaction mixture was stirred for 2 h at 25 °C, and then poured into ice and extracted with ether (2 × 10 mL). The organic extracts were combined, washed with brine, dried over anhydrous MgSO4, filtered and concentrated to give acyl azide 15, which was used without further purification.
1-(3-(1-Isocyanatoethyl)phenyl) ethanone (16)
Acyl azide 15 (80 mg, 0.42 mmol) was refluxed in benzene (5 mL) of 1.5 h and then the solvent was removed under vacuum to give isocyanate 16 in quantitative yield, which was used without further purification.
1-(1-(3-Acetylphenyl)ethyl)-3-(4-chlorophenyl)urea (17)
Compound 17 (71%) was prepared following the general procedure for 6. 1H NMR (DMSO-d6, 400 MHz) δ 1.35 (d, J = 6.8 Hz, 3H), 2.55 (s, 3H), 4.95 (pent, J = 6.8 Hz, 1H), 5.68 (d, J = 7.2 Hz, 1H), 7.12-7.17 (m, 4H), 7.32-7.36 (m, 2H), 7.44 (d, J = 8.0 Hz, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.84 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 23.20, 26.97, 49.77, 120.94, 124.96, 127,84, 128.14, 129.14, 129.16, 131.40, 137.47, 137.58, 145.23, 155.23, 199.09; ESI-HRMS for C17H18N2O2Cl (M+H)+ calcd. 317.1057, found 317.1060.
3-(2-Methyl-1,-3-dioxolan-2-yl)benzonitrile (19)
To the mixture of ethylene glycol (0.22 mL, 4 mmol), 3-acetylbenzonitrile 18 (300 mg, 2.04 mmol) and benzene (10 mL) in a Dean-Stark apparatus was added a catalytic amount of p-TSA (0.1 equiv). The reaction mixture was heated at 110 °C for 4 h. The benzene was removed under reduced pressure and the residue was purified by column chromatography using ethylacetate-hexane as an eluent to give 19 (293 mg, 76%). 1H NMR (CDCl3, 400 MHz) δ 2.62 (s, 4H), 7.59 (t, J = 7.6 Hz, 1H), 7.82 (d, J = 7.6 Hz, 1H)), 8.15 (d, J = 8 Hz, 1H), 8.21 (s, 1H).
3-(2-Methyl-1,3-dioxolan 2 yl)phenylmethanamine (20)
A solution of cyano ketal 19 (290 mg, 1.53 mmol) in dry THF (10 mL) was cooled to 0 °C under a nitrogen atmosphere. Then a solution of 2M lithium aluminum hydride (3 mmol) in THF was added over a 10-min period. The reaction mixture was stirred for 1.5 h and then ethyl acetate was added followed by slow addition of water to decompose the excess LAH. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in chloroform, washed with brine, dried over anhydrous MgSO4, filtered and concentrated. The residue was purified by column chromatography using chloroform and methanol as an eluent to give 20 (176 mg, yield 60%). 1H NMR (CDCl3, 400 MHz) δ 1.66 (s, 3H), 3.79 (t, J = 6 Hz, 2H), 3.88 (s, 2H), 4.04 (t, J = 6 Hz, 2H), 7.24-7.42 (m, 4H).
4-Nitrophenyl-3-(2-methyl-1,3-dioxolan 2 yl)benzylcarbamate (21)
To a solution of benzylamine 20 (176 mg, 0.911 mmol) and N,N-diisopropylethylamine (313 cL, 1.8 mmol) in 4 mL of 1:1 CH2Cl2/THF was added a solution of 4-nitrophenylchloroformate (366 mg, 1.82 mmol) in 2 mL of 1:1 CH2Cl2/THF. After stirring the reaction mixture at room temperature for 24 h, it was diluted with dichloromethane and washed sequentially with saturated NaHCO3, water and brine. The organic layer was dried over anhydrous MgSO4, filtered and concentrated. The residue was purified by column chromatography eluting with ethylacetate/hexane to give 21 (267 mg, 82%).
1-(4-Chlorophenyl)-3-(3-(2-methyl-1,3-dioxolan-2-yl)benzyl)urea (22)
4-Nitrophenyl-N-benzylcarbamate 21 (267 mg, 0.767 mmol) was added to a solution of 4-chloroaniline (97 mg, 0.767 mmol) and triethylamine in dichloromethane (5 mL). The mixture was stirred at room temperature until starting materials were consumed. The reaction mixture was then diluted with dichloromethane (50 mL) and washed with aq. NaOH, water and brine. The organic layer was dried over anhydrous MgSO4, filtered and concentrated The residue was purified by column chromatography eluting with methanol/chloroform to give 22 (165 mg, 71%). 1H NMR (CDCl3, 400 MHz) δ 1.58 (s, 3H), 3.71 (t, J = 6.8 Hz, 2H), 3.99 (t, J = 6.8 Hz, 2H), 4.36 (d, J = 5.6 Hz, 2H), 6.78 (s, 1H), 7.16-7.18 (m, 4H), 7.23 - 7.26 (m, 1H), 7.35 (d, J = 5.6 Hz, 2H).
1-(3-Acetylbenzyl)-3-(4-chlorophenyl) urea (23)
A 2N HCl solution was added to a solution of 22 (165 mg, 0.479 mmol) in THF (2 mL). The mixture was refluxed for several hours until the starting materials were consumed. The reaction mixture was allowed to cool to room temperature, quenched with solid NaHCO3, and then the volatiles were removed under reduced pressure. The residue was diluted with ethyl acetate and then washed with water. The organic layer was dried over anhydrous MgSO4, filtered and concentrated. The residue was purified by chromatography to give 23 (144 mg, 100%)1H NMR (DMSO-d6, 400 MHz) δ 4.44 (d, J = 5.6 Hz, 2H), 5.37 (t, J = 3.7 Hz, 1H), 6.83 (s, 1H), 7.20-7.26 (5H, m), 7.40 (t, J = 7.6 Hz, 1H), 7.50 (d, J = 7.2 Hz, 1H), 7.82 (d, J = 7.6 Hz, 1H), 7.85 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 27.45, 43.13, 119.85, 125.21, 127.11, 127.59, 129.13, 129.37, 132.61, 137.49, 140.07, 141.68, 155.73, 198.58; ESI-HRMS for C16H16N2O2Cl (M+H)+ calcd. 303.0900, found 303.0907.
3-Acetyl-α, α-dimethylbenzylamine (24)
3-Acetyl-α, α-dimethylbenzyl isocyanate 3 (1000 mg, 4.92 mmol) in 8N HCl (30 mL) were refluxed for 30 min. The reaction mixture was cooled to 0 °C and then washed with diethyl ether. The aqueous portion was neutralized with a 10% NaOH solution and extracted with diethyl ether (3 × 20 mL). The combined organic layers were washed with brine, dried over anhydrous MgSO4, filtered and concentrated. The residue was purified by chromatography eluting with methanol/chloroform to give 24 as a yellow oil (478 mg, 55%). 1H NMR (CDCl3, 400 MHz) δ 1.57 (s, 6H), 2.61 (s, 3H), 3.72 (s, 2H), 7.43 (t, J = 7.6 Hz, 1H), 7.75 (d, J = 7.6 Hz, 1H), 7.82 (d, J =7.6 Hz, 1H), 8.14 (s, 1H).
4-Chlorophenyl (2-(3-acetylphenyl)propan-2-yl)carbamate (25)
4-Chlorophenyl chloroformate (317 cL, 2.24 mmol) in dichloromethane (2 μL) was added to a mixture of 3-acetyl-α, α-dimethylbenzylamine 24 and diisopropylethylamine (390 μL, 2.24 mmol). The reaction mixture was stirred for 2 h. It was then diluted with dichloromethane, washed with 1N HCl, and then brine. The organic layer was dried over anhydrous magnesium sulfate, filtered and concentrated. The residue was purified by column chromatography using hexane/ethyl acetate as eluent to give 25 (594 mg, 81%). 1H NMR (CDCl3, 400 MHz) δ 1.73 (s, 6H), 2.60 (s, 3H), 5.65 (s, 1H), 7.02 (d, J = 8.4 Hz, 1H), 7.25 (d, J = 6 Hz, 2H), 7.44 (t, J = 8 Hz, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.82 (d, J = 7.6 Hz, 1H), 8.07 (1H, s); 13C NMR (DMSO-d6, 100 MHz) δ 27.45, 29.79, 55.50, 124.23, 124.55, 127.31, 129.28, 129.55, 129.79, 130.39, 137.30, 148.70, 150.32, 152.97, 198.64; ESI-HRMS for C18H19NO3Cl (M+H)+ calcd. 332.1053, found 332.1059.
N-(2-(3-Acetylphenyl)propan-2-yl)-2-(4-chlorophenyl)acetamide (26)
4-Chlorophenylacetylchloride (106 mg, 0.56 mmol) in dichloromethane was added to a solution of 3-acetyl-α, α-dimethylbenzylamine 24 (100 mg, 0.56 mmol) and triethylamine (120 μL, 0.86 mmol) in dichloromethane over a period of 5 to 10 min at 0 °C. The reaction mixture was stirred at room temperature for 2 h. The mixture was diluted with dichloromethane (20 mL) and washed with 1N HCl, water and brine. The organic layer was dried over anhydrous MgSO4, filtered and concentrated. The residue was purified by column chromatography using hexane/ethyl acetate as eluent to give 26 (146 mg, 80%). 1H NMR (CDCl3, 400 MHz) δ 1.61 (s, 6H), 2.53 (s, 3H), 3.46 (s, 2H), 5.68 (s, 1H), 7.19 (d, J = 7.2 Hz, 2H), 7.30-7.38 (m, 3H), 7.46 (d, J = 8 Hz, 1H), 7.75 (d, J =7.2 Hz, 1H), 7.87 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 27.26, 29.89, 42.54, 55.34, 124.71, 126.76, 128.74, 128.98, 130.23, 131.45, 131.63, 136.22, 137.19, 148.96, 169.54, 198.59; ESI-HRMS for C19H21NO2Cl (M+H)+ calcd. 330.1261, found 330.1270.
3-Acetyl-α, α-dimethylbenzylalcohol (27)
A solution of 3-acetyl-α α-dimethylbenzylamine (500 mg, 2.82 mmol) in chloroform was cooled to 0 °C. Chlorosulfonic acid (109 mg, 0.94 mmol) was added drop wise over a period of 15 min. During this time a white precipitate formed. Stirring was continued for another 10 min. The reaction mixture was filtered to yield the corresponding substituted N-benzylsulfonic acid (253 mg, 70% with respect to chlorosulfonic acid), which was used without further purification.
The N-benzylsulfonic acid (169 mg, 0.658 mmol) was suspended in 2 mL of water and then KNO2 (168 mg, 1.97 mmol, 3 equiv) was added. An almost clear solution was immediately formed. To this solution was added 2 mL sulfate buffer (pH = 2.8) and the resulting mixture was stirred for 2 hours. The reaction mixture was diluted with 4 mL water and extracted with ethyl acetate (3 × 10 mL). The combined organic layers were washed with water. The organic layer was dried over anhydrous MgSO4, filtered and concentrated. The residue was purified by column chromatography eluting with ethyl acetate/hexane to obtained 26 (26 mg, ~30%). 1H NMR (CDCl3, 400 MHz) δ 1.55 (s, 6H), 2.55 (s, 3H), 7.37 (t, J = 8 Hz, 1H), 7.66 (d, J = 7.2 Hz, 1H), 7.76 (d, J = 7.6 Hz, 1H), 8.04 (s, 1H). 13C NMR (CDCl3, 400 MHz) δ 26.9, 31.9, 72.5, 124.4, 126.9, 128.6, 129.6, 137.1, 150.0, 198.8.
2-(3-Acetylphenyl) propan-2-yl (4-chlorophenyl)carbamate (28)
To a solution of 3-acetyl-α, α-dimethylbenzylalcohol (27, 21 mg, 0.117 mmol) in 2 mL dry benzene was added 4-chlorophenyl isocyante, followed by 17 μL triethyamine. The reaction mixture was heated to 70 °C for 3 h. The reaction mixture was allowed to cool to room temperature and then diluted with ethyl acetate (20 mL), washed with 1N HCl, brine and water. The organic layer was dried over anhydrous magnesium sulfate, filtered and evaporated. The residue was purified by column chromatography eluting with ethyl acetate/hexane yielding 28 (23 mg, 62%). 1H NMR (CDCl3, 400 MHz) δ 1.84 (s, 6H), 2.60 (s, 3H), 6.71 (s, 1H), 7.20 (d, J = 8.8 Hz, 2H), 7.27 (d, J = 8.4 Hz, 2H), 7.45 (t, J = 8.0 Hz), 7.62 (d, J = 7.6 Hz, 1H), 7.84 (d, J = 7.6 Hz, 1H), 8.02 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 26.96, 29.08, 81.73, 119.8, 127.74, 128.90, 129.15, 129.26, 129.92, 136.67, 137.39, 146.71, 151.89, 198.29; ESI-HRMS for C18H18ClNO3 (M+Na)+ calcd. 354.0873, found 354.0872.
3-Phenyl α, α-dimethylbenzylamine (30)
To a room temperature stirring solution of 3-cyanobiphenyl (200 mg, 1.11 mmol) in 5 mL of diethyl ether was added 1M methylmagnesium bromide in butyl ether (3.33 mmol). The reaction mixture was stirred for 30 min and then 326 μL (1.11 mmol) of (i-PrO)4Ti was added. The solution became dark-brown and was stirred overnight before being treated with 10% solution NaOH (4 mL). The suspension was filtered to remove precipitated inorganic material, which washed with dichloromethane. The organic layer was separated and the aqueous layer was extracted with dichloromethane. The combined organic solutions were washed with water, brine and then dried over anhydrous MgSO4, filtered and evaporated to dryness. The residue was purified by column chromatography eluting with methanol/chloroform to give 30 (133 mg, 57%). 1H NMR (CDCl3, 400 MHz) δ 1.57 (s, 6H), 2.61 (bs, 2H), 7.34 -7.50 (m, 6H), 7.60 (d, J =7.2 Hz, 2H), 7.73 (s, 1H); 13C NMR (CDCl3, 400 MHz) δ 32.7, 53.0, 123.97, 123.98, 125.4, 127.4, 127.5, 128.9, 141.4, 141.7, 150.4.
3-Phenyl α-cyclopropylbenzylamine (31)
Ethyl magnesium bromide (3M) in diethyl ether (1.841 mmol, 2.2 eq.) was added at −78 °C to a solution of 3-cyanobiphenyl (150 mg, 0.838 mmol) and Ti(Oi-Pr)4 (0.269 mL, 0.920 mmol) in 5 mL of Et2O. The yellow solution was stirred for 10 min and then the solution was allowed to warm to room temperature and continued to be stirred for 1 h before BF3•OEt2 (0.206 mL, 1.76 mmol) was added. Stirring was continued for an additional hour. The reaction mixture was quenched with 3 mL of 1 N HCl and then washed with ether (15 mL). The aqueous layer was basified with 10% NaOH (10 mL) and extracted with diethyl ether. The organic layer was dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel eluting with methanol/chloroform to yield 31 (98 mg, 52%). 1H NMR (CDCl3, 400 MHz) δ 1.1 (t, J = 4 Hz, 2H), 1.04 (t, J = 4 Hz, 2H), 7.25 -7.79 (m, 1H), 7.35-7.46 (m, 5H), 7.52 (s, 1H), 7.58 (d, J = 7.2 Hz,1H).
4-Nitrophenyl(2-([1, 1′-biphenyl]-3-yl) propan-2-yl)carbamate (33)
Prepared following the same procedure as 21. Yield 71%. 1H NMR (CDCl3, 400 MHz) δ 1.62 (s, 6H), 6.81 (d, J = 8.8 Hz, 2H), 7.26-7.66 (m, 10H), 8.11 (d, J = 8.8 Hz, 2H).
4-Nitrophenyl(1-([1,1′-biphenyl]-3-yl)cyclopropyl)carbamate (34)
Prepared following the same procedure as 21. Yield 68%. 1H NMR (CDCl3, 400 MHz) δ 1.39-1.40 (m, 2H), 1.42-1.45 (m, 2H), 6.89 (d, J = 9.2 Hz, 2H), 7.31-7.49 (m, 6H), 7.54-7.58 (m, 3H), 8.16 (d, J = 9.2 Hz, 2H).
4-Nitrophenyl (2-phenylpropan-2-yl)carbamate (35)
Prepared folloeing the same procedure as 21. Yield 62%. 1HNMR (CDCl3, 400 MHz) δ 1.73 (s, 6H), 5.6 (bs, 1H), 6.79 (d, J = 8.8 Hz, 2H), 7.23-7.25 (m, 2H), 7.34 (t, J = 7.6 Hz, 1H), 8.09 (d, 2H, J = 9.2 Hz), 8.16 (d, J = 8.8 Hz, 1H).
5-(3-(2-([1, 1′-biphenyl]-3-yl) propan-2-yl)ureido)-2-chlorobenzamide (36a)
Prepared following the same procedure as 22. Yield 45%; 1H NMR (CDCl3, 400 MHz) δ 1.74 (s, 6H), 5.63 (s, 1H), 5.96 (s, 1H), 6.62 (s, 1H), 7.18 (d, J = 8.8 Hz, 1H), 7.28 (d, J = 2.4, 1H), 7.31-7.35 (m, 1H), 7.39 -7.48 (m, 6H), 7.55 (d, J = 7.2 Hz, 2H), 7.68 (s, 1H), 7.77 (d, J = 8.8 Hz, 1H); 13C NMR (CD3OD, 100 MHz) δ 29.1, 54.9, 118.3, 120.6, 122.8, 123.4, 123.7, 124.8, 126.8, 127.0, 128.5, 130.1, 132.5, 136.0, 141.3, 141.6, 155.1, 171.0; ESI-HRMS for C23H23N3O2Cl (M+H)+ calcd. 408.1479, found 408.1485.
1-(2-([1,1′-Biphenyl]-4-yl)propan-2-yl)-3-(4-chlorophenyl)urea (36b)
Synthesized following the synthetic procedure in scheme 6 starting with 4-phenylbenzonitrile. Yield 68%; 1H NMR (CDCl3, 400 MHz) δ 1.73 (s, 6H), 5.53 (s, 1H), 6.76 (d, J = 8.8 Hz, 1H), 7.18 (s, 1H), 7.23 (d, J= 8.8 Hz, 1H), 7.28 (d, J = 7.2 Hz, 1H), 7.35-7.38 (m, 2H), 7.45 -7.53 (m, 6H), 8.06 (d, J = 8.8 Hz, 1H), 8.14 (d, J = 8.4 Hz, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 30.28, 54.79, 116.49, 119.52, 126.07, 126.89, 126.99, 127.18, 129.11, 129.58, 138.48, 140.02, 140.66, 148.20, 154.48; ESI-HRMS for C22H22N2OCl (M+H)+ calcd. 365.1421, found 365.1428.
5-(3-(1-([1,1′-Biphenyl]-3-yl)cyclopropyl)ureido)-2-chlorobenzamide (37)
Prepared following the same procedure as 22. Yield 62%; 1H NMR (DMSO-d6, 400 MHz) δ 1.23 (t, 2H, J = 6.8 Hz), 1.30 (t, 2H, J = 8 Hz), 7.22 (d, J = 7.2 Hz, 2H), 7.28 (d, J = 8.8 Hz, 1H), 7.35-7.47 (m, 5H), 7.53 ( t, J = 2.4 Hz, 2H), 7.61 (d, J = 7.6 Hz, 2H), 7.80 (s, 1H), 8.70 (s, 1H), 8.70 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 19.2, 34.9, 118.1, 121.6, 123.7, 124.6, 124.8, 127.0, 127.4, 128.0, 129.3, 129.5, 130.3, 137.8, 139.8, 140.7, 141.1, 145.5, 155.5, 168.8; ESI-HRMS for C23H20N3O2Cl (M+H)+ calcd. 406.1322, found 406.1323.
1-(2-Oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)-3-(2-phenylpropan 2 yl)urea (38)
1.52 (s, 6H), 6.38 (d, J = 6 Hz, 1H), 6.56 ( t, J = 7.2 Hz, 1H), 6.66 (t, J = 7.2 Hz, 1H), 7.10-7.15 (m, 2H), 7.21-7.25 (m, 2H), 7.31-7.33 (m, 2H), 8.16 (s, 1H), 10.26 (s, 1H), 10.35 (s, 1H) ); 13C NMR (DMSO-d6, 100 MHz) δ 30.4, 54.8, 94.6, 99.9, 108.9, 110.7, 124.5, 125.4, 126.4, 128.5, 130.4, 134.8, 149.2, 154.8, 156.1; ESI-HRMS for C17H18N4O2 (M+H)+ calcd. 311.1508, found 311.1511.
Biological assays
Determination of IC50 values
Inhibition of recombinant CpIMPDH, purified from E. coli,12 was assessed by monitoring the production of NADH by fluorescence at varying inhibitor concentrations (25 pM - 5 cM). IMPDH was incubated with inhibitor for 5 min at room temperature prior to addition of substrates. The following conditions were used: 50 mM Tris-HCl, pH 8.0, 100 mM KCl, 3 mM EDTA, 1 mM dithiothreitol (assay buffer) at 25 °C, 10 nM CpIMPDH, 300 VM NAD and 150 VM IMP. To characterize the non-specific binding of inhibitors, assays were also carried out in the presence of 0.05% BSA (fatty acid free). IC50 values were calculated for each inhibitor according to Equation 1 using the SigmaPlot program (SPSS, Inc.):
| (Eq.1) |
where νi is initial velocity in the presence of inhibitor (I) and νo is the initial velocity in the absence of inhibitor. Inhibition at each inhibitor concentration was measured in quadruplicate and averaged; this value was used as νi. The IC50 values were determined three times; the average and standard deviations are reported.
Determination of antiparastic activity
Antiparasitic activity was tested by monitoring the growth of a of T. gondii strain (Toxo/CpIMPDH) that relies on CpIMPDH. Wild-type T. gondii (Toxo/WT) relies on a eukaryotic IMPDH that should be resistant to CpIMPDH inhibitors. Both parasites express yellow fluorescent protein, which allows growth to be easily monitored. Parasites were cultured on hTERT immortalized human foreskin fibroblasts cells in 96 well plates and fluorescence was measured daily with a SpectraMax M22/M2e (Molecular Devices) plate reader (Ex 485, Em 530) for 6-7 days. Growth inhibition was calculated on a day within the exponential growth phase21.
Stability assays
Mouse microsomal and plasma stability experiments were performed by Cyprotex Discovery (Watertown, MA).
Supplementary Material
Table 6.
Antiparasitic activity of selected urea compounds. Assays as described in Methods 21. Unless otherwise stated all values are the average 3 independent determinations.
| Compound | EC50 (μM) | Selectivitya | |
|---|---|---|---|
| Toxo/WT | Toxo/CpIMPDH | ||
| 5a | 6 ± 4 | 0.77 ± 0.06 | 8 |
| 5b | > 25 | 0.18 ± 0.02 | > 140 |
| 5c | > 25 | 0.2 ± 0.01 | > 120 |
| 5d | 0.402 ± 0.002 | 0.38 ± 0.01 | 1.0 |
| 5g | 20 ± 2 b | 7.2 ± 0.4 b | 3 |
| 5h | 4 ± 2 | 0.22 ± 0.04 | 20 |
| 5j | 2.3 ± 0.2 b | 0.018 ± 0.002 b | 127 |
| 5k | 15 ± 3 | 0.23 ± 0.04 | 65 |
| 5l | 19 ± 6 | 0.18 ± 0.03 | 100 |
| 5m | 9 ± 1 | 0.09 ± 0.03 | 100 |
| 5n | 8 ± 1 | 0.2 ± 0.2 | 50 |
| 5o | > 25 | 0.20 ± 0.03 | > 120 |
| 5q | 7.6 ± 0.8 | 0.02 ± 0.01 | ≥ 80 |
| 5r | 3.0 ± 0.1 b | < 0.2 | > 15 |
| 5s | 2 ± 1 | 1.1 ± 0.4 | 2.3 |
| 5t | 3 ± 1 | 3.4 ± 0.3 | 0.9 |
| 6b | 3.2 ± 0.7 | 0.2 ± 0.1 | 16 |
| 6c | > 25 | 0.38 ± 0.05 b | > 66 |
| 7a | 2.7 ± 0.2 | 0.01 ± 0.01 | 250 |
| 7b | 4 ± 2 | 0.006 ± 0.005 | 670 |
| 7c | > 25 | 1.9 ± 0.6 | > 12 |
| 7d | 5 ± 4 | 0.08 ± 0.01 | 63 |
| 7e | 14 ± 6 | 0.058 ± 0.003 | 230 |
| 8a | 1.4 ± 0.4 | 0.016 ± 0.008 | 86 |
| 8b | >25 | 0.2 ± 0.1 | >120 |
| 8c | 2.3 ± 0.8 | 0.013 ± 0.009 | 180 |
| 9 | 10.1 ± 1.9 | 0.51 ± 0.007 | 20 |
| 10 | 19 ± 6 | 0.9 ± 0.4 | 20 |
| 17 | 8 ± 4 | 2.3 ± 0.6 | 4 |
| 23 | 7 ± 5 | 11 ± 3 | 0.6 |
| 36a | 6 ± 3 | 0.21 ± 0.09 | 30 |
| 37 | 9 ± 6 | 0.6 ± 0.2 | 15 |
Selectivity is the ratio of EC50 Toxo/CpIMPDH to EC50 Toxo/WT.
Two determinations.
Acknowledgment
This work was supported by funding from the National Institute of Allergy and Infectious Diseases (U01AI075466) to LH. GDC thanks the New England Regional Center of Excellence for Biodefense and Emerging Infectious Diseases (NERCE/BEID) and the Harvard NeuroDiscovery Center for financial support. BS is a Georgia Research Alliance Distinguished Investigator.
Abbreviations
- Cp
Cryptosporidium parvum
- BSA
bovine serum albumin
- DIPEA
diisopropylethylamine
- hTERT
human telomerase reverse transcriptase
- IMP
inosine 5′-monophosphate
- IMPDH
inosine 5′-monophosphate dehydrogenase
- N.D.
not determined
- p-TSA
p-toluenesulfonic acid
- TEA
triethylamine
- Toxo
Toxoplasma
- XMP
xanthosine 5′-monophosphate
Footnotes
Supporting Information Available: Detailed information for HPLC methods and compound purity are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Guerrant RL, Oriá RB, Moore SR, Oriá MOB, Lima AAM. Malnutrition as an enteric infectious disease with long-term effects on child development. Nutr. Rev. 2008;66:487–505. doi: 10.1111/j.1753-4887.2008.00082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2(a).Snelling WJ, Xiao L, Ortega-Pierres G, Lowery CJ, Moore JE, Rao JR, Smyth S, Millar BC, Rooney PJ, Matsuda M, Kenny F, Xu J, Dooley JSG. Cryptosporidiosis in developing countries. J. Infect. Dev. Ctries. 2007;1:242–256. [PubMed] [Google Scholar]; (b) Huang DB, White AC. An updated review on Cryptosporidium and Giardia. Gastroenterol Clin North Am. 2006;35:291–314. doi: 10.1016/j.gtc.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Fayer R. Cryptosporidium: A water-borne zoonotic parasite. Vet. Parasitol. 2004;126:37–56. doi: 10.1016/j.vetpar.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 4(a).Samie A, Bessong PO, Obi CL, Sevilleja JE, Stroup S, Houpt E, Guerrant RL. Cryptosporidium species: Preliminary descriptions of the prevalence and genotype distribution among school children and hospital patients in the Venda region, Limpopo Province, South Africa. Exp. Parasitol. 2006;114:314–322. doi: 10.1016/j.exppara.2006.04.007. [DOI] [PubMed] [Google Scholar]; (b) Ajjampur SS, Rajendran P, Ramani S, Banerjee I, Monica B, Sankaran P, Rosario V, Arumugam R, Sarkar R, Ward H, Kang G. Closing the diarrhoea diagnostic gap in Indian children by the application of molecular techniques. J. Med. Microbiol. 2008;57:1364–1368. doi: 10.1099/jmm.0.2008/003319-0. [DOI] [PubMed] [Google Scholar]
- 5(a).Carey CM, Lee H, Trevors JT. Biology, persistence and detection of Cryptosporidium parvum and Cryptosporidium hominis oocyst. Water Res. 2004;38:818–862. doi: 10.1016/j.watres.2003.10.012. [DOI] [PubMed] [Google Scholar]; (b) Mac Kenzie WR, Hoxie NJ, Proctor ME, Gradus MS, Blair KA, Peterson DE, Kazmierczak JJ, Addiss DG, Fox KR, Rose JB, Davis JP. A massive outbreak in Milwaukee of Cryptosporidium infection transmitted through the public water supply. N. Engl. J. Med. 1994;331:161–167. doi: 10.1056/NEJM199407213310304. [DOI] [PubMed] [Google Scholar]; (c) Medema G, Teunis P, Blokker M, Deere D, Davison A, Charles P, Loret J-F. Risk assessment of Cryptosporidium in drinking water. World Health Organization; 2009. [Google Scholar]
- 6.DuPont HL, Chappell CL, Sterling CR, Okhuysen PC, Rose JB, Jakubowski W. The infectivity of Cryptosporidium parvum in healthy volunteers. N Engl J Med. 1995;332:855–859. doi: 10.1056/NEJM199503303321304. [DOI] [PubMed] [Google Scholar]
- 7(a).Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, Deng M, Liu C, Widmer G, Tzipori S, Buck GA, Xu P, Bankier AT, Dear PH, Konfortov BA, Spriggs HF, Iyer L, Anantharaman V, Aravind L, Kapur V. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science. 2004;304:441–445. doi: 10.1126/science.1094786. [DOI] [PubMed] [Google Scholar]; (b) Xu P, Widmer G, Wang Y, Ozaki LS, Alves JM, Serrano MG, Puiu D, Manque P, Akiyoshi D, Mackey AJ, Pearson WR, Dear PH, Bankier AT, Peterson DL, Abrahamsen MS, Kapur V, Tzipori S, Buck GA. The genome of Cryptosporidium hominis. Nature. 2004;431:1107–1112. doi: 10.1038/nature02977. [DOI] [PubMed] [Google Scholar]; (c) Umejiego NN, Li C, Riera T, Hedstrom L, Striepen B. Cryptosporidium parvum IMP dehydrogenase: Identification of functional, structural and dynamic properties that can be exploited for drug design. J Biol Chem. 2004;279:40320–40327. doi: 10.1074/jbc.M407121200. [DOI] [PubMed] [Google Scholar]
- 8.Hedstrom L. IMP dehydrogenase: structure, mechanism and inhibition. Chem. Rev. 2009;109:2903–2928. doi: 10.1021/cr900021w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Striepen B, White MW, Li C, Guerini MN, Malik SB, Logsdon JM, Jr., Liu C, Abrahamsen MS. Genetic complementation in apicomplexan parasites. Proc Natl Acad Sci U S A. 2002;99:6304–6309. doi: 10.1073/pnas.092525699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Umejiego NN, Gollapalli D, Sharling L, Volftsun A, Lu J, Benjamin NN, Stroupe AH, Riera TV, Striepen B, Hedstrom L. Targeting a prokaryotic protein in a eukaryotic pathogen: identification of lead compounds against cryptosporidiosis. Chem Biol. 2008;15:70–77. doi: 10.1016/j.chembiol.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11(a).Chen L, Pankiewicz KW. Recent development of IMP dehydrogenase inhibitors for the treatment of cancer. Curr Opin Drug Discov Devel. 2007;10:403–412. [PubMed] [Google Scholar]; (b) Ratcliffe AJ. Inosine 5′-monophosphate dehydrogenase inhibitors for the treatment of autoimmune diseases. Curr Opin Drug Discov Devel. 2006;9:595–605. [PubMed] [Google Scholar]
- 12.Boularot A, Giglione C, Petit S, Duroc Y, Alves de Sousa R, Larue V, Cresteil T, Dardel F, Artaud I, Meinnel T. Discovery and refinement of a new structural class of potent peptide deformylase inhibitors. J. Med. Chem. 2006;50:10–20. doi: 10.1021/jm060910c. [DOI] [PubMed] [Google Scholar]
- 13.Hase TA, Salonen K. A non-organometallic method for the synthesis of methyl ketones from acyl chlorides. Syn. Commun. 1980;10:221–224. [Google Scholar]
- 14.Özcan S, Balci M. The chemistry of homophthalic acid: A new synthetic strategy for construction of substituted isocoumarin and indole skeletons. Tetrahedron. 2008;64:5531–5540. [Google Scholar]
- 15.Salmoria GV, Neves A, Dall’Oglio EL, Zucco C. Preparation of aromatic ethers and dioxolanes under microwave irradiation. Syn. Commun. 2001;31:3323–3328. [Google Scholar]
- 16.Diss ML, Kennan AJ. Facile production of mono-substituted urea side chains in solid phase peptide synthesis. Biopolymers. 2007;86:276–281. doi: 10.1002/bip.20737. [DOI] [PubMed] [Google Scholar]
- 17.White EH, Li M, Lu S. Alkyldiazonium ion pairs and deamination. 46. N-nitrososulfamates: sources of carbonium ions in aqueous media and substrates in solid-state decompositions. J. Org. Chem. 1992;57:1252–1258. [Google Scholar]
- 18.Tomashenko OA, Sokolov VV, Tomashevskii AA, Potekhin AA, de Meijere A. Addition of organometallic reagents to nitriles promoted by titanium(IV) isopropoxide as a procedure of synthesis of primary tert-alkylamines. Russ. J.Org. Chem. 2007;43:1421–1426. [Google Scholar]
- 19.Bertus P, Szymoniak J. A direct synthesis of 1-aryl- and 1-alkenylcyclopropylamines from aryl and alkenyl nitriles. J. Org. Chem. 2003;68:7133–7136. doi: 10.1021/jo034710+. [DOI] [PubMed] [Google Scholar]
- 20.Smith DA, Di L, Kerns EH. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat Rev Drug Discov. 2010;9:929–939. doi: 10.1038/nrd3287. [DOI] [PubMed] [Google Scholar]
- 21.Sharling L, Liu X, Gollapalli DR, Maurya SK, Hedstrom L, Striepen B. A Screening pipeline for antiparasitic agents targeting Cryptosporidium inosine monophosphate dehydrogenase. PLoS Negl Trop Dis. 2010;4:e794. doi: 10.1371/journal.pntd.0000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22(a).Maurya SK, Gollapalli DR, Kirubakaran S, Zhang M, Johnson CR, Benjamin NN, Hedstrom L, Cuny GD. Triazole inhibitors of Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase. J Med Chem. 2009;52:4623–4630. doi: 10.1021/jm900410u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) MacPherson IS, Kirubakaran S, Gorla SK, Riera TV, D’Aquino JA, Zhang M, Lu J, Cuny GD, Hedstrom L. The structural basis of Cryptosporidium-specific IMP dehydrogenase inhibitor selectivity. J. Am. Chem. Soc. 2010;132:1230–1231. doi: 10.1021/ja909947a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kirubakaran S, Gorla SK, Sharling L, Zhang M, Liu X, Ray SS, MacPherson IS, Striepen B, Hedstrom L, Cuny GD. Structure–activity relationship study of selective benzimidazole-based inhibitors of Cryptosporidium parvum IMPDH. Bioorg. Med. Chem. Lett. 2012;22:1985–1988. doi: 10.1016/j.bmcl.2012.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.