

SUMMARY
Triplex structure-forming GAA/TTC repeats pose a dual threat to the eukaryotic genome integrity. Their potential to expand can lead to gene inactivation, the cause of Friedreich’s ataxia disease in humans. In model systems, long GAA/TTC tracts also act as chromosomal fragile sites that can trigger gross chromosomal rearrangements. The mechanisms that regulate the metabolism of GAA/TTC repeats are poorly understood. We have developed an experimental system in the yeast Saccharomyces cerevisiae that allows us to systematically identify genes crucial for maintaining the repeat stability. Two major groups of mutants defective in DNA replication or transcription initiation are found to be prone to fragility and large-scale expansions. We demonstrate that problems imposed by the repeats during DNA replication in actively dividing cells and during transcription initiation in nondividing cells can culminate in genome instability. We propose that similar mechanisms can mediate detrimental metabolism of GAA/TTC tracts in human cells.
INTRODUCTION
Triplex-forming GAA/TTC trinucleotide repeat tracts are a common sequence motif present in many prokaryotic and eukaryotic genomes (Clark et al., 2006; Kassai-Jáger et al., 2008). They are particularly abundant in mammalian genomes including humans where GAA/TTC loci were found to be highly polymorphic (Clark et al., 2004). Repeat tracts can both contract and expand, the latter feature is responsible for the inactivation of the FXN gene causing Friedreich’s ataxia (FRDA) (Campuzano et al., 1996). FRDA patients are carriers of expanded repeats ranging from 66 to 1,700 copies, whereas normal individuals bear <65 repeats (Campuzano et al., 1996; Clark et al., 2004 and references therein). It has been demonstrated that premutation (34–65 repeats) and mutant (>66 repeats) alleles are unstable in both proliferating and terminally differentiated cells wherein the tissue type is one of the determinants of instability (Clark et al., 2007; De Biase et al., 2007a, 2007b). Although in most somatic cells contractions are a predominant class of tract length variations, neuronal cells exhibit an expansion bias. The ability of GAA/TTC repeats to undergo tract length variations is not only confined to human cells. Long tracts are also highly unstable in bacteria, yeast, and mice with contractions being the most predominant class in microbial systems (Bourn et al., 2009; Clark et al., 2007; Kim et al., 2008; Krasilnikova and Mirkin, 2004; Pollard et al., 2004).
Besides being prone for size variations, expanded GAA/TTC tracts were also found to induce recombination and gross chromosomal rearrangements (GCRs) in bacteria and yeast (Kim et al., 2008; Tang et al., 2011; Vetcher and Wells, 2004). In yeast, this ability of repeats to compromise genome integrity is attributed to double-strand break (DSB) formation. We demonstrated that replication is a factor in GAA/TTC-mediated fragility and rearrangements (Kim et al., 2008). In contrast, Tang et al. (2011) found that mitotic crossover stimulated by GAA/TTC tracts results from DSB formation on unreplicated chromosomes, where the breakage was deduced to occur at the G1 stage of the cell cycle.
Overall, size variations in GAA/TTC tracts and repeat-associated fragility happen in both actively dividing and nondividing cells. This likely reflects the complexity of the processes that affect repeat metabolism where DNA replication, transcription, repair, and chromatin organization are all contributing factors to the genetic instability. The homopurine/homopyrimidine nature of GAA/TTC tracts along with their intrinsic mirror symmetry provide the repeats the ability to adopt triplex (or H-DNA) secondary structure (reviewed in Frank-Kamenetskii and Mirkin, 1995). The formation of triplex structure is considered to be one of the main triggering events for repeat-associated instability. Triplex formation is facilitated in single-stranded regions that are natural intermediates during replication, transcription, and repair. Consequently, stable secondary structure, which is an abnormal template for DNA and RNA synthesis, can hinder DNA and RNA polymerase progression (Kim et al., 2008; Krasilnikova et al., 2007 and references therein). The attempt to repair or bypass these impediments eventually results in destabilization of the repeats (Bourn et al., 2009; Kim et al., 2008; Shishkin et al., 2009).
In yeast, using two-dimensional (2D) gel electrophoresis, we found that long GAA/TTC tracts can stall the progression of the replication fork in a plasmid and in the chromosome when the GAA strand is a template for lagging strand synthesis (Kim et al., 2008; Krasilnikova and Mirkin, 2004). The propensity of the repeats to block the replication fork also correlates with their ability to stimulate chromosomal breakage and rearrangements (Kim et al., 2008). At the same time, GAA/TTC expansion potential is not dependent on the orientation of the repeats relative to replication origin (Shishkin et al., 2009). However, expansions are stimulated in the replication-checkpoint surveillance mutants tof1 and csm3, and compromised in sgs1, rad5, and rad6 mutants defective in repair and postreplicative template switch. These data point strongly toward the existence of replication problems imposed by the repetitive tracts on leading as well as on lagging strands in actively dividing cells.
GAA/TTC tracts can efficiently block transcription elongation both in vitro and in vivo, presumably due to triplex formation (Krasilnikova et al., 2007 and references therein). Also, it has been shown that expanded alleles in the first intron of the FXN gene can promote spreading of repressive chromatin marks that can reach the promoter and affect transcription initiation (reviewed in Kumari and Usdin, 2012). Both deficiencies in elongation and initiation of transcription are implicated in the FRDA pathology. At the same time, a change in the level of transcription across repeats has a strong effect on repeat instability. In human cell lines, induction of transcription promotes deletions (Soragni et al., 2008). Increasing or decreasing the basal level of transcription antagonizes expansions (Ditch et al., 2009). The emerging mechanism of the repeat instability associated with transcription is the accumulation of persistent DNA-RNA hybrids (R-loops) in the tract regions resulting from RNA polymerase stalling (Belotserkovskii et al., 2010; Grabczyk et al., 2007; McIvor et al., 2010; Reddy et al., 2011). R-loops are known recombinagenic substrates (reviewed in Aguilera and Gómez-González, 2008), and the processing of these structures has been implicated in destabilization of GAA/TTC repeats (McIvor et al., 2010). Repeat instability documented in nonreplicating cells can be explained by transcription-associated mechanisms. Collision of DNA polymerase with trapped RNA polymerase at R-loops can also be a factor in generation of breaks and size variations in actively-dividing cells (Rindler and Bidichandani, 2011).
In this study, we carried out a systematic and unbiased genome-wide screen in yeast Saccharomyces cerevisiae to identify genes that govern GAA/TTC repeat instability. A complete set of 4,786 deletion mutations and 800 essential genes, for which expression is either regulated by doxycycline or compromised due to mRNA perturbation, was tested. The screen identified 33 mutants that destabilize the repeats. We determined that most of the mutations that augment GAA/TTC fragility also increase the rate of repeat expansions, indicating that both types of instability can be driven by a common initiating event. We found that defects in proteins that constitute the core of the replisome and replication-pausing checkpoint surveillance strongly increase GAA/TTC fragility and expansions. A dysfunction of origin and telomere also stimulates breakage and expansions. Surprisingly, another major class of mutants exhibiting hyperinstability belongs to transcription initiation. Consistently, we have found that GAA/TTC repeats serve as promoters and recruit transcription initiation factors. Our findings suggest that problems imposed by the triplex structure during DNA replication are a major triggering event in chromosomal fragility and size variations operating in actively dividing cells. At the same time, proper transcription initiation at GAA/TTC tracts is a guardian of repeat stability in both dividing and nondividing cells. We propose that similar mechanisms can mediate detrimental metabolism of GAA/TTC tracts in human cells.
RESULTS
Genome-wide Screen of Mutants that Augment GAA/TTC Fragility
To identify mutants exhibiting an increased level of fragility mediated by GAA/TTC repeats, we carried out genome-wide screening of three collections of strains purchased from Open Biosystems. A total of 4,786 deletion mutations (YKO collection) and 860 essential genes, whose expression is either regulated by doxycycline (yTHC collection) or compromised due to mRNA perturbation (DAmP collection), were tested. The scheme for combining mutant alleles marked with kanMX cassette and GAA/TTC tracts is based on the approach developed by Tong et al. (2001) with modifications (Figures 1A and 1B; Experimental Procedures). Hyper-GCR mutants were identified based on the increased number of papillae in a visual comparison to wild-type strain (Figure 1C). Because the GCR rate induced by (GAA)230 repeats (~2 × 10−6) is approximately ten times higher than the mutation rate (~3 × 10−7) at the CAN1 locus in wild-type strains, the screen preferentially selects for GCR inducers or strong mutators.
Figure 1. Genome-wide Screen Methodology.

(A) Query strain to uncover genotypes prone to GAA/TTC fragility. The 230 GAA/TTC repeats were positioned on the left arm of chromosome V with GAA as a template for lagging strand synthesis. The arrangement of LYS2, hphMX cassette, and CAN1 on chromosome V are shown. The location of HIS3 ORF under control of MATa-specific promoter and rpl28-Q38K allele on chromosomes IV and VII are depicted.
(B) Schematics of the screen. MATα query strains carrying GAA/TTC tract were crossed with MATa tester strains from YKO, yTHC, and DAmP libraries. Diploids were selected by replica plating on media containing both G418 and Hygromycin B. After sporulation of diploids, MATa haploids were selected using histidine drop-out media containing cycloheximide. Haploids harboring both GAA/TTC tract and mutations of interests were further selected by G418 and Hygromycin B containing media. Cells were transferred to canavanine-containing media to score for arm-loss events.
(C) Example plate for the screen. Columns are duplicates of query strains. Each row is one tester strain. The level of arm loss in TET-RFA2 (hyperfragile), Δmrc1 (hyperfragile), TET-SEC4 (no change in fragility), and wild-type are shown.
(D) Experimental assay to verify results from the screen. ADE2 is placed between CAN1 and LYS2. Mutations in CAN1 will manifest as CanRAde+ white colonies, whereas arm loss events will give rise to CanRAde− red papillae. See also Table S1.
To verify the effect of hyper-GCR mutants and to distinguish them from strong mutators at the CAN1 locus, we introduced the alleles into strains carrying the GCR assay described in Kim et al. (2008). The ADE2 marker placed between CAN1 and LYS2 helps to differentiate between DSB-mediated arm loss and mutations in CAN1. In this assay, GCR isolates are manifested as canavanine-resistant red colonies whereas mutations in CAN1 give rise to canavanine-resistant white colonies (Figure 1D). The effect of the mutations was tested in the strains containing 120 and 230 copies of GAA/TTC repeats and in the strains lacking repeats.
Hyperfragility Mutants Identified in the Screen
The screen identified 11 hyperfragility mutants from the YKO collection (Table S1 available online). After recreating these deletions in the color-based GCR assay, we excluded Δtsa1 from further analysis because this mutant exhibited a strong mutator phenotype but not increased arm loss (data not shown). The remaining ten mutants fall into two groups: DSB repair and replication. The first group consists of mutants deficient in Mre11-Rad50-Xrs2 (MRX complex) and Sae2 activities. The MRX complex as well as Sae2 are nucleases involved in DSB end resection. In addition, MRX provides a bridge between the two broken molecules during DSB repair (reviewed in Mimitou and Symington, 2009). Previously we have demonstrated that breaks at GAA/TTC tracts are predominantly repaired via break-induced replication involving GAA-rich stretches on nonhomologous chromosomes (Kim et al., 2008). It is therefore conceivable that the lack of extensive resection in mrx or sae2 mutants will allow for improved recovery of broken chromosome V due to increased probability of recombination between GAA regions. Moreover, in the absence of the MRX bridge, the acentric broken fragment of chromosome V can be lost more efficiently than in wild-type strains. Consistent with this, the GCR rates were ~6-fold higher in mrx mutants than in sae2 (Table 1). Hence, it is likely that a deficiency in MRX complex and Sae2 is not related to stability of the repeats, but rather reflects a change in the repair dynamics of GAA/TTC-mediated breaks.
Table 1.
Effect of Mutants Identified in the Genome-wide Screen on GAA-Induced GCRs
| Genetic Background | Rate of GCR (×10−7) | |||||
|---|---|---|---|---|---|---|
| (GAA)0 | (GAA)120 | (GAA)230 | ||||
| Wild-type | 0.03 (0.02–0.04)a | 3 (3–4) | 20 (10–30) | |||
| Double-strand break repair genes | ||||||
| Δmre11 | 36 (27–42) | 1,200b | 126 (110–140) | 42 | 350 (240–400) | 18 |
| Δsae2 | 0.9 (0.2–1) | 30 | 17 (13–24) | 6 | 60 (50–70) | 3 |
| Replication genes | ||||||
| TET-RFA2 | 4 (3–6) | 133 | 272 (93–380) | 91 | 740 (490–910) | 37 |
| TET-POL12 | 4 (1–8) | 133 | 65 (46–72) | 22 | 280 (70–580) | 14 |
| TET-PRI2 | 1 (1–2) | 33 | 49 (39–51) | 16 | 630 (550–910) | 32 |
| TET-POL3 | 0.5 (0.2–0.7) | 17 | 30 (23–40) | 10 | 150 (140–200) | 8 |
| TET-POL2 | 0.2 (0.2–0.5) | 7 | 7 (5–8) | 2 | 80 (70–100) | 4 |
| TET-POL30 | 0.6 (0.4–1) | 20 | 32 (21–40) | 11 | 370 (290–540) | 19 |
| TET-RFC2 | 0.2 (0.1–0.4) | 7 | 30 (9–41) | 10 | 280 (250–860) | 14 |
| TET-DNA2 | 0.5 (0.3–0.7) | 17 | 16 (12–21) | 5 | 170 (150–200) | 9 |
| TET-MCM4 | 2 (1–3) | 67 | 28 (20–31) | 9 | 150 (90–210) | 8 |
| TET-ORC4 | 3 (2–4) | 100 | 26 (19–33) | 9 | 160 (130–220) | 8 |
| Δrad27 | 12 (7–20) | 400 | 32 (26–46) | 11 | ND | ND |
| Δrtt101 | 3 (2–3) | 100 | 15 (12–16) | 5 | 140 (120–360) | 7 |
| Δmms1 | 0.14 (0.1–0.4) | 5 | 16 (12–21) | 5 | 200 (120–830) | 10 |
| Δmrc1 | 4 (3–8) | 133 | 58 (45–66) | 19 | 290 (210–350) | 15 |
| Δtof1 | 2 (0.9–3) | 67 | 35 (29–53) | 12 | 210 (170–230) | 11 |
| Telomere maintenance genes | ||||||
| TET-TEN1 | 0.2 (0.2–0.5) | 7 | 16 (5–28) | 5 | 160 (90–300) | 8 |
| TET-CDC13 | 2 (0.8–2) | 67 | 13 (10–18) | 4 | 220 (180–260) | 11 |
| Transcription initiation genes | ||||||
| TET-TAF4 | 0.2 (0.07–0.2) | 7 | 132 (105–170) | 44 | 460 (390–660) | 23 |
| TET-TAF9 | 0.1 (0.05–0.2) | 3 | 23 (9–31) | 8 | 190 (130–370) | 10 |
| TET-TOA1 | 0.6 (0.4–1) | 20 | 36 (17–44) | 12 | 210 (200–270) | 11 |
| TET-SUA7 | 0.2 (0.1–0.4) | 7 | 86 (60–120) | 29 | 390 (370–460) | 20 |
| TET-TFG1 | 0.5 (0.2–1) | 17 | 23 (17–27) | 8 | 370 (310–450) | 19 |
| TET-SPN1 | 0.3 (0.2–0.4) | 10 | 10 (7–11) | 3 | 160 (100–240) | 8 |
ND, not determined. See also Tables S1, S2, and S3.
Numbers in parentheses correspond to the 95% confidence intervals.
Fold increase in GCR rates in mutants compared to wild-type strains.
The second group includes mutations in Rad27, the flap-endonuclease, Rtt101-Mms1-Mms22, the proteins involved in the repair of stalled replication forks, and Tof1-Csm3-Mrc1, the replication-pausing checkpoint surveillance complex. Disruption of genes that encode for these proteins caused a 5- to 19-fold increase in chromosome V arm loss in strains carrying 120 and 230 GAA/TTC repeats. These data indicate that unperturbed replication progression and checkpoint sensing of the replication problems imposed by triplex structure are critical for the maintenance of GAA/TTC tracts. The list of alleles of the essential genes that altered GAA/TTC fragility (see below) also corroborates this conclusion.
The effect of the decrease in expression of the essential genes on GAA/TTC fragility was assessed using the yTHC and DAmP collections. Screening using the yTHC collection was more successful in identifying hypomorphic alleles that destabilize the repeats. Although during the initial screen doxycycline was used to downregulate expression of the genes, we noticed that the identified strains with TET-ORFs exhibited hyperfragility even without the addition of the drug to the medium (Table S3). It has been shown that tetO7-driven expression leads to an increased level of mRNA production (Bellí et al., 1998). However, we found that increase in transcription does not result in elevated protein levels and the TET-ORFs behave as recessive alleles (N.S. and K.L., unpublished data; Table S2). Therefore, we consider TET-ORFs to be mutant alleles (henceforth referred to as TET alleles). Overall, these mutants can be grouped into three main categories.
First, in agreement with results from the screening of YKO library, 16 hyperfragility alleles from the essential gene collections belonged to the DNA replication pathway (Table S1). They fall into eight distinct complexes or proteins including polymerase α-primase complex, lagging strand polymerase δ, nuclease/helicase Dna2, single-strand DNA binding protein RPA, clamp loader RFC, processivity clamp PCNA, replicative helicase MCM, and origin recognition complex ORC. Although Pol2, the leading strand polymerase ε, was not identified in the initial screen using the library, we found that replacing POL2 promoter with the doxycycline-regulatable promoter leads to hyperfragility. The effect of the mutant alleles on the GAA/TTC-induced GCRs varied from 2- to 91-fold (Table 1). These results indicate that a defect in replication machinery can tremendously affect the fragility potential of the repeats and intact synthesis of both leading and lagging strands is required for preventing the breakage. It is interesting to note the effect of the TET-ORC alleles. The LYS2 region is replicated from the ARS507 origin located ~26 kb away. It suggests that the dysfunction of origin firing or improper assembly of the replisome in these mutants affects the efficiency of breakage at a distant site.
Second, ten1-DAmP hypomorphic allele was found to increase GAA/TTC-mediated GCRs. Ten1 forms a complex with Cdc13 and Stn1 that protects telomeric ends in yeast (reviewed in Giraud-Panis et al., 2010). To determine if the hyperfragility of ten1-DAmP comes from a defect in expression of solely the TEN1 gene or results from the malfunction of the complex, we placed TEN1 and CDC13 under control of the tetO7-promoter. Both TET alleles caused a 4- to 11-fold increase in the fragility of 120 and 230 GAA/TTC repeats (Table 1). This suggests that proper maintenance of telomeres, the closest telomere being ~40 kb away from the repeat locus, is important to prevent DSB formation.
Third, besides TET alleles of the essential genes associated with DNA replication, another major group of hyperfragile alleles comprised of genes encoding for transcription initiation proteins such as components of TFIID, TFIIA, TFIIB, and TFIIF complexes (reviewed in Hahn and Young, 2011) (Tables 1 and S1). The effect of transcription initiation mutants on GAA/TTC fragility ranged from an 8- to 44-fold increase over the wild-type. In addition, the TET-SPN1 hyperfragile allele that codes for a factor that functions in both the recruitment of RNA polymerase II to the promoter and the elongation steps of transcription (Lindstrom et al., 2003), was identified. Although we acknowledge that the screen could have missed some mutants, especially those with mild effect on GCR rate, these data point toward two major pathways that underlie GAA/TTC fragility in yeast: replication and transcription initiation.
TET-TAF4 and TET-RFA2 Strains Exhibit Increased DSB Formation
In order to determine if the increase in GCR levels observed in mutants results from elevated levels of chromosomal breakage, we analyzed the accumulation of DSB intermediates in repeat-containing strains. No breaks were detected in the wild-type strain containing 20 or 230 copies of GAA/TTC repeats when detection of the breakage was carried out in yeast cultures grown overnight. Two mutants with the highest levels of GCR events from replication and transcription initiation categories, TET-TAF4 and TET-RFA2, respectively, accumulated DSBs (Figure 2). Densitometry analysis revealed that 12% and 21% of chromosomes V were broken in TET-TAF4 and in TET-RFA2 strains, correspondingly.
Figure 2. Physical Detection of GAA/TTC-Induced DSBs.

Location of GAA/TTC tracts on chromosome V is depicted. Intact chromosome V is ~585 kb, DSB at the tract results in a 43 kb broken fragment. Chromosomal DNA was separated by CHEF. Both intact and broken chromosomal V were detected by southern blot hybridization using a HPA3-specific probe. Lanes: 1, wild-type strain with (GAA)20; 2, wild-type strain with (GAA)230; 3, TET-TAF4 strain with (GAA)230; 4, TET-RFA2 strain with (GAA)230.
GAA/TTC Tracts Can Promote Transcription
In the screen for hyperfragile mutants, we have identified alleles for proteins specifically involved in initiation but not in elongation or termination steps of transcription. It has also been shown that GAA/TTC repeats exclude nucleosomes in vitro, perhaps due to the AT-richness of the tracts (Ruan and Wang, 2008). In eukaryotic genomes, AT-rich regions are often loci of open chromatin where transcription can start (reviewed in Hahn and Young, 2011). Based on these observations, we proposed that GAA/TTC tracts can recruit transcription initiation complexes and serve as noncanonical promoter elements in yeast. To test this hypothesis, we replaced the native promoter of the chromosomal TRP1 gene with GAA/TTC tracts of different lengths. The repeats were placed 56 bps upstream from the ATG codon of TRP1 in the orientation where during transcription GAA strand will be the sense strand. As expected, strains containing promoter-less TRP1 ORF did not grow on media lacking tryptophan (Figure S1). Five GAA/TTC repeats led to a weak growth of yeast on selective media. Strains with (GAA)20 and (GAA)120 exhibited normal growth comparable to the strain bearing TRP1 expressed from its native promoter. Interestingly, growth was inhibited in strains with (GAA)230 and (GAA)400. Plating serial dilutions on TRP1 counter-selective media containing 5-fluoroanthranilic acid resulted in reverse growth phenotypes: strains with 5, 120, 230, and 400 repeats grew better than strains containing 20 repeats. Moreover, 20 and 120 repeats placed in another orientation relative to the ATG codon (where TTC strand is the sense strand) also drove expression of TRP1 (data not shown). Consistent with these data, chromatin immunoprecipitation (ChIP) of TAP-tagged Sua7 protein demonstrated that this TFIIB transcription initiation factor is associated with (GAA)120 tracts with an efficiency comparable to binding to the native TRP1 promoter.
Fragility Is Increased in Nondividing Cells in a Time-Dependent Manner and Is Amplified in TET-TAF4 Strains
Increased GAA/TTC fragility in replisome-defective mutants suggests that DSBs occur during DNA replication. At the same time, the involvement of transcription-initiation factors in GAA/TTC fragility predicts the existence of a second pathway for breakage where DSBs can be formed outside the S phase of the cell cycle. To check this premise experimentally, we estimated GCR frequencies in wild-type and TET-TAF4 strains carrying 120 copies of GAA/TTC repeats in cultures that actively divide during the log phase of growth and in those that are arrested at stationary phase. Yeast were cultured from <20 cells to saturation (~1 × 108/ml) in liquid YPD and instead of rates, the frequencies of GCRs were calculated (Figures 3A and 3B). Wild-type and TET-TAF4 strains stop dividing ~43 and 49 hr after inoculation, respectively. Cells were held at this stage up to ~70 hr. The level of fragility in wild-type and mutant strains increased significantly after they reached stationary phase in comparison to actively dividing cultures (Figure 3B; Table S4). Holding yeast cultures at the arrested stage gradually augmented GAA/TTC fragility. GCR frequencies measured in cultures held arrested for 70 hr were ~70-fold higher than those in log-phase cultures for both wild-type and TET-TAF4 mutants. Overall, fragility frequencies in TET-TAF4 mutants were 5- to 30-fold higher than in wild-type in both logarithmic and stationary phases.
Figure 3. GAA/TTC Fragility Increases in Nondividing Cells.

(A) Growth curves of wild-type and TET-TAF4 strains. Wild-type and TET-TAF4 strains enter stationary phase at ~43 and 49 hr after inoculation, correspondingly. Error bars indicate SEM.
(B) GAA/TTC fragility frequencies of wild-type and TET-TAF4 strain in dividing and nondividing cells. Values are the median frequencies obtained from fluctuation tests of at least eight cultures carried out at the indicated time-points. Error bars represent 95% confidence intervals.
(C) Detection of DSBs in wild-type and TET-TAF4 strains at different time-points during stationary phase. Zero hour corresponds to the time when strains stopped growing. Twenty hour and 70 hr indicate the time each strain spent in stationary phase. Lanes: 1, wild-type strain with (GAA)20; 2, wild-type strain with (GAA)230; 3, TET-TAF4 strain with (GAA)230. Positions of DSBs and unbroken chromosome V are indicated by arrows. See also Table S4 and Figure S1.
To obtain direct evidence for time-dependent chromosomal breakage in nondividing cells, accumulation of DSB intermediates in the log phase and the arrested cultures was monitored (Figure 3C). No broken molecules were detected in wild-type strains carrying 20 or 230 GAA/TTC repeats at any analyzed time points. Consistent with the measurements of the GCR rates, in the TET-TAF4 (GAA)230 strain, there was an increase in the amount of broken molecules when cells spent more time in the stationary phase. The fraction of DSBs at the beginning of the stationary phase was 3%, which increased to 13% and 18% at 20 hr and 70 hr after arrest, respectively.
These data demonstrate that fragility occurs and is amplified in nondividing cells. Elevated fragility in TET-TAF4 compared to wild-type strains in stationary phase suggests that intact transcription initiation is important in controlling GAA/TTC stability in nondividing cells.
Mutations Conferring Hyperfragility Also Induce Large- Scale Repeat Expansions
Formation of triplex structure by GAA/TTC repeats was proposed to be a triggering event for both fragility and tract length variations (Kim et al., 2008; Shishkin et al., 2009). Hence, we reasoned that the identified mutants predisposed for GAA/TTC breakage should also be prone to repeat instability including large-scale expansions. To address this experimentally, we introduced a selectable cassette containing 100 GAA/TTC repeats (Shishkin et al., 2009; Experimental Procedures) into the strains with hyperfragile alleles. The repeat tract is inserted into an artificial intron in the URA3 gene located 1 kb from the ARS306 on chromosome III. Expansion of repeats beyond 130 copies leads to the inactivation of RNA splicing, blocking URA3 expression. These events give rise to colonies resistant to 5-fluoroorotic acid (5-FOAR).
We measured the rates of expansions in colonies that grew for 3 days on YPD plates. A defect in Mre11-Rad50-Xrs2 and Sae2 that caused increase in GCRs events did not change the rate of expansions (Table 2 and data not shown). This is in agreement with our interpretation of the effect of these mutations on the GCRs efficiency: mrx and sae2 mutants do not affect the stability of the repeats, but influence the repair step during GCR generation. Remarkably, all the other identified hyperfragile alleles profoundly affected the ability of GAA/TTC tracts to expand. The degree of impact of the mutant alleles on fragility and expansion also generally correlated. Malfunction of telomere maintenance caused a modest 4- to 11-fold increase in GCRs (Table 1) and 12- to 14-fold elevation of the expansion rates (Table 2). Along with this, the rate of expansions was elevated 171-fold in the most fragile mutant from DNA replication group, TET-RFA2 and 58-fold in the most fragile mutant from transcription initiation group, TET-TAF4. To verify that the 5-FOAR colonies arose due to GAA/TTC expansions, but not due to changes in URA3 expression levels in transcription initiation mutants, expansions at the repeat locus were confirmed by PCR in both wild-type and TET-TAF4 strains. In addition, analysis of URA3 expression using reverse transcriptase PCR showed that wild-type and TET-TAF4 strains are comparable (data not shown).
Table 2.
Rates of Expansions of (GAA)100 in Wild-type and Hyper-GCR Mutants
| Genetic Background | Expansion Rate (×10−5) | Fold Increase |
|---|---|---|
| Wild-type | 1 (0.6–1)a | 1b |
| Double-strand break repair genes | ||
| Δsae2 | 1 (0.6–2) | 1 |
| Replication genes | ||
| TET-RFA2 | 171 (84–330) | 171 |
| TET-POL12 | 55 (49–82) | 55 |
| TET-PRI2 | 110 (77–190) | 110 |
| TET-POL3 | 11 (7–25) | 11 |
| TET-POL2 | 5 (2–12) | 5 |
| TET-POL30 | 26 (16–30) | 26 |
| TET-RFC2 | 26 (13–66) | 26 |
| TET-DNA2 | 8 (4–12) | 8 |
| TET-MCM4 | 23 (17–36) | 23 |
| TET-OCR4 | 18 (4–90) | 18 |
| Δrad27 | 20 (9–35) | 20 |
| Δrtt101 | 4 (2–31) | 4 |
| Δmms1 | 3 (2–4) | 3 |
| Δtof1 | 10 (13–24) | 10 |
| Telomere maintenance genes | ||
| TET-TEN1 | 12 (9–17) | 12 |
| TET-CDC13 | 14 (10–29) | 14 |
| Transcription initiation genes | ||
| TET-TAF4 | 58 (40–68) | 58 |
| TET-TAF9 | 37 (32–46) | 37 |
| TET-TOA1 | 14 (8–23) | 14 |
| TET-SUA7 | 19 (12–23) | 19 |
| TET-TFG1 | 22 (17–57) | 22 |
| TET-SPN1 | 2 (1–3) | 2 |
Numbers in parentheses correspond to the 95% confidence intervals.
Fold increase in rates of expansions in mutants compared to the wild-type strain.
As demonstrated above, GAA/TTC-induced DSBs accumulate during stationary phase and intact transcription initiation counteracts the fragility. Transcription initiation mutants are also prone to tract expansions. Based on these data, we asked whether large-scale repeat expansions occur in nondividing cells during stationary phase. We estimated the expansion frequencies in actively dividing and starvation-arrested cells in wild-type and TET-TAF4 strains (Figure 4; Table S5). Interestingly, the expansion frequency in TET-TAF4 strains increased at the point of transition from actively dividing to the stationary stage. However, unlike fragility dynamics, expansions were not elevated upon holding cultures in the stationary stage, indicating that replication is required for this process. In fact, there was a slight decrease in the expansion frequencies at later time points in the stationary phase, which is likely due to accumulation of deletions.
Figure 4. Expansion Dynamics of (GAA)100 Repeats in Actively Dividing and Nondividing Cells in Wild-type and TET-TAF4 Strains.
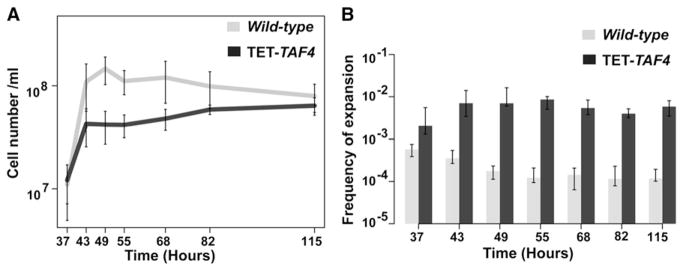
(A) Growth curves for wild-type and TET-TAF4 strains. Error bars represent SEM.
(B) Frequencies of expansion of (GAA)100 in dividing and arrested cells. Values are the median frequencies obtained from fluctuation tests of at least eight cultures. Error bars represent 95% confidence intervals. See also Table S5.
Together, these studies reveal replication and transcription initiation as the two major determinants of GAA/TTC stability in yeast. Both fragility and expansion are increased in replication and transcription initiation mutants, indicating existence of common intermediates that can drive both types of instability. Although fragility can happen in both dividing and nondividing cells, large-scale expansions are dependent on DNA replication.
DISCUSSION
In this work, we have isolated 33 mutants prone to GAA/TTC fragility and large-scale expansions in yeast. Moreover, we uncovered the role for GAA/TTC tracts as initiators of transcription. The identity of the mutants, the ability of repeats to promote transcription and the analysis of breakage and expansion dynamics point toward mechanisms that govern GAA/TTC instability during and outside of S phase. Below we will discuss the results of the genome-wide screen according to the category of the revealed mutants.
Defect in DNA Replication
Intact replication machinery and replication-pausing checkpoint surveillance are required to maintain GAA/TTC repeat stability (Tables 1, 2, and S1). A model of how a defect in DNA replication can influence the propensity of GAA/TTC tracts to break and expand is presented on Figure 5I. It is likely that a deficiency in the structural components of the replisome such as Polα-Primase, Polε, Polδ, RFC, PCNA, MCM, and Tof1-Csm3-Mrc1, results in the generation of long single-stranded regions on the template for lagging or leading strand synthesis. This creates optimal conditions for triplex formation. Conceivably, depletion of Dna2, Fen1, and PCNA impairs Okazaki fragment maturation and leaves an unprocessed flap that could be folded into a triplex. The secondary structure may be destroyed by components of the replication machinery such as the single-strand DNA-binding protein RPA (Figure 5Ia). Otherwise, the triplex could create a strong block for DNA synthesis. Consistent with this, using 2D gel analysis we found that in TET-RFA2 strains that exhibit the highest levels of fragility and expansions, replication progression through (GAA)120 tract is impaired (Figure S2). Triplex or triplex-arrested fork might be sensed and removed by the Rtt101-Mms1-Mms22 complex. Because Tof1-Csm3-Mrc1 complex also carries out checkpoint function (reviewed in Tourrière and Pasero, 2007), we can not exclude that triplex-mediated arrest can also be under the surveillance of this replication-pausing machinery. If undetected, the triplex can cause template switch resulting in expansions (Figure 5Ib). Alternatively, the secondary structure can be attacked by nucleases resulting in DSBs (Figure 5Ic).
Figure 5. Model for GAA/TTC Fragility and Expansion in Dividing and Nondividing Cells.

(I) Replication-associated pathway for GAA/TTC (red line) instability. Hoogsteen base pairing in the triplex is indicated as blue dots. DNA replication helicase (green ring), DNA polymerase (solid gray oval), and attacking nuclease (solid orange pacman) are shown.
(II) Transcription-associated pathway for GAA/TTC instability. Transcription initiation factors (solid brown ovals), the newly synthesized RNA (blue line) forming abnormal RNA-DNA hybrid (R-loop), and RNA polymerase II (solid purple ovals) are depicted. A detailed description is presented in the text. See also Figure S2.
From the list of mutants associated with DNA replication that affect GAA/TTC stability it is important to pinpoint the deficiency in the ORC complex. The ORC complex is required for the initiation of DNA replication but it does not travel along with the replisome (reviewed in Prasanth et al., 2004). The increase in breakage and expansions observed in these mutants can be explained in several ways. First, the ORC complex might be loaded at the GAA/TTC tract. This interaction could be promoted either by exposure of ssDNA due to triplex structure formation or the AT rich nature of the repeats. Interestingly, it has been demonstrated in Cos-1 cells that GAA/TTC tract can stimulate initiation of alternative replication on plasmid (Chandok et al., 2012). Based on this observation, it is conceivable that binding of the OCR complex might destroy the triplex structure thereby stabilize the repeats. Second, ORC deficiency might lead to the assembly of a noncanonical and faulty replisome. Improper replication progression can result in the accumulation of persistent single-stranded regions where triplex structures can be formed. Third, in orc mutants, the GAA/TTC locus might not be replicated from the closest active origin, ARS507. It is possible that the replication machinery traveling from distant origins might be compromised and less processive. This can mimic what is happening when structural components of the replisome are hampered. The third explanation echoes data obtained in human cells where it was shown that fragility of FRA3B and FRA16D sites is suppressed when replication is initiated in these regions and is increased when the fragile sites are replicated by forks traveling from distant origins (Letessier et al., 2011).
Defect in Telomere Maintenance
Somewhat surprisingly, in strains carrying ten1-DAmP, TET-TEN1, and TET-CDC13 alleles, rates of GAA/TTC fragility and expansions increased. Cdc13, Stn1, and Ten1 form a complex that plays a role in chromosome end protection, telomere replication, and regulation of telomerase (reviewed in Giraud-Panis et al., 2010). In cdc13 mutants, telomere uncapping leads to C-rich strand resection, accumulation of single-stranded DNA and activation of the Rad53 and Chk1-dependent checkpoint response (reviewed in Longhese, 2008). One possible explanation among others for the effect of telomere dysfunction on repeat instability could be the sequestration of the replication-pausing checkpoint complex to the uncapped telomeres to counteract Exo1-mediated resection. It has been shown that telomere length is decreased in Δmrc1 mutants; and Δmrc1cdc13-1, Δtof1cdc13-1, or Δcsm3cdc13-1 double mutants show a synthetic growth defect (Grandin and Charbonneau, 2007; Tsolou and Lydall, 2007). Moreover, the growth defect and the amount of degradation are reduced in Δmrc1cdc13-1 strains when Exo1 nuclease is removed (Tsolou and Lydall, 2007). These data indicate that the Mrc1-Tof1-Csm3 complex protects uncapped telomeres by inhibiting DNA resection. Hence, it is possible that telomere uncapping can sequester the replication-pausing complex from the replication fork to single-stranded regions in ten1-DAmP, TET-TEN1, and TET-CDC13 strains thereby creating a paucity of Mrc1-Tof1-Csm3 in the replisome. As discussed above, this deficiency can cause accumulation of single-stranded DNA regions at the replication fork or compromise checkpoint response when the replisome encounters the triplex barrier. Also, we can not exclude the possibility of Ten1-Stn1-Cdc13 binding the repeats or the triplex structure and influencing the tract stability.
Defect in Transcription Initiation
Transcription initiation mutants are the second largest group of mutants that predispose repeats for both breakage and large-scale expansions. In yeast, transcription initiation and start site selection by RNA polymerase II require the activity of several general transcription factors, including TFIIA, TFIIB, TFIID, and TFIIF complexes. TFIID is specifically important for transcription from TATA-less promoters that contain AT-rich regulatory sequences (reviewed in Hahn and Young, 2011). GAA/TTC tracts, similar to poly-AT sequences, bind nucleosomes poorly (Ruan and Wang, 2008; Russell et al., 1983; Struhl, 1985) and thus likely generate nucleosome-depleted regions, a feature of many sites in yeast chromosomes where transcription is initiated (Nagalakshmi et al., 2008; Xu et al., 2009). In this study, we found that Sua7 binds to GAA/TTC tracts (Figure S1D). Moreover, GAA/TTC repeats serve as a promoter element and drive transcription of the TRP1 ORF. These data demonstrate that in yeast, GAA/TTC sequences are target sites for binding of the transcription factors that trigger mRNA synthesis. Interestingly, although the underlying mechanism is not known, in Mycoplasma gallisepticum, GAA/TTC tracts located upstream of the putative promoter for the M9 gene also promote transcription in a repeat length-dependent manner (Glew et al., 2000 and references therein). In human fibroblast cells, FXN antisense transcript was detected, which is elevated in FRDA patients with expanded alleles, suggesting that these repeats might play a role in transcription initiation (De Biase et al., 2009).
We found that fragility, in contrast to expansions, occurs in nondividing cells that are preferentially arrested at the G1 stage of the cell cycle. This observation is consistent with data obtained by Tang et al. (2011), who demonstrated that mitotic crossover induced by GAA tracts in diploid cells is likely due to DSB at the G1 stage. Moreover, we showed that fragility and expansions are elevated in transcription initiation mutants during active division and fragility is further amplified in stationary phase. How can a deficiency in transcription initiation machineries destabilize repeat tracts? One possible explanation is that triplex formation outside of the S phase, likely during the G1 stage of the cell cycle, can be counteracted by transcription initiation factors binding to the GAA/TTC region. In mutants defective in transcription initiation, the triplex can persist and be attacked by a nuclease leading to DSB. If the structure survives until S phase, template switch (as described above) could result in large-scale expansions. This model leaves the question of how a triplex can be formed in nondividing cells and does not explain why breaks accumulate whereas cells are arrested in the stationary phase. Another explanation that accommodates results from other studies includes accelerated accumulation of R-loops in the GAA/TTC region when transcription initiation is abnormal (Figure 5II). It has been shown that GAA/TTC tracts are poor substrates for RNA polymerase II progression (Grabczyk and Usdin, 2000; Krasilnikova et al., 2007) and readily accumulate recombinagenic R-loops (McIvor et al., 2010; Reddy et al., 2011). A lack of components of TFIIA, TFIIB, TFIID, and TFIIF complexes can lead to the loading of the hampered transcription elongation machinery. In mutants, abortive transcription can lead to futile cycles of RNA synthesis inside GAA/TTC tracts, RNA polymerase trapping, and more efficient R-loop production culminating in increased DNA breakage in nondividing cells (Figure 5IIa). In dividing cells, trapped RNA polymerase has been shown to be an obstacle for replication progression (Mirkin et al., 2006). Collision of the replication machinery with RNA polymerase at the noncanonical promoter can cause removal of this barrier and unwinding of R-loops (Figure 5IIb). Alternatively, replication bypass via template switch can generate large-scale expansions (Figure 5IIc) or the block and persistent R-loops can culminate in DSBs (Figure 5IId). There are several observations that are in agreement with this model. First, breaks are higher in cells arrested in stationary phase than in cells that actively divide (Figure 3). This indicates that intact replication acts as a preventing force for fragility. Second, there is a time-dependent accumulation of GAA/TTC-mediated DSBs during stationary phase. This points toward a dynamic process promoting fragility where transcription is the most likely mechanism. Third, unlike fragility, large-scale expansions do not occur in nondividing cells and require DNA replication (Figure 4). Fourth, increase in the size of repeats from 20 to 400 leads to a decrease in TRP1 expression level, indicating that longer tracts are more problematic templates for RNA polymerase progression, or longer R-loops prevent efficient translation.
Conclusions and Implications for the Human Genome Stability
We found several pathways that determine GAA/TTC stability and operate in dividing and nondividing yeast cells. Intact replication machinery is required to prevent increased breakage and expansion of the repeat tracts in dividing cells. In humans, it would indicate that carriers of the hypomorphic alleles of replication-associated genes should be highly predisposed for acquiring GAA/TTC expansions as well as for repeat-mediated- chromosomal breakage and rearrangements. Remarkably, we found that proper origin firing and telomere metabolism are also important for maintaining stability of the repeats. Dysfunctions of these processes can contribute to tissue- and cell-dependent instability in human cells. It is becoming evident that cells from different tissue types differ in replication initiation patterns and replication timing (Hansen et al., 2010; Ryba et al., 2010). Therefore, the potential of GAA/TTC tracts for instability can be dependent on the location of the repeat tract in the particular tissue-specific replicon. We also propose that GAA/TTC stability should be compromised in cells experiencing telomere crisis such as aging or cancer cells. Our finding that GAA/TTC tracts attract transcription factors and promote mRNA synthesis has dual implications for the metabolism of repeat tracts in human cells. First, R-loop formation in the expanded tracts might not require GAA/TTC region to be transcribed from an outside promoter. This conjecture also suggests that if tracts are located inside an ORF, GAA/TTC repeats can produce antisense RNA. As proposed by Tufarelli (2006) and McIvor et al. (2010), R-loops and/or antisense transcripts can trigger silencing and might explain accumulation of repressing chromatin markers in regions that flank expanded GAA/TTC repeats (reviewed in Kumari and Usdin, 2012). Second, unlike yeast, in human cells, transcription initiation factors such as Taf proteins can be coded by several genes where particular alleles are expressed in different tissues during differentiation and development (D’Alessio et al., 2009). If these alternate complexes have a different efficiency of transcription initiation in GAA/TTC tracts, this can also contribute to tissue-dependent stability of the repeats.
In humans, repeats can expand in nondividing neuronal cells (De Biase et al., 2007a) and in confluent embryonic kidney cell lines where it was demonstrated that transcription promotes expansions (Ditch et al., 2009). We found that although fragility of repeats is increased in nondividing yeast cells and involves transcription process, large-scale expansions require cell proliferation. The question that remains to be answered is whether transcription-associated fragility in nondividing cells can result in large-scale expansions. The obvious difference between the yeast system used in this study and human cells is ploidy. Whether fragility in arrested yeast cells promotes expansions when a homologous chromosome carrying an additional copy of GAA/TTC tracts is present remains to be tested. The identification of the nuclease that creates breaks in repeats in nondividing cells is also an important avenue for future studies.
EXPERIMENTAL PROCEDURES
Yeast Strains
The three collections of tester strains: yTHC, DAmP, YKO strains, were purchased from Open Biosystems. The genotype of the query strain, HMK246, used in the screen is: MATα, ura3-Δ, leu2-Δ, his3-Δ, lys2-Δ, rpl28-Q38K, mfa1Δ::MFA1pr-HIS3, V34205::lys2::(GAA)230, V29617::hphMX. The strains whose fragility was assessed have the following genotype: MATa, bar1-Δ, trp1-Δ, his3-Δ, ura3-Δ, leu2-Δ, ade2-Δ, lys2-Δ, V34205::ADE2, lys2::(GAA)n. The details of the constructions of all strains used in this study are given in the Supplemental Experimental Procedures.
Schematics of the Genome-wide Screen
The scheme was adapted from Tong et al. (2001) with modifications. MATα query strains marked with hphMX and MATa tester strains carrying kanMX were patched on YPD plates overnight. Query strains were patched in duplicates. Strains were crossed on YPD overnight and selected on medium containing 300 mg/l G418 and 300 mg/l hygromycin B for diploids. Sporulation was induced by replica plating cells to sporulation medium for 5 days. Yeast were transferred to histidine drop-out plates supplemented with 5 mg/l cycloheximide for 2 days. Because the query strain carries a mating type-specific reporter MFA1pr-HIS3 and a recessive mutation rpl28-Q38K that provides resistance to cycloheximide, this procedure allows selecting for MATa haploid progeny. This step was repeated to assure the selection of haploids. Haploids harboring both GAA/TTC tracts and mutated genes were selected on medium containing G418 and hygromycin B. Cells were then either replica plated to canavanine plates (for YKO, yTHC, and DAmP libraries), or first to medium containing 2 μg/ml doxycycline and then to canavanine plates (for yTHC library). After incubation for 3 days, the effect of the mutants was evaluated based on the amount of revertants on canavanine plates compared with the wild-type level.
Measurement of GAA/TTC Fragility and Expansion Rates
Strains were grown on YPD plates for 3 days; a minimum of 14 independent colonies for each strain were taken for fluctuation tests to calculate the rates of fragility or expansions. Because GAA/TTC repeats are prone to size variations, only the colonies with the correct tract sizes (prescreened by PCR) were used for calculation. See details in the Supplemental Experimental Procedures.
To monitor GAA/TTC fragility and expansion in actively dividing and nondividing cells, approximately five yeast cells were inoculated in 5 ml YPD. Samples were taken at indicated time-points for fluctuation tests. A minimum of eight cultures were used in the experiments for each strain. Frequencies instead of rates were compared between the actively dividing and arrested cells.
DSB Detection
Cells were embedded into agarose plugs (0.8% agarose) at a concentration of 24 × 108 cells/ml. Contour-clamped homogeneous electric field (CHEF) gel was used to separate the broken left arm of chromosome V from the intact chromosome V. Chromosomes were separated at 14°C in a 1% agarose gel in 0.5× Tris/Borate/EDTA (TBE) for 28 hr at 6 V/cm (Bio-Rad CHEF Mapper XA). The included angle was 120°, the initial and final switching times were 12.56 s and 17.35 s, respectively. The gel was transferred to a nylon membrane at 6 V in 0.5× TBE for 5 hr using the Genie electrophoresis blotter (Idea Scientific). HPA3-specific probe was used for Southern hybridization at 67°C overnight. The membrane was washed twice in washing buffer (0.1× SSC, 0.1% SDS) at 70°C and was exposed to Kodak BioMax film. The hybridization signals were quantified using the Carestream Molecular Imaging System software.
Analysis of the Ability of GAA/TTC Repeats to Serve as Promoter Elements
Strains were grown on YPD for 3 days. The sizes of the repeat tracts were verified before 10-fold serial dilutions were spotted on plates. The plates were incubated at 30°C for 1 day on SDC and -Trp media and for 4 days on 0.75 g/l 5-FAA-containing media.
2D Gel Analysis, ChIP, and qPCR
Replication intermediates were detected using 2D gel analysis and southern blot hybridization. ChIP and qPCR were employed to determine binding of Sua7 to the GAA/TTC tracts. All details for these procedures are described in the Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
We thank Dr. N. Degtyareva, Dr. B. Hammer, and Z. Sheng for critical reading of the manuscript and helpful discussions. This work was supported by award number R01GM0825950 from NIGMS/NIH and MCB-0818122 from NSF to K.S.L. and R01GM60987 from NIGMS/NIH to S.M.M. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIGMS/NIH or the NSF.
References
- Aguilera A, Gómez-González B. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet. 2008;9:204–217. doi: 10.1038/nrg2268. [DOI] [PubMed] [Google Scholar]
- Bellí G, Garí E, Aldea M, Herrero E. Functional analysis of yeast essential genes using a promoter-substitution cassette and the tetracycline-regulatable dual expression system. Yeast. 1998;14:1127–1138. doi: 10.1002/(SICI)1097-0061(19980915)14:12<1127::AID-YEA300>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Belotserkovskii BP, Liu R, Tornaletti S, Krasilnikova MM, Mirkin SM, Hanawalt PC. Mechanisms and implications of transcription blockage by guanine-rich DNA sequences. Proc Natl Acad Sci USA. 2010;107:12816–12821. doi: 10.1073/pnas.1007580107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourn RL, Rindler PM, Pollard LM, Bidichandani SI. E. coli mismatch repair acts downstream of replication fork stalling to stabilize the expanded (GAA.TTC)(n) sequence. Mutat Res. 2009;661:71–77. doi: 10.1016/j.mrfmmm.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- Chandok GS, Patel MP, Mirkin SM, Krasilnikova MM. Effects of Friedreich’s ataxia GAA repeats on DNA replication in mammalian cells. Nucleic Acids Res. 2012;40:3964–3974. doi: 10.1093/nar/gks021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RM, Dalgliesh GL, Endres D, Gomez M, Taylor J, Bidichandani SI. Expansion of GAA triplet repeats in the human genome: unique origin of the FRDA mutation at the center of an Alu. Genomics. 2004;83:373–383. doi: 10.1016/j.ygeno.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Clark RM, Bhaskar SS, Miyahara M, Dalgliesh GL, Bidichandani SI. Expansion of GAA trinucleotide repeats in mammals. Genomics. 2006;87:57–67. doi: 10.1016/j.ygeno.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Clark RM, De Biase I, Malykhina AP, Al-Mahdawi S, Pook M, Bidichandani SI. The GAA triplet-repeat is unstable in the context of the human FXN locus and displays age-dependent expansions in cerebellum and DRG in a transgenic mouse model. Hum Genet. 2007;120:633–640. doi: 10.1007/s00439-006-0249-3. [DOI] [PubMed] [Google Scholar]
- D’Alessio JA, Wright KJ, Tjian R. Shifting players and paradigms in cell-specific transcription. Mol Cell. 2009;36:924–931. doi: 10.1016/j.molcel.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Biase I, Rasmussen A, Endres D, Al-Mahdawi S, Monticelli A, Cocozza S, Pook M, Bidichandani SI. Progressive GAA expansions in dorsal root ganglia of Friedreich’s ataxia patients. Ann Neurol. 2007a;61:55–60. doi: 10.1002/ana.21052. [DOI] [PubMed] [Google Scholar]
- De Biase I, Rasmussen A, Monticelli A, Al-Mahdawi S, Pook M, Cocozza S, Bidichandani SI. Somatic instability of the expanded GAA triplet-repeat sequence in Friedreich ataxia progresses throughout life. Genomics. 2007b;90:1–5. doi: 10.1016/j.ygeno.2007.04.001. [DOI] [PubMed] [Google Scholar]
- De Biase I, Chutake YK, Rindler PM, Bidichandani SI. Epigenetic silencing in Friedreich ataxia is associated with depletion of CTCF (CCCTC-binding factor) and antisense transcription. PLoS One. 2009;4:e7914. doi: 10.1371/journal.pone.0007914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditch S, Sammarco MC, Banerjee A, Grabczyk E. Progressive GAA.TTC repeat expansion in human cell lines. PLoS Genet. 2009;5:e1000704. doi: 10.1371/journal.pgen.1000704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank-Kamenetskii MD, Mirkin SM. Triplex DNA structures. Annu Rev Biochem. 1995;64:65–95. doi: 10.1146/annurev.bi.64.070195.000433. [DOI] [PubMed] [Google Scholar]
- Giraud-Panis MJ, Teixeira MT, Géli V, Gilson E. CST meets shelterin to keep telomeres in check. Mol Cell. 2010;39:665–676. doi: 10.1016/j.molcel.2010.08.024. [DOI] [PubMed] [Google Scholar]
- Glew MD, Browning GF, Markham PF, Walker ID. pMGA phenotypic variation in Mycoplasma gallisepticum occurs in vivo and is mediated by trinucleotide repeat length variation. Infect Immun. 2000;68:6027–6033. doi: 10.1128/iai.68.10.6027-6033.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabczyk E, Usdin K. The GAA*TTC triplet repeat expanded in Friedreich’s ataxia impedes transcription elongation by T7 RNA polymerase in a length and supercoil dependent manner. Nucleic Acids Res. 2000;28:2815–2822. doi: 10.1093/nar/28.14.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabczyk E, Mancuso M, Sammarco MC. A persistent RNA.DNA hybrid formed by transcription of the Friedreich ataxia triplet repeat in live bacteria, and by T7 RNAP in vitro. Nucleic Acids Res. 2007;35:5351–5359. doi: 10.1093/nar/gkm589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandin N, Charbonneau M. Mrc1, a non-essential DNA replication protein, is required for telomere end protection following loss of capping by Cdc13, Yku or telomerase. Mol Genet Genomics. 2007;277:685–699. doi: 10.1007/s00438-007-0218-0. [DOI] [PubMed] [Google Scholar]
- Hahn S, Young ET. Transcriptional regulation in Saccharomyces cerevisiae: transcription factor regulation and function, mechanisms of initiation, and roles of activators and coactivators. Genetics. 2011;189:705–736. doi: 10.1534/genetics.111.127019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci USA. 2010;107:139–144. doi: 10.1073/pnas.0912402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassai-Jáger E, Ortutay C, Tóth G, Vellai T, Gáspári Z. Distribution and evolution of short tandem repeats in closely related bacterial genomes. Gene. 2008;410:18–25. doi: 10.1016/j.gene.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Kim HM, Narayanan V, Mieczkowski PA, Petes TD, Krasilnikova MM, Mirkin SM, Lobachev KS. Chromosome fragility at GAA tracts in yeast depends on repeat orientation and requires mismatch repair. EMBO J. 2008;27:2896–2906. doi: 10.1038/emboj.2008.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasilnikova MM, Mirkin SM. Replication stalling at Friedreich’s ataxia (GAA)n repeats in vivo. Mol Cell Biol. 2004;24:2286–2295. doi: 10.1128/MCB.24.6.2286-2295.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasilnikova MM, Kireeva ML, Petrovic V, Knijnikova N, Kashlev M, Mirkin SM. Effects of Friedreich’s ataxia (GAA)n*(TTC)n repeats on RNA synthesis and stability. Nucleic Acids Res. 2007;35:1075–1084. doi: 10.1093/nar/gkl1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari D, Usdin K. Is Friedreich ataxia an epigenetic disorder? Clin. Epigenetics. 2012;4:2. doi: 10.1186/1868-7083-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letessier A, Millot GA, Koundrioukoff S, Lachagès AM, Vogt N, Hansen RS, Malfoy B, Brison O, Debatisse M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature. 2011;470:120–123. doi: 10.1038/nature09745. [DOI] [PubMed] [Google Scholar]
- Lindstrom DL, Squazzo SL, Muster N, Burckin TA, Wachter KC, Emigh CA, McCleery JA, Yates JR, 3rd, Hartzog GA. Dual roles for Spt5 in pre-mRNA processing and transcription elongation revealed by identification of Spt5-associated proteins. Mol Cell Biol. 2003;23:1368–1378. doi: 10.1128/MCB.23.4.1368-1378.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhese MP. DNA damage response at functional and dysfunctional telomeres. Genes Dev. 2008;22:125–140. doi: 10.1101/gad.1626908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIvor EI, Polak U, Napierala M. New insights into repeat instability: role of RNA•DNA hybrids. RNA Biol. 2010;7:551–558. doi: 10.4161/rna.7.5.12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. DNA end resection: many nucleases make light work. DNA Repair (Amst ) 2009;8:983–995. doi: 10.1016/j.dnarep.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin EV, Castro Roa D, Nudler E, Mirkin SM. Transcription regulatory elements are punctuation marks for DNA replication. Proc Natl Acad Sci USA. 2006;103:7276–7281. doi: 10.1073/pnas.0601127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard LM, Sharma R, Gómez M, Shah S, Delatycki MB, Pianese L, Monticelli A, Keats BJ, Bidichandani SI. Replication-mediated instability of the GAA triplet repeat mutation in Friedreich ataxia. Nucleic Acids Res. 2004;32:5962–5971. doi: 10.1093/nar/gkh933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanth SG, Méndez J, Prasanth KV, Stillman B. Dynamics of pre-replication complex proteins during the cell division cycle. Philos Trans R Soc Lond B Biol Sci. 2004;359:7–16. doi: 10.1098/rstb.2003.1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy K, Tam M, Bowater RP, Barber M, Tomlinson M, Nichol Edamura K, Wang YH, Pearson CE. Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Res. 2011;39:1749–1762. doi: 10.1093/nar/gkq935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rindler PM, Bidichandani SI. Role of transcript and interplay between transcription and replication in triplet-repeat instability in mammalian cells. Nucleic Acids Res. 2011;39:526–535. doi: 10.1093/nar/gkq788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan H, Wang YH. Friedreich’s ataxia GAA.TTC duplex and GAA.GAA.TTC triplex structures exclude nucleosome assembly. J Mol Biol. 2008;383:292–300. doi: 10.1016/j.jmb.2008.08.053. [DOI] [PubMed] [Google Scholar]
- Russell DW, Smith M, Cox D, Williamson VM, Young ET. DNA sequences of two yeast promoter-up mutants. Nature. 1983;304:652–654. doi: 10.1038/304652a0. [DOI] [PubMed] [Google Scholar]
- Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, Schulz TC, Robins AJ, Dalton S, Gilbert DM. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010;20:761–770. doi: 10.1101/gr.099655.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishkin AA, Voineagu I, Matera R, Cherng N, Chernet BT, Krasilnikova MM, Narayanan V, Lobachev KS, Mirkin SM. Large-scale expansions of Friedreich’s ataxia GAA repeats in yeast. Mol Cell. 2009;35:82–92. doi: 10.1016/j.molcel.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soragni E, Herman D, Dent SY, Gottesfeld JM, Wells RD, Napierala M. Long intronic GAA*TTC repeats induce epigenetic changes and reporter gene silencing in a molecular model of Friedreich ataxia. Nucleic Acids Res. 2008;36:6056–6065. doi: 10.1093/nar/gkn604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K. Naturally occurring poly(dA-dT) sequences are upstream promoter elements for constitutive transcription in yeast. Proc Natl Acad Sci USA. 1985;82:8419–8423. doi: 10.1073/pnas.82.24.8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Dominska M, Greenwell PW, Harvanek Z, Lobachev KS, Kim HM, Narayanan V, Mirkin SM, Petes TD. Friedreich’s ataxia (GAA)n•(TTC)n repeats strongly stimulate mitotic crossovers in Saccharomyces cerevisiae. PLoS Genet. 2011;7:e1001270. doi: 10.1371/journal.pgen.1001270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Pagé N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- Tourrière H, Pasero P. Maintenance of fork integrity at damaged DNA and natural pause sites. DNA Repair (Amst ) 2007;6:900–913. doi: 10.1016/j.dnarep.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Tsolou A, Lydall D. Mrc1 protects uncapped budding yeast telomeres from exonuclease EXO1. DNA Repair (Amst ) 2007;6:1607–1617. doi: 10.1016/j.dnarep.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufarelli C. The silence RNA keeps: cis mechanisms of RNA mediated epigenetic silencing in mammals. Philos Trans R Soc Lond B Biol Sci. 2006;361:67–79. doi: 10.1098/rstb.2005.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetcher AA, Wells RD. Sticky DNA formation in vivo alters the plasmid dimer/monomer ratio. J Biol Chem. 2004;279:6434–6443. doi: 10.1074/jbc.M309595200. [DOI] [PubMed] [Google Scholar]
- Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Münster S, Camblong J, Guffanti E, Stutz F, Huber W, Steinmetz LM. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009;457:1033–1037. doi: 10.1038/nature07728. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.