

Abstract
The Sir2 family of enzymes or sirtuins are known as nicotinamide adenine dinucleotide (NAD)-dependent deacetylases1 and have been implicated in the regulation of transcription, genome stability, metabolism, and lifespan2, 3. However, four of the seven mammalian sirtuins have very weak deacetylase activity in vitro. Here we show that human Sirt6 efficiently removes long chain fatty acyl groups, such as myristoyl, from lysine residues. The crystal structure of Sirt6 reveals a large hydrophobic pocket that can accommodate long chain fatty acyl groups. We demonstrate further that Sirt6 promotes the secretion of tumor necrosis factor α (TNFα) by removing the fatty acyl modification on K19 and K20 of TNFα. Protein lysine fatty acylation has been known to occur in mammalian cells, but the function and regulatory mechanisms of this modification were unknown. Our data suggest that protein lysine fatty acylation is a novel mechanism that regulates protein secretion. The discovery of Sirt6 as an enzyme that controls protein lysine fatty acylation provides new opportunities to investigate the physiological function of the previously ignored protein posttranslational modification.
It was recently demonstrated that Sirt5, one of the four sirtuins with weak deacetylase activity, preferentially hydrolyze succinyl and malonyl lysine.4, 5 The discovery of novel Sirt5 activity suggested that other sirtuins with weak deacetylase activity may utilize alternative substrates as well. We therefore set out to investigate whether Sirt6 has any new enzymatic activity. Sirt6 has been reported to be important for DNA repair, transcriptional regulation of genes important for metabolism and immune responses, and for life span.6–10 In most cases, the biological functions have been linked to the sequence-specific deacetylase activity of Sirt6 on histone H3 K9 and K56, but not other peptide sequences.9, 11, 12 The deacetylase activity of Sirt6 on H3 K9 and K56 can indeed be detected in our assay. However, the catalytic efficiency is low (Table 1), suggesting that the deacetylase activity may only account for a part of its biological function.
Table 1.
Catalytic efficiencies of Sirt6 on different acyl peptides.
| acyl peptide | kcat (s−1) | Km (μM) | Kcat/Km (s−1M−1) |
|---|---|---|---|
| H3 K9 acetyl | 0.0039 ± 0.0006 | 810 ± 160 | 4.8 |
| H3 K9 butyryl | 0.0021 ± 0.0004 | 200 ± 120 | 10 |
| H3 K9 octanoyl | 0.0046 ± 0.0005 | 40 ± 10 | 1.2 × 102 |
| H3 K9 myristoyl | 0.0049 ± 0.0004 | 3.4 ± 0.9 | 1.4 × 103 |
| H3 K9 palmitoyl | 0.0027 ± 0.0002 | 0.9 ± 0.4 | 3.0 × 103 |
| TNF K19 myristoyl | 0.0020 ± 0.0002 | 2.4 ± 0.6 | 8.3 × 102 |
| TNF K20 myristoyl | 0.0050 ± 0.0004 | 4.5 ± 1.1 | 1.1 × 103 |
To discover possible novel activity of Sirt6, we synthesized H3 K9 peptides with different acyl groups (acetyl, malonyl, succinyl, butyryl, myristoyl, and palmitoyl) and assayed these peptides with recombinant Sirt6. Sirt6 can hydrolyze long chain fatty acyl groups efficiently (Figure 1A). The Kcat/Km for demyristoylation (1400 s−1M−1) is ~300-fold better than that for deacetylation (4.8 s−1M−1). The increased catalytic efficiency comes mainly from the decrease in Km. For deacetylation, the Km is 810 μM, but for demyristoylation, the Km is 3.4 μM. The activity of Sirt6 (a Class IV sirtuin) is similar to that of PfSir2A,5 which was classified as a Class III sirtuin13 but lacks the conserved Arg and Tyr residues that recognize negatively charge succinyl or malonyl groups.4 This emphasizes that it is important to examine the activity of each sirtuin experimentally as bioinformatics predictions may not be sufficient.
Figure 1.

Sirt6 prefers to hydrolyze long chain fatty acyl lysine in vitro. (A) HPLC traces showing Sirt6-catalyzed hydrolysis of different acyl peptides based on the H3K9 sequence. (B) H2B K12 myristoyl peptide can be hydrolyzed by Sirt6 while the corresponding acetyl peptide cannot. Reactions were carried out with 50 μM peptide, 1 μM Sirt6, 20 mM Tris pH 8.0, 0.5 mM NAD, and 1 mM DTT at 37°C for 30 min.
The conclusion that Sirt6 is better at hydrolyzing long chain fatty acyl groups was also supported by its ability to hydrolyze myristoyl group from other peptide sequences. The deacetylase activity of Sirt6 was reported to be sequence-specific. Only H3 K9 and H3 K56 acetyl peptides, but not other peptides, were reported to be deacetylated by Sirt6.9, 11, 12 To test whether the more efficient demyristoylation activity allows Sirt6 to utilize more peptide substrates, we synthesized acetyl and myristoyl peptides based on the H2B K12 sequence. Consistent with previous report, the hydrolysis of the acetyl peptide by Sirt6 was undetectable. In contrast, the hydrolysis of the corresponding myristoyl peptide could be readily detected (Figure 1B). These results further support Sirt6 prefers long chain fatty acyl groups.
To determine the structural basis for enhanced Sirt6 activity with fatty acyl peptides, we obtained a crystal structure of Sirt6 in complex with a H3 K9 myristoyl peptide and ADP-ribose (ADPR) at 2.2 Å resolution (PDB 3ZG6, Figure 2A, Figure S1). The overall structure is similar to the published Sirt6 structure28 (PDB 3K35) (Figure S1). Residues 2–10 and 166–174 of Sirt6 are visible in the H3 K9 myristoyl bound structure, while the corresponding regions are missing in the published Sirt6 structure without peptide bound 28 (Figure S2). The way that H3 K9 myristoyl and ADPR bind to Sirt6 is similar to that seen in other ternary complex structures of other sirtuins, such as Sir2Tm in complex with the p53 acetyl peptide and NAD (Figure S3).14 The peptide interacts with Sirt6 via many hydrogen bonding interactions. Similar to what was observed with other sirtuins, including Sirt5, most of the hydrogen bonds come from main chain C=O and N-H of the myristoyl peptide, with the only side chain hydrogen-bonding coming from Trp11 (Figure S4). Therefore, the selectivity for peptide sequences is not high, which is consistent with our enzymology data. The myristoyl group is located in a hydrophobic pocket (Figure 2B) formed by hydrophobic residues from several flexible loops, including Ala11, Pro60, Phe62, Trp69, Pro78, Phe80, Phe84, Val113, Leu130, Leu184, and Ile217. The substrate-binding sites in other available human sirtuin structures do not possess such a big hydrophobic pocket (Figure S5). The structural data thus are consistent with the biochemical data and provide a reasonable explanation for Sirt6’s preference for long chain fatty acyl groups.
Figure 2.
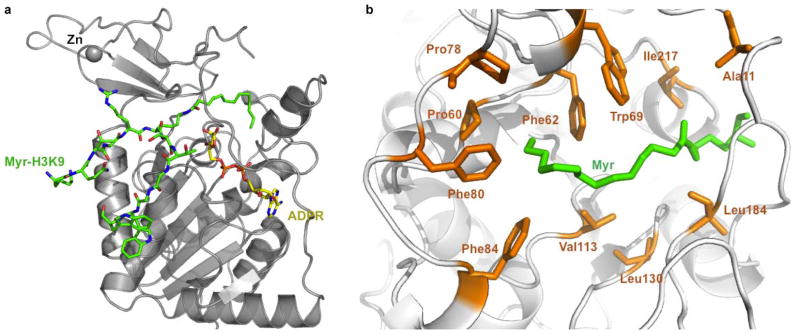
Structure basis for Sirt6 activity with long chain fatty acyl groups. (A) Overall structure of Sirt6 with myristoyl H3K9 (Myr-H3K9, green) peptide and ADP-ribose (ADPR, yellow) bound. (B) Hydrophobic residues in Sirt6 that accommodate the myristoyl (Myr) group.
The next important question was whether this activity is physiologically relevant. There are a few proteins known to be modified by fatty acyl groups on lysine, although the function of the modification is unclear.15, 16 One of the fatty acylated proteins is TNFα,16 a type II membrane protein with a single transmembrane domain linking the N-terminal intracellular domain and C-terminal extracellular domain.17 When cleaved by proteases, the extracellular domain is released. The released TNFα can then bind its receptors and induce various signaling pathways.17 It was reported that Sirt6 can regulate the synthesis of TNFα, but the mechanism was unclear.18, 19 We hypothesized that Sirt6 may regulate TNFα secretion via defatty-acylation.
To test this, we measured the fatty acylation level on TNFα in Sirt6 wild type (WT) and knockout (KO) mouse embryonic fibroblast (MEF) cells. Flag-tagged TNFα was transfected into the cells. The cells were cultured in the presence of an alkyne-tagged fatty acid analog, Alk14, which can covalently label fatty acylated proteins.20, 21 TNFα was immunoprecipitated and conjugated to rhodamine-azide (Rh-N3) using click chemistry (Figure 3A). A protein will be fluorescently labeled if it is fatty acylated by Alk14. Because Alk14 can also label cysteine residues,21, 22 we treated TNFα with hydroxylamine to remove cysteine fatty acylation (Figure 3A). As shown in Figure 3B, TNFα from Sirt6 KO MEF cells had significantly increased Alk14 labeling compared to TNFα from Sirt6 WT MEF cells, suggesting that Sirt6 regulates the fatty acylation level on TNFα. When human Sirt6 WT was overexpressed in Sirt6 KO MEF cells, TNFα had lower fatty acylation level than TNFα from cells without overexpression of human Sirt6, while overexpression of human Sirt6 H133Y catalytic mutant did not have much effect on TNFα fatty acylation (Figure 3C), suggesting that enzymatic activity of Sirt6 is required for controlling TNFα fatty acylation.
Figure 3.

Sirt6 regulates TNFα fatty acylation and secretion. (A) Method of using Alk14 to detect TNFα fatty acylation. (B) Sirt6 controls TNFα fatty acylation on K19 and K20. (C) H133 of Sirt6 is required for TNFα defatty-acylation. (D) Sirt6 defatty-acylates TNFα in vitro. (E) Sirt6 regulates secretion of TNFα in MEF cells. n = 6; ***P < 0.001. (F-I) Sirt6 regulates fatty acylation level and secretion of endogenous TNFα in THP-1 cells (F and G; n = 3; **P < 0.01) and bone marrow-derived macrophages (H and I; n = 5; *P < 0.02). Secretion data were expressed as mean ± sd.
To further confirm that fatty acylation occur on lysine residues of TNFα, we mutated K19 and K20, the reported sites of myristoylation on TNFα,16 to Arg (labeled as “KR” mutant in Figure 3B). The labeling intensities of the mutant from Sirt6 KO MEF cells dropped to background level (Figure 3B), confirming that K19 and K20 are the major fatty acylation sites. Furthermore, TNFα isolated from Sirt6 KO MEF cells can be defatty-acylated in vitro in an NAD-dependent manner (Figure 3D). Synthetic TNFα K19 and K20 myristoyl peptides can be efficiently hydrolyzed by Sirt6 (Table 1). To rule out that TNFα could also be regulated by lysine acetylation, we examined the acetylation level using a pan-specific acetyl lysine antibody. No acetylation was detected on K19 and K20 of TNFα (Figure S6). Thus, Sirt6 regulates the fatty acylation level, but not acetylation level on K19 and K20 of TNFα.
We next investigated the function of TNFα fatty acylation on K19 and K20. It was reported that the amount of TNFα detected in the media of Sirt6 KO cells is less than that in the media of Sirt6 WT cells19. This was attributed to the regulation of TNFα synthesis by Sirt619. Given that Sirt6 regulates lysine fatty acylation, we hypothesized Sirt6 may regulate the secretion of TNFα. To test this, Flag-tagged TNFα was transiently transfected into Sirt6 WT and KO MEF cells. The amount of secreted TNFα in the medium and the amount of TNFα in the cells were measured by ELISA (Figure S7). The percentage of secreted TNFα was then calculated. TNFα secretion efficiency was lower in Sirt6 KO MEF cells than in Sirt6 WT MEF cells (Figure 3E). The secretion efficiency is due to lysine fatty acylation because the secretion efficiency of TNFα KR mutant was not affected by Sirt6 knockout. These data suggested that lysine fatty acylation regulates TNFα secretion and Sirt6 promotes TNFα secretion by defatty-acylation.
We further investigated whether endogenous TNFα are also regulated by Sirt6. For this purpose, we used both human THP-1 cells and bone marrow-derived mouse macrophages. Two Sirt6 knockdown (KD) THP-1 cell lines and one control KD THP-1 cell line were generated (Figure 3F). TNFα from Sirt6 KD cells contained more fatty acylation than TNFα from the control KD cells (Figure 3F). The amount of secreted TNFα in the medium and the amount of TNFα in the cells were measured (Figure S8) and the percentage of secreted TNFα was calculated (Figure 3G). The secretion efficiency of TNFα was lower in Sirt6 KD THP-1 cells than in control KD cells, especially when the cells were supplemented with palmitic acid (Figure 3G). Similar results were obtained for TNFα in mouse macrophages. TNFα in Sirt6 WT macrophages had less lysine fatty acylation than in Sirt6 KO macrophages (Figure 3H), while TNFα secretion efficiency in Sirt6 WT macrophages was higher than in Sirt6 KO macrophages (Figure 3I).
Sirt6 was reported to be mainly localized in the nucleus. The regulation of TNFα secretion by Sirt6 suggested that Sirt6 might be present in secretive organelles, such as the endoplasmic reticulum (ER). We indeed detected Sirt6 in the ER fraction of MEF and THP-1 cell lysates (Figure S9). In Sirt6 KD THP-1 cells or Sirt6 KO MEF cells, less or no Sirt6 was detected in the ER fraction compared with Sirt6 WT cells (Figure S9). The existence of Sirt6 in ER provided further supports for the conclusion that Sirt6 regulates TNFα secretion through defatty-acylation of TNFα.
In summary, our enzymological and structural studies suggest that human Sirt6, which has weak deacetylase activities in vitro, catalyzes the hydrolysis of fatty acyl lysine modifications more efficiently. Sirt6 regulates the fatty acylation level on K19 and K20 of TNFα and this modulates the secretion of TNFα. It has been reported that Sirt6 regulates the acetylation level on histone H3 K9 and K56. Our results are not in conflict with these reports because it is possible that the association with chromatin can regulate the activity of Sirt6. Alternatively, fatty acylation of lysine residues may occur to histones21, 22 and Sirt6 can regulate both acetylation and fatty acylation of histones. The significance of our finding is several folds. First, it reveals a novel physiological activity for Sirt6. Second, it demonstrated for the first time that lysine fatty acylation, a previously ignored protein posttranslational modification, plays an important role in regulating protein secretion. Other proteins (Figure S10), such as insulin-like growth factor 1 (IGF1) may be regulated by similar mechanisms6, 23. Third, the discovery of Sirt6 activity on protein lysine fatty acylation provides an avenue to further investigate this under-recognized protein modification15, 16 and may reveal interesting connections to the well-known cysteine palmitoylation and N-terminal glycine myristoylation24.
Methods
Reagents
Mouse monoclonal anti-Flag M2 antibody conjugated with HRP, anti-FlagM2 affinity gel, and human/mouse Sirt6 antibody (S4322) were from Sigma. Human TNFα antibody (D5G9) and mouse TNFα antibody (D2D4) were from Cell Signaling Technology. TNFα affibody immobilized on agarose (ab31909) and human Sirt6 antibody (ab88494) were from Abcam. Lamin A/C antibody (636), β-actin antibody (C4), and GRP 78 antibody (H-129) were from Santa Cruz Biotechnology. The rabbit pan-specific anti-acetyl lysine antibody was from ImmuneChem Pharmaceuticals Inc. The goat anti-rabbit and mouse IgG conjugated with HRP and protein A/G plus-agarose were from Santa Cruz Biotechnology. Human and mouse TNFα ELISA kits were from eBioscience. Brefeldin A (BFA), phorbol 12-myristate 13-acetate (PMA), lipopolysaccharides from Escherichia coli 0111:B4 (LPS) and palmitic acid were purchased from Sigma. Alk14 was synthesized according to reported procedures20. Sirt6 WT or KO MEF cells were generated as previously reported.7 THP-1 cells were purchased from ATCC.
Cloning, expression, and purification of full length Sirt6 for activity assay
The open reading frame of full length human Sirt6(1-355) was inserted into a pET28a vector between the BamHI and NotI sites. This plasmid was transformed in the E. coli Arctic Express (DE3) cells. The cells were cultured at 37°C in 2×YT culture medium (5 g of NaCl, 16 g of bactotrypton, and 10 g of yeast extract per liter). Isopropyl-β-D-1-thiogalactopyranoside (IPTG. 0.2 mM) was use to induce expression when OD600 was 0.6, and the culture was grown for 20 hr at 289 K. Cells were harvested by centrifugation at 7,330 g for 10 min and then re-suspended in lysis buffer (20 mM Tris-HCl, pH 7.2, 500 mM NaCl and 2% glycerol). Cells were lysed using a cell disrupter. After centrifugation at 29,300 g for 25 min at 277 K, the supernatant was loaded onto a nickel column (QFF-sepharose, Amersham Biosciences) pre-equilibrated with 20 mM Tris-HCl pH 7.2 with 500 mM NaCl. The protein was eluted with a linear gradient of imidazole (0 – 500 mM). The desired fractions were pooled, concentrated and buffer exchanged to cation exchange buffer (20 mM Tris pH 7.2, 80 mM NaCl, 5% glycerol). The protein was loaded onto a cation exchange column (Amersham Biosciences) and eluted with 1 M NaCl, 20 mM Tris-HCl, pH 7.2, 2% glycerol. The purified protein was stored at −80 °C.
Cloning, expression, and purification of truncated Sirt6 for crystallization
Human Sirt6 (1-294) was inserted into a pET28a vector between the NdeI and NotI sites. The protein was overproduced at 37°C in E. coli Rosetta (DE3) strain using 2×YT medium. The expression and purification method was the same as that used for the full length Sirt6 except that after Ni column purification the protein was further purified by gel filtration using a Superdex75 column (Amersham Biosciences). The protein was eluted with 20 mM Tris-HCl, pH 7.0, 100 mM NaCl. After concentrated to 8 mg/mL, the target protein was frozen at −80°C.
Synthesis of acyl peptides
The peptides were synthesized and purified as described previously5 The identity of the peptides was confirmed using LCQ Fleet ThermoFisher Mass Spectrometer. The acetyl, butyryl peptides were dissolved in 25% (v/v) DMSO in water. The longer chain fatty acyl peptides were dissolved in DMSO. The concentrations of peptides were determined at 280 nm using extinctions coefficient of the two tryptophans attached at the C termini of the peptides.
Deacylation activity assay
The activity of Sirt6 was analyzed using reverse phase HPLC on a Kinetex XB-C18 column (100A, 75 mm × 4.60 mm, 2.6 μm, Phenomenex). Sirt6 full length (1 μM) was incubated in a reaction mixture (60 μL) containing 20 mM Tris pH 8.0, 1 mM DTT, 0.5 mM NAD and 50 μM H3 K9 acyl peptides at 37°C for 30 min. Total DMSO content in the reaction was < 2.5% unless mentioned otherwise. The reactions were quenched with 60 μL of 0.5N HCl in methanol. The reactions were then monitored by HPLC as described below in the kinetics assay.
Kinetics assay for acetyl and butyryl peptides
Peptide concentrations were varied from 0 to 250 μM for H3 K9 butyryl peptide and from 0 to 600 μM for the acetyl peptide. The reaction mixtures (60 μL with 2 mM NAD, 1 mM DTT, 20 mM Tris, pH 8.0, 4 μM Sirt6 full length, and acyl peptides at various concentrations) were incubated for 30 min at 37°C. The reaction was stopped using 60 μL of 0.5 N HCl in methanol. The reaction mixtures were spun at 18,000 g for 10 min and were analyzed on a Kinetex XB-C18 column (100A, 100 mm × 4.60 mm, 2.6 μm, Phenomenex). The gradient of 20–40% B (acetonitrile with 0.1% TFA) in 17 min at 0.5 mL/min was used.
Kinetics assay for long chain fatty acyl peptides
The peptide concentrations were varied from 1 to 20 μM. The reactions (60 μL with 2 mM NAD, 1 mM DTT, 20 mM Tris pH 8.0, 0.2 μM recombinant Sirt6 full length, and acyl peptides at different concentrations) was incubated for 15 min at 37°C. The reaction was stopped using 60 μL of 0.5 N HCl in methanol. The reaction mixtures were spun at 18,000 g for 10 min and were analyzed on Kinetex XB-C18 column (100A, 75 mm × 4.60 mm, 2.6 μm, Phenomenex). The gradient of 0–55% B in 10 min at 0.5 ml//min was used. The product and the substrate peaks were quantified using absorbance at 280 nm and converted to initial rates, which were then plotted against the acyl peptide concentrations and fitted using Kaleidagraph.
Crystallization, X-ray data collection and structure determination
Crystals of complex Sirt6 with H3K9-Myr and ADPR were obtained by hanging drop vapor-diffusion method at 291 K using commercial screens from Hampton Research. Each drop consisting of 1 μL of 10 mg/mL protein complex solution (20 mM Tris-HCl, pH 7.4, 100 mM NaCl, 5 mM DTT) and 1 μL reservoir solution was equilibrated against 400 μL reservoir solution. The qualified crystals of Sirt6 grew with a cube profile within 1 week with a reservoir containing 12% PEG6K, 0.1 M MES, pH 6.5. The mixture of 30% glycerol with reservoir solution above was used as cryogenic liquor. The X-ray diffraction data were collected at 100 K in a liquid nitrogen gas stream at the Shanghai Synchrotron Radiation Facility BL17U. 180 frames were collected with a 1° oscillation and the data were indexed and integrated using the program HKL200025. The complex structure of Sirt6 with H3 K9 myristoyl peptide and ADPR was solved by molecular replacement using the program Molrep from the CCP4 Suite26 with the published Sirt6 structure (PDB: 3K35)27 as the search model. Refinement and model building were performed with REFMAC5 and COOT from CCP4. The X-ray diffraction data collection and structure refinement statistics are shown in Table S1.
Generation of Sirt6 KO MEF cells with human Sirt6 WT and H133Y mutant knock-in
Human Sirt6 WT or H133Y cDNA was inserted to lentiviral vector (engineered from pSIN-EF2-OCT4 vector with OCT4 deletion to convert into gateway destination vector, and provided by Dr. Chengliang Zhang from Professor Richard Cerione’s lab in Cornell University) using Gateway Cloning. After co-transfection of Sirt6 WT or H133Y lentiviral plasmid, pCMV-dR8.2, and pMD2.G into 293T cells, the medium was collected to infect Sirt6 KO MEF cells. The Sirt6 WT or H133Y knock-in cells were selected using 1.5 μg/mL puromycin in complete cell culture medium. Overexpressed Sirt6 WT and H133Y mutant have a C-terminal V5 tag.
Cloning of TNFα
For construction of human TNFα expression vector to express wild-type TNFα protein (TNFα_wt) with N-terminal Flag tag and C-terminal HA tag, human full length TNFα cDNA were generated by PCR and inserted into pCMV4A vector at EcoR V and Xho I sites. The plasmid of TNFα double lysine mutant (K19R, K20R), TNFα_KR, was made by overlap extension PCR.
Transfection of TNFα into MEF cells
Sirt6 WT and KO MEF cells were maintained in DMEM medium containing glucose and L-glutamine (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen). The pCMV vectors containing target genes were transfected into cells using FuGene 6 (Promega) according to the manufacture’s protocol. Empty pCMV vector was transfected into cells as negative control. This transfection method was also applied to Sirt6 KO MEF cells with human Sirt6 WT or H133Y mutant knock-in for TNFα overexpression.
Labeling of TNFα in MEF cells with Alk14
Sirt6 WT or KO MEF cells were cultured with fresh medium containing 20 μM Alk14 and 5 μg/mL brefeldin A for 12 hours after transient transfection of TNFα. Cells without Alk14 or without overexpression of TNFα were used as negative controls. Cells were collected by cell scraper and re-suspended in 1× PBS buffer. After centrifugation at 500 g for 5 min at 4°C, the cell pellet was dissolved in lysis buffer (25 mM Tris, pH 7.4, 150 mM NaCl, 10% glycerol, 1% Nonidet P-40) with protease inhibitor cocktail (Sigma). The supernatant was collected after centrifugation at 16000 g for 10 min at 4°C, and used for western blotting, immunoprecipitation, or TNFα ELISA experiment.
Generation of stable Sirt6 KD THP-1 cells
Sirt6 shRNA lentiviral plasmids in pLKO.1-puro vector were purchased from Sigma. Sirt6 shRNA #1 (TRCN0000378253) ccggcagtacgtccgagacacagtc ctcgaggactgtgctcggacgtactgtttttg and shRNA #2 (TRCN0000232528) ccgggaagaatgtgccaagtgtaagctc gagcttacacttggcacattcttctttttg were used. After co-transfection of Sirt6 shRNA plasmid, pCMV-dR8.2, and pMD2.G into 293T cells, the medium was collected to infect THP-1 cells. The Sirt6 KD cells were selected using 1.5 μg/mL puromycin in complete cell culture medium. THP-1 cells infected with lentivirus containing control shRNA plasmid were carried out similarly.
Labeling of TNFα in THP-1 cells with Alk14
THP-1 cells (with control shRNA, Sirt6 shRNA #1 orSirt6 shRNA #2, at 0.6 × 106 cells/mL) were treated with PMA (200 ng/mL) for 24 hours. Then cells were cultured with fresh medium containing Alk14 (50 μM) and LPS (1 μg/mL). After 1 hour, brefeldin A was added to culture medium (5 μg/mL). Cells were collected after 10 hours, and dissolved in lysis buffer (25 mM Tris, pH 7.4, 150 mM NaCl, 10% glycerol, 1% Nonidet P-40) with protease inhibitor cocktail (Sigma). The supernatant was collected after centrifugation at 16000 g for 10 min at 4°C and used for western blotting or immunoprecipitation.
Isolation of Sirt6 WT and KO macrophages and TNFα secretion
Bone marrow-derived macrophages were isolated from Sirt6 WT and KO mice as described28, 29 with some modifications. Briefly, femurs and tibiae were dissected and marrow tissue eluted by irrigation with DMEM. Cells were cultured in petri dishes containing complete media (DMEM+20% FCS+10ng/ml recombinant murine M-CSF+ penicillin/streptomycin) for 7–10 days at 37°C in a humidified 5% CO2 atmosphere. For TNFα mRNA expression analysis, 1.5×105 macrophages were plated in 6-well plates containing complete media and incubated overnight. Next day, LPS (100 ng/ml) was added and the cells were cultured for 12 hr. For TNFα protein expression and secretion, cells were plated as above and, after 12 hr of LPS stimulation, the supernatants were collected and used for ELISA analysis. To measure TNFα fatty acylation levels, 1.5×106 macrophages were plated in 6-cm plates in complete media and incubated overnight. Next day, cells were treated using the same procedure as described above for THP-1 cells.
Western blot for TNFα
Protein samples were separated by 18% SDS/PAGE and transferred to PVDF membrane. The membrane was blocked with 5% BSA in TBST (25 mM Tris, pH 7.4, 150 mM NaCl, 0.1% Tween-20), incubated with antibodies in TBST, and developed in ECL Plus western blotting detection reagents (GE Healthcare). The chemiluminescence signal was recorded by Storm 860 Imager (Amersham Biosciences) and analyzed with ImageQuant TL v2005.
Immunoprecipitation of Flag-TNFα using anti-Flag affinity gel
Cell lysate (250–400 μg from Sirt6 WT or KO MEF cells) was incubated with 20 μL suspension of anti-Flag M2 affinity gel for 2 hours at 4°C. After centrifugation at 500 g for 2 min at 4°C, the affinity gel was washed three times with 500 μL washing buffer (25 mM Tris, pH 7.4, 150 mM NaCl, 0.2% Nonidet P-40) and used for later experiments.
Immunoprecipitation of endogenous TNFα from THP-1 cells
Cell lysate (~500 μg) was incubated with 20 μL suspension of TNFα affibody immobilized agarose for 4 hours at 4°C. After centrifugation at 500 g for 2 min at 4°C, the agarose was washed three times with 500 μL washing buffer (25 mM Tris, pH 7.4, 150 mM NaCl, 0.2% Nonidet P-40) and used for later experiments.
Immunoprecipitation of endogenous TNFα from bone-marrow-derived mouse macrophage
Cell lysate (~100 μg from Sirt6 WT or KO macrophages) was incubated with 2 μg mouse TNFα antibody for 2 hours at 4°C. Then the mixture was incubated with 40 μL suspension of protein A/G plus-agarose for 8 hours at 4°C. After centrifugation at 500 g for 2 min at 4°C, the agarose was washed three times with 500 μL washing buffer (25 mM Tris, pH 7.4, 150 mM NaCl, 0.2% Nonidet P-40) and used for later experiments.
Detection of fatty acylation on TNFα by fluorescence
After immunoprecipitation, the gel/agarose was washed with and then re-suspended in 10 μL buffer (25 mM Tris, pH 7.4, 50 mM NaCl, 1% Nonidet P-40) for click chemistry. Rh-N3 (in DMF) was added to the above suspension to a final concentration of 200 μM, followed by the addition of Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (in DMF, final concentration 600 μM), CuSO4 (in water, final concentration 2 mM), and TCEP (in water, final concentration 2 mM). The click chemistry was allowed to proceed at room temperature for 60 min. The reaction mixture was mixed with 2× protein loading buffer and heated at 95°C for 10 min. After centrifugation at 16000 g for 2 min at room temperature, the supernatant was collected and heated with hydroxylamine (pH 7.2, 60 mM) at 95°C for 7 min. The samples were resolved by SDS-PAGE using 18% acrylamide gel. Rhodamine fluorescence signal was recorded by Typhoon 9400 Variable Mode Imager (GE Healthcare Life Sciences) with setting of Green (532 nm)/580BP30 PMT 500 V (normal sensitivity), and analyzed by ImageQuant TL v2005.
Detection of acetyl-lysine on TNFα by Western blot using anti-acetyl lysine antibody
Sirt6 KO MEF cell lysate (~30 mg), with/without overexpression of TNFα_WT or TNFα_KR, was incubated with 40 μL suspension of anti-Flag M2 affinity gel for 2 hours at 4°C. After centrifugation at 500 g for 2 min at 4°C, the affinity gel was washed three times with 500 μL washing buffer (25 mM Tris, pH 7.4, 150 mM NaCl, 0.2% Nonidet P-40) and then heated with 2× protein loading buffer at 100°C for 10 min. The samples were resolved by SDS-PAGE using 18% acrylamide gel and examined by western-blotting using anti-acetyl-lysine antibody. Acetylated BSA was used for positive control to demonstrate acetyl-lysine signal by western-blotting. After recording acetyl-lysine signal, PVDF membrane was washed with water and stained with Coomassie Blue to detect TNFα protein. Another western-blotting using anti-Flag antibody was also carried out to demonstrate the equal loading of TNFα_wt and TNFα_KR.
Defatty-acylation of TNFα by Sirt6 in vitro
TNFα_wt was immunoprecipated from lysate of Sirt6 KO MEF cells (~1 mg) overexpressing Flag-TNFa and cultured with Alk-14 using 40 μL suspension of anti-Flag M2 affinity gel following procedure describe above. After washing, the gel was divided into four equal aliquots. Each aliquot was re-suspended in 10 μL assay buffer (50 mM Tris, pH 7.4, 100 mM NaCl, 2 mM MgCl2, 1% Nonidet P-40) with 20 μM of Sirt6 or BSA and with or without 2 mM NAD and incubated at 37°C for 1 hour. Then detection of fatty acylation by fluorescence after hydroxylamine treatment was carried out with each aliquot as described earlier.
Secretion of TNFα and TNFα_KR from Sirt6 WT and KO MEF Cells
After transient transfection of TNFα_wt or TNFα_KR, Sirt6 WT and KO MEF cells were further incubated with fresh medium for 12 hours. Then culture medium and cells were collected separately for detection of TNFα by using human TNFα ELISA kit. Secretion efficiency was calculated by TNFα amount in culture medium versus total amount of TNFα in culture medium and cells. Six independent experiments were carried out.
TNFα secretion from THP-1 cells
Stable shRNA infected THP-1 cells (at 0.6 × 106 cells/mL) were treated with PMA (200 ng/mL) for 20 hours. (A) Without palmitic acid treatment: cells were incubated with fresh medium containing LPS (100 ng/mL). After 2 hours, culture medium and cells were collected for human TNFα ELISA assay. (B) With palmitic acid treatment: cells were incubated with palmitic acid (final 50 μM) in culture medium for 2 hours, and then with fresh medium containing LPS (100 ng/mL) and palmitic acid (50 μM). After 2 hours, culture medium and cells were collected for human TNFα ELISA assay. Three independent experiments were carried out.
ER fractions from THP-1 cells and Sirt6 WT/KO MEF cells
ER fraction was obtained from THP-1 cells, Sirt6 WT/KO MEF cells, or Sirt6 KO MEF cells with human Sirt6 WT or H133Y knock-in, using an isopycnic flotation method.30 ER fraction was solubilized in lysis buffer (25 mM Tris, pH 7.4, 150 mM NaCl, 10% glycerol, 1% Nonidet P-40) with protease inhibitor cocktail (Sigma). The supernatant was collected after centrifugation at 16000 g for 10 min at 4°C, and used for western blotting.
Statistical Analysis
Data were expressed as mean ± s.d. (standard deviation, shown as error bars). Differences were examined by two-tailed Student’s t test between two groups; and *P < 0.02, **P < 0.01, ***P < 0.001.
Supplementary Material
Acknowledgments
This work was supported in part by NIH R01GM086703 (H.L.), Hong Kong GRF766510 (Q.H.) and NIH R01GM087544 (H.C.H). We thank Dr. Chengliang Zhang for help with the cloning of Sirt6 WT and H133Y to generate lentiviral particles and the staff at the Shanghai Synchrotron Radiation Facility for assistance during the data collection.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author contributions. H.J. designed and carried out all biochemical experiments involving TNFα and synthesized Rh-N3. S.K. synthesized acyl peptides and carried out all enzymology experiments of Sirt6. Y.W. carried out all crystallography experiments. G.C. and B.H. synthesized Alk14. G.C. and H.C.H. provided expertise on the labeling experiments using Alk14. C.S. and R.M. generated the MEF cells and bone marrow-derived macrophages from Sirt6 WT and KO mice. J.D., R.K., and E.G. contributed to the cloning, expression and purification of Sirt6. Q.H. directed the structural studies and wrote the structural part of the manuscript. H.L. directed the biochemical studies, coordinated the collaborations among different labs, and wrote the manuscript with help from H.J., S.K., Y.W., R.M., H.C.H., and Q.H.
References
- 1.Imai S-i, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 2.Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu Rev Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 3.Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Du J, et al. Sirt5 is an NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu AY, et al. Plasmodium falciparum Sir2A preferentially hydrolyzes medium and long chain fatty acyl lysine. ACS Chem Biol. 2011;7:155–159. doi: 10.1021/cb200230x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mostoslavsky R, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 7.Zhong L, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1α. Cell. 2010;140:280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao C, et al. SIRT6 deficiency results in severe hypoglycemia by enhancing both basal and insulin-stimulated glucose uptake in mice. J Biol Chem. 2010;285:36776–36784. doi: 10.1074/jbc.M110.168039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michishita E, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452:492–496. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanfi Y, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–221. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- 11.Yang B, Zwaans BMM, Eckersdorff M, Lombard DB. The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle. 2009;8:2662– 2663. doi: 10.4161/cc.8.16.9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Michishita E, et al. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle. 2009;8:2664– 2666. doi: 10.4161/cc.8.16.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 14.Hoff KG, Avalos JL, Sens K, Wolberger C. Insights into the sirtuin mechanism from ternary complexes containing NAD+ and acetylated peptide. Structure. 2006;14:1231–1240. doi: 10.1016/j.str.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Stevenson FT, et al. The 31-kDa precursor of interleukin 1 alpha is myristoylated on specific lysines within the 16-kDa N-terminal propiece. Proc Natl Acad Sci U S A. 1993;90:7245–7249. doi: 10.1073/pnas.90.15.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stevenson FT, Bursten SL, Locksley RM, Lovett DH. Myristyl acylation of the tumor necrosis factor alpha precursor on specific lysine residues. J Exp Med. 1992;176:1053–1062. doi: 10.1084/jem.176.4.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 18.Bruzzone S, et al. Catastrophic NAD depletion in activated T lymphocytes through Nampt inhibition reduces demyelination and disability in EAE. PLoS ONE. 2009;4:e7897. doi: 10.1371/journal.pone.0007897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Gool F, et al. Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin-dependent manner. Nat Med. 2009;15:206–210. doi: 10.1038/nm.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Charron G, et al. Robust fluorescent detection of protein fatty-acylation with chemical reporters. J Am Chem Soc. 2009;131:4967–4975. doi: 10.1021/ja810122f. [DOI] [PubMed] [Google Scholar]
- 21.Wilson JP, et al. Proteomic analysis of fatty-acylated proteins in mammalian cells with chemical reporters reveals S-acylation of histone H3 variants. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin BR, Cravatt BF. Large-scale profiling of protein palmitoylation in mammalian cells. Nat Meth. 2009;6:135–138. doi: 10.1038/nmeth.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwer B, et al. Neural sirtuin 6 (Sirt6) ablation attenuates somatic growth and causes obesity. Proc Natl Acad Sci U S A. 2010;107:21790–21794. doi: 10.1073/pnas.1016306107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walsh CT. Posttranslational Modification of Proteins: Expanding Nature’s Inventory. Roberts and Company Publishers; Englewood, Colorado: 2005. [Google Scholar]
- 25.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 26.Collaborative. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 27.Pan PW, et al. Structure and biochemical functions of SIRT6. J Biol Chem. 2011;286:14575–14587. doi: 10.1074/jbc.M111.218990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sebastian C, et al. Deacetylase activity is required for STAT5-dependent GM-CSF functional activity in macrophages and differentiation to dendritic cells. J Immunol. 2008;180:5898–906. doi: 10.4049/jimmunol.180.9.5898. [DOI] [PubMed] [Google Scholar]
- 29.Sebastian C, et al. Deacetylase activity is required for STAT5-dependent GM-CSF functional activity in macrophages and differentiation to dendritic cells. J Immunol. 2008;180:5898–5906. doi: 10.4049/jimmunol.180.9.5898. [DOI] [PubMed] [Google Scholar]
- 30.Stephens SB, et al. Analysis of mRNA partitioning between the cytosol and endoplasmic reticulum compartments of mammalian cells. Methods Mol Biol. 2008;419:197–214. doi: 10.1007/978-1-59745-033-1_14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.