

Abstract
We have synthesized an oxaliplatin derivative using N,N′-dimethyl-1,2-diaminocyclohexane (Me2dach) as the diamine ligand. The complex (S,R,R,S)-Pt(Me2dach)(oxalate), where S,R,R,S represents the chiralities at N,C,C,N, respectively, was prepared and characterized by 1H NMR spectroscopy, COSY, NOESY, and HMQC. Oxaliplatin reacts with N-acetylmethionine (N-AcMet) to form [Pt(dach)(N-AcMet-S)2] and [Pt(dach)(N-AcMet-S,N)], with the former favored at higher molar ratios of N-AcMet. In contrast, Pt(Me2dach)(oxalate) reacts to form [Pt(Me2dach)(N-AcMet-S,O)]+ even in the presence of excess N-AcMet. Molecular mechanics calculations are consistent with significant steric clashes in models of [Pt(Me2dach)(N-AcMet-S)2]. When N-AcMet was reacted with an excess of each platinum complex, the rate of N-AcMet decrease was very similar for both complexes. Thus, the methyl groups at the nitrogen atoms had little to no effect on the addition of the sulfur atom of a single N-acetylmethionine, but they prevented chelation of the amide nitrogen or coordination of a second N-acetylmethionine residue.
Keywords: Nuclear magnetic resonance, platinum, oxaliplatin, amino acids, methionine
1. Introduction
The anticancer drugs cisplatin and oxaliplatin are known to interact with both DNA and protein targets in vitro and in vivo. Whereas interaction with DNA is thought to result in the cytotoxicity, interaction with amino acids and proteins could be involved in resistance pathways,[1] oxidative stress,[2] and/or cellular uptake.[3] The amino acid methionine is a key amino acid target, with small molecule studies indicating that reaction with methionine or related thioether molecules is kinetically favored over reaction with guanine when the amine ligands are relatively small.[4] Reactions of platinum(II) complexes with proteins may have additional applications including their use as selective protein cleavage agents[5–7] or as specific enzyme inhibitors[8, 9]. Thus, it is important to understand the factors that influence reactivity with particular biologically relevant targets.
Addition of alkyl groups to the amine nitrogen atoms can affect the reaction of cisplatin analogs with methionine. A platinum complex with the bulky triamine ligand N,N,N′,N′,N″-pentamethyldiethylenetriamine (Me5dien) reacted faster with 5′-GMP than with N-acetylmethionine due to additional steric hindrance in reactions involving the latter.[10] More recently, the coordination of one N-AcMet residue to [Pt(Me4en)(D2O)2]2+ [Me4en = N,N,N′,N′-tetramethylethylenediamine] was determined to be approximately 16-fold slower than coordination of N-AcMet to [Pt(en)(D2O)2]2+.[11] The significant slowing was attributed at least in part to steric hindrance. By contrast, the rate of reaction with N-AcHis was not affected by the additional size of the Me4en ligand; thus, the size of the amine ligand could impact which amino acid residues are preferentially targeted.
Previously, it was found that [Pt(Me4en)(D2O)2]2+ and [Pt(Et2en)(D2O)2]2+ [Et2en = N,N-diethylethylenediamine] reacted with only one Met or N-AcMet residue; chelates involving the sulfur atom and a carboxylate oxygen atom of N-AcMet or the sulfur and nitrogen atoms of Met were stable products.[12, 13] Upon reaction between the platinum complex and the methionine sulfur atom, the remaining available coordination site would be cis to the methionine sulfur atom and also cis to a bulky amine ligand, and thus steric hindrance prevented coordination of a second methionine ligand or the chelation of the amide nitrogen of N-AcMet.
Oxaliplatin (Figure 1), a third generation platinum(II) anticancer drug, utilizes a (R,R)-1,2-diaminocyclohexane (dach) ligand in addition to an oxalate leaving ligand. The rate constants for reaction with methionine were found to be similar for [Pt(en)(H2O)2]2+ and [Pt(dach)(H2O)2]2+ complexes,[14] suggesting that the additional carbons of the dach ligand do not interfere with coordination of an incoming methionine ligand. However, the stereochemistry of the carbons of the dach ligand should limit the ligand flexibility, and such chirality has been previously found to influence chirality introduced at the nitrogen atoms of diamine ligands[15, 16]. Thus, we hypothesized that the stereochemistry of the dach ligand would influence the resulting chirality at the nitrogen atoms when secondary amines were present.
Figure 1.

Representations of (R,R)-Pt(dach)(ox) [oxaliplatin] and (S,R,R,S)-Pt(Me2dach)(ox).
In the current study, we have utilized commercially available N,N′-dimethyl-1,2-diaminocyclohexane (Me2dach) as a diamine ligand containing secondary amine nitrogen atoms (Figure 1). This ligand is an analog of the dach ligand utilized in oxaliplatin, with one methyl group added to each nitrogen atom for additional bulk that would be near the available coordination sites. Since previous studies probing reaction at methionine[10–13, 17] have generally utilized platinum(II) complexes with primary amine nitrogen atoms (en, dien) or tertiary amine nitrogen atoms (Me4en, Me5dien), we were interested to see how the presence of a secondary amine nitrogen atom influenced the reaction with methionine.
2. Materials and Methods
Silver nitrate (Sigma-Aldrich), oxalic acid (Acros), potassium tetrachloroplatinate (Sigma-Aldrich), (R,R)-1,2-diaminocyclohexane (Sigma-Aldrich), (R,R)-N,N′-dimethyl-1,2-diaminocyclohexane (Sigma-Aldrich), and N-acetylmethionine (Sigma-Aldrich) were used as received.
2.1. Silver oxalate
Silver oxalate was prepared by a combination of 1 g of silver nitrate and 350 mg of oxalic acid in 20 mL of water. The reaction was stirred in an amber vial for ~30 minutes and the insoluble silver oxalate was collected by filtration and washed with water.
2.2. Pt(dach)(ox) (oxaliplatin)
In a typical reaction, 64 mg (0.56 mmol) of (R,R)-1,2-diaminocyclohexane (dach) was dissolved in 5 mL of methanol and added dropwise to an aqueous solution (5 mL) of 232 mg (0.56 mmol) of K2PtCl4. The reaction was stirred overnight and a yellow precipitate of Pt(dach)Cl2 was collected and washed with water, ethanol, and ether. and silver oxalate were Yield 128.8 mg (61%). Equimolar (0.17 mmol) amounts of Pt(dach)Cl2 combined in ~35 mL of water in an amber vial and stirred overnight. The sample was filtered to remove the AgCl precipitate and rotovapped with heating to ~35 °C to isolate Pt(dach)(ox). Yield 27.9 mg (41%) 1H NMR (D2O): 2.33 ppm (1 H, dd), 2.04 ppm (1 H, broad d), 1.56 ppm (1 H, m), 1.30 ppm (1 H, m), 1.15 ppm (1 H, m).
2.3. Pt(Me2dach)(ox)
In a typical reaction, 56.8 mg (0.4 mmol) of (R,R)-N,N′-dimethyl-1,2-diaminocyclohexane (Me2dach) in 5 mL methanol was added dropwise to an aqueous solution (5 mL) of 166 mg (0.4 mmol) K2PtCl4. After stirring overnight, a yellow precipitate was collected by vacuum filtration (Pt(Me2dach)Cl2) and washed with water, ethanol, and ether. Yield 100 mg (61%). An equimolar amount (75 mg) of silver oxalate was added to the Pt(Me2dach)Cl2 and the mixture was stirred in an amber vial for at least 24 hours. The sample was filtered to remove AgCl precipitate to form Pt(Me2dach)(ox) with some [Pt(Me2dach)(H2O)2]2+, which converted to Pt(Me2dach)(ox) when rotovapped with heating to ~35 °C. Yield: 72.9 mg (69%). Mass spectrometry: 426.2 (M+H+), 448.3 (M+Na+). 1H NMR (D2O): 2.53 ppm (3 H, s), 2.30 (1 H, br), 2.27 (1 H, br), 1.62 (1 H, br), 1.12 (2 H, br). See Table 1 for 1H and 13C NMR assignments.
Table 1.
Assignments of 1H and 13C NMR resonances of Pt(Me2dach)(ox) in D2O. All values in ppm.
| C-H1,2 | C-H3,6 | C-H4,5 | N-CH3 | |
|---|---|---|---|---|
| 1H | 2.27 | 1.12 (ax) 2.30 (eq) |
1.12 (ax) 1.62 (eq) |
2.53 |
| 13C | 70.83 | 28.23 | 24.39 | 38.68 |
2.4. NMR spectroscopy
1H, 13C, and 195Pt NMR data were collected on a JEOL 500 MHz NMR instrument. HMQC experiments utilized a J-coupling value of 140 Hz, and NOESY experiments utilized a 500 ms mixing time. Spectra were referenced to the residual HOD signal (1H), TMS (13C), or K2PtCl6 (195Pt).
Stock solutions (typically 10 mM) of the appropriate platinum compound and N-acetylmethionine were individually prepared in D2O and the pH (uncorrected) adjusted to 4. Aliquots of each individual solution were then combined in D2O to give the appropriate concentrations.
2.5. Molecular mechanics
Molecular mechanics and dynamics calculations were performed as described previously[13] using an AMBER force field modified to include parameters for platinum[18] and methionine.[12] For each configuration and conformation, dynamics were typically run for 250 ps at 300 K with structures saved every 1 ps; these structures were then subjected to energy minimization.
3. Results
3.1 Determination of stereochemistry of Pt(Me2dach)(ox)
The 1H NMR spectrum of [Pt(Me2dach)(ox)] shows only one methyl group at 2.53 ppm (Table 1), indicating that only one diastereomer is formed and that it is C2-symmetrical. Since both chiral carbons have the R stereochemistry, the stereochemistry of the N atoms must be both R or both S.
HMQC was used to definitively identify C1,2 and H1,2 since these were the only ring carbons with only one hydrogen attached (see Figure 1 for numbering). The lack of a NOESY or COSY cross peak of the H1,2 resonance to the signal at 1.62 ppm led to the assignment of the 1.62 ppm signal as one of the H4,5 signals. The HMQC was then used to assign the remaining H4,5 signal. The H3,6 signals were assigned based on the remaining 13C signal in the HMQC spectrum.
A NOESY spectrum acquired in DMSO-d6 revealed a cross peak between the N-H resonance at 6.38 ppm and one H3,6 resonance. No NOESY was observed between the N-H resonance and the H1,2 resonances. Molecular mechanics models of (S,R,R,S)- and (R,R,R,R)-Pt(Me2dach)Cl2 revealed that the former would have ~2.4 Å between N-H and the axial H3,6 whereas the latter would have a similar distance between the N-H and the H1,2 atoms. Thus, the stereochemistry of the isomer was assigned to (S,R,R,S)-Pt(Me2dach)(ox).
3.2 Characterization of the final products of the reaction of N-AcMet with Pt(dach)(ox) and Pt(Me2dach)(ox)
When N-AcMet and Pt(dach)(ox) were combined in a 2:1 ratio, broad resonances at 2.01, 2.56, and 2.58 ppm were visible after 1 day (Figure 2); the 2.01 ppm signal is in an area typical for Ac-CH3 resonances of N-AcMet whereas the other two are characteristic of S-CH3 signals in which the platinum atom has coordinated to the sulfur atom. When the sample was heated, the broad resonances at 2.56 and 2.58 ppm coalesced into one signal and began to sharpen (Figure 2). A 195Pt NMR spectrum showed a peak at −3739 ppm; this chemical shift is in the −3600 to −3800 range that is seen for a PtN2S2 coordination environment.[12, 19, 20] The final product was therefore assigned to [Pt(dach)(N-AcMet-S)2].
Figure 2.

Partial 1H NMR spectra of 3.33 mM Pt(dach)(ox) and 6.67 mM N-AcMet. Each spectrum has the y-axis scaled for the largest peak. The * indicates the S-CH3 signals of the product.
When an equimolar solution of Pt(dach)(ox) and N-AcMet was monitored by NMR spectroscopy, a resonance at 2.00 ppm and a broad resonance at 2.34 ppm, just downfield of the H1,2 resonance of oxaliplatin at 2.33 ppm, grew over the first 2 h but eventually disappeared by 24 h (Figure 3). These resonances were assigned to the intermediate [Pt(dach)(N-AcMet-S)(ox-O)]-. The dominant products after 24 hours had S-CH3 resonances at 2.30, 2.37 (two nearly overlapping resonances), and 2.43 ppm and overlapping Ac-CH3 resonances at 2.01 ppm. The shifts of these S-CH3 resonances are very similar to those observed previously for [Pt(en)(N-AcMet-S,N)] complexes; four products were possible due to chirality at the S atom and the possibility of cis/trans isomers around the amide bond.[17] Thus, we assign the major products of the equimolar Pt(dach)(ox) reaction with N-AcMet to the isomers of [Pt(dach)(N-AcMet-S,N)], with the platinum coordinated to the sulfur atom and the (deprotonated) amide nitrogen atom. The 195Pt NMR spectrum showed signals at −3215 and −3260, which are in the range expected for a PtN3S coordination environment.[19, 20] Small broad signals at 2.56 and 2.58 ppm in the 1H NMR spectrum were also observed, and these sharpened with the temperature was increased; thus some [Pt(dach)(N-AcMet-S)2] also formed in this reaction.
Figure 3.

Partial 1H NMR spectra of 3.33 mM Pt(dach)(ox) and 3.33 mM N-AcMet. Each spectrum has the y-axis scaled for the largest peak. The * indicates the S-CH3 signals of the chelates.
The final NMR spectrum of the reaction of N-AcMet with Pt(Me2dach)(ox) showed three doublet of doublets in the range of 5.7–5.9 ppm (Figure 4) regardless of the molar ratios of the N-AcMet or platinum complex. These signals are characteristic of the α-hydrogen of N-AcMet residues chelated via the sulfur and carboxyl oxygen atoms[12, 13]; HMQC results showed a corresponding 13C chemical shift of 54.8 ppm, consistent with the α-carbon of N-AcMet[17]. The 195Pt NMR spectrum showed peaks at ca. −2870 and −2890 ppm. This is within the range of −2600 to −2900 ppm that is typical for a PtN2SO coordination environment[12, 13] but is downfield of the typical range for the PtN3S coordination environment (−3000 to −3300 ppm) and significantly downfield of the observed shifts for [Pt(dach)(N-AcMet-S,N)]. The significant downfield shift of the Hα signals and the 195Pt NMR shift indicate that the products are assignable to [Pt(Me2dach)(N-AcMet-S,O)]+.
Figure 4.
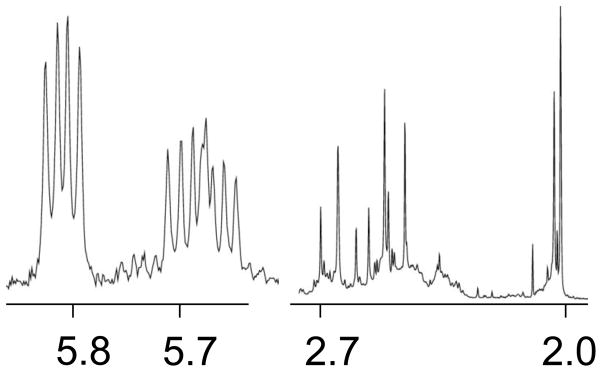
Partial 1H NMR spectrum of 10 mM Pt(Me2dach)(ox) and 10 mM N-AcMet. Each side of the spectrum has the y-axis scaled for the largest peak.
When the temperature was raised, several of the resonances broadened, indicating multiple species are in chemical exchange (Figure 5). A similar broadening was noted for [Pt(Et2en)(N-AcMet-S,O)]+ previously[13] and was attributed to the differences in the R and S chiralities at the sulfur atom upon coordination to the platinum.
Figure 5.

Partial 1H NMR spectra of [Pt(Me2dach)(N-AcMet-S,O)]+ at several temperatures. All spectra are referenced to the HOD signal adjusted for temperature.
3.3 Molecular mechanics calculations
Previously, molecular mechanics calculations have been used to model [Pt(Me4en)(9-EtG)2] and [Pt(Me4en)(Met)2] complexes in order to understand better why the latter complexes do not form in solution[12]. The minimum energy structure of the latter was >21 kcal/mol higher in energy than the minimum energy of the former; by comparison, the minimum energy of cis-[Pt(NH3)2(Met)2] was only 7 kcal/mol higher than the minimum energy of cis-[Pt(NH3)2(9-EtG)2].
In the present study, we determined the minimum energy of various conformations of [Pt(dach)(9-EtG)2], [Pt(dach)(Met)2], [Pt(Me2dach)(9-EtG)2], and [Pt(Me2dach)(Met)2] (Table 2). The methionine complex was <6 kcal/mol higher in energy than the guanine complex when the dach ligand was utilized, which is comparable to the calculated difference between analogous complexes with cisplatin. In contrast, the methionine complex was >17 kcal/mol higher in energy for complexes having the Me2dach ligand; this value is closer to the ~21 kcal/mol seen for the Me4en ligand previously. The structures of [Pt(Me2dach)(Met)2] had significant distortion of the platinum coordination plane due to steric clashes (Supplementary Material).
Table 2.
Calculated minimum energies of [PtA2(9-EtG)2] and [PtA2(Met)2] complexes, where A2 = dach, Me2dach, cis-Pt(NH3)2, or Me4en. All energies in kcal/mol.
| Pt ligand | PtA2G2 energy | PtA2Met2 energy | Difference |
|---|---|---|---|
| dach | −17.0 | −11.5 | 5.5 |
| Me2dach | −11.7 | 5.5 | 17.2 |
| cis-Pt(NH3)2a | −17.7 | −10.6 | 7.1 |
| Me4ena | −5.4 | 16.1 | 21.5 |
Reference [12]
Molecular mechanics calculations were also performed on [Pt(Me2dach)(Met-S,O)]+ with either the R or S chirality at the sulfur atom and a variety of conformations of the 7-membered chelate ring. Overall, we found three structures that were significantly lower in energy (by more than 4 kcal/mol) than other structures and that had relatively few steric clashes (Supplementary material). Thus, the observation of three sulfur, oxygen chelates in the molecular mechanics is consistent with the experimental observation of three sets of NMR resonances for [Pt(Me2dach)(N-AcMet-S,O)]+.
3.4 Comparison of the reactivity of Pt(dach)(ox) and Pt(Me2dach)(ox) with N-AcMet
In order to compare the relative reaction rates of Pt(dach)(ox) and Pt(Me2dach)(ox) with N-AcMet, we used 5 mM concentrations of each platinum complex and 1 mM concentrations of N-AcMet; these conditions favor coordination of only one N-AcMet to Pt(dach)(ox). Because both mono products and chelates are possible in each reaction and the signals from the unreacted platinum interfere with many of the key product signals, the reaction rates were monitored primarily by observing the decrease in the free N-AcMet signals. Both Pt(dach)(ox) and Pt(Me2dach)(ox) react with a half-life of approximately 1 hour under these conditions (Figure 6). By 4 hours, the unreacted N-AcMet was nearly depleted in both samples. In contrast, [Pt(Me4en)(D2O)2]2+ reacted ~16 times more slowly with N-AcMet than did [Pt(en)(D2O)2]2+.[11] Thus, the presence of a secondary amine N cis to the coordination site in Pt(Me2dach)(ox) did not significantly affect the rate of the first N-AcMet coordination.
Figure 6.

Partial 1H NMR spectra of 5 mM Pt(dach)(ox) (top) or Pt(Me2dach)(ox) (bottom) reacted with 1 mM N-AcMet. For a given platinum complex, the spectra at each time point are scaled by the same factor. The unreacted N-AcMet signals are the two singlets of approximately equal size at ~2.0 and 2.1 ppm indicated by * in the spectra at 15 min. In the top spectrum, the broad peaks seen in all spectra are from the dach ligand of unreacted Pt(dach)(ox) and the large singlet increasing in size over time is the acetyl CH3 signal of product. The broad signals in the bottom spectra are acetyl CH3 product signals.
4. Discussion
The (R,R)-N,N′-dimethyl-1,2-diaminocyclohexane (Me2dach) ligand is commercially available and coordinates to platinum to produce one diastereomer of Pt(Me2dach)(ox), with the stereochemistry of (S,R,R,S) at the N,C,C,N atoms, respectively. Chirality in the carbon atoms of a ring system has previously been shown to influence the nitrogen stereochemistry[15, 16]. The use of the Me2dach ligand leads to a relatively simple synthesis of a platinum(II) diamine complex with a C2-symmetrical ligand having chirality at the nitrogen atoms.
A platinum(II) triamine complex with the bulky Me5dien ligand was previously shown to slow reaction of N-acetylmethionine dramatically relative to 5′-GMP, and thus we speculated that large amine ligands on platinum(II) complexes could lead to less reaction with proteins relative to DNA.[10] The Me5dien ligand represents an extreme case, with tertiary amines present at both coordination sites cis to the available coordination site. Previously, a platinum(II) complex with the DNSH-dien ligand, which has a primary amine and a bulky sulfonamido ligand cis to the available coordination site, was found to react with 5′-GMP and Met at roughly equal rates.[21] More recently, research on phenanthriplatin, a platinum(II) complex with a bulky anthracene-like ligand cis to the available coordination site found that reaction with N-AcMet and 5′-dGMP occurred at similar rates; this platinum(II) complex had higher activity than less bulky monofunctional complexes such as pyriplatin, and reduced reaction with proteins relative to DNA was proposed as a possible reason.[22]
When excess N-AcMet is present, the final product of the reaction with Pt(dach)(ox) is [Pt(dach)(N-AcMet-S)2] (Figure 7, left). When excess or equimolar Pt(dach)(ox) is present, [Pt(dach)(N-AcMet-S,N)] is the dominant final product (Figure 7, middle). Previously, reaction of Pt(dach)(ox) with Met has also been shown to result in a S,N chelate.[23] Also, [Pt(en)(N-AcMet-S,N)] has been an observed product in reactions with N-AcMet.[17]
Figure 7.

Representations of [Pt(dach)(N-AcMet-S)2], [Pt(dach)(N-AcMet-S,N)], and [Pt(Me2dach)(N-AcMet-S,O)]+ complexes, respectively.
Several reports have indicated that Pt(dach)(ox) reacts with Met to form [Pt(dach)(Met-S,N)] as a major species[23–25] even in the presence of excess methionine[23]. However, N-AcMet forms some [Pt(dach)(N-AcMet-S)2] even at equimolar ratios (Figure 3) and as a dominant product when excess N-AcMet is present (Figure 2). The amine nitrogen of Met would be expected to be a better ligand than the amide nitrogen of N-AcMet; thus, the observation of the S,N chelate in the presence of excess Met may be due to a faster chelation of the amine nitrogen compared with the amide nitrogen in N-AcMet.
The presence of a tertiary amine nitrogen cis to the available coordination site prevented coordination of a second methionine or chelation of the amide nitrogen of N-AcMet to complexes utilizing the Me4en or Et2en ligands; steric clashes between the acetyl group or the second methionine and the methyl or ethyl groups of the amine ligand were significant.[12, 13] Thus, chelates involving the sulfur atom and the carboxyl oxygen atom were observed. In the present study, [Pt(Me2dach)(N-AcMet-S,O)]+ (Figure 7, right) is a stable product even in the presence of excess N-AcMet. Therefore, the secondary amine atoms of the Me2dach also cause significant steric clashes to prevent coordination of the amide nitrogen and/or second methionine residue. Molecular mechanics calculations (Table 2) support the presence of more severe steric clashes between methionine residues and the Me2dach ligand compared with the dach ligand. Our previous studies[11–13] establish that coordination of a methionine trans to a tertiary amine nitrogen atom effectively blocks coordination of a second methionine residue cis to the methionine. Our current studies indicate that coordination trans to a secondary amine nitrogen also prevents the second methionine residue from reacting. A recent study found that a modified dach ligand with a 2-butyl substituent on one of the nitrogen atoms of an oxaliplatin derivative led to a relatively high antitumor activity and low toxicity for the platinum(II) complex.[26] Agarose gel electrophoresis experiments suggested that the complex could be binding to the DNA in a monofunctional fashion. Thus, the presence of a large substituent on a secondary amine could affect DNA reactivity.
Three different S,O products were observed by 1H NMR spectroscopy, and the signals from these complexes broadened at higher temperature, indicating chemical exchange. Two different chiralities at the sulfur atom would lead to diastereomers that could be distinguished by NMR spectroscopy; additionally, several possible conformations of the 7-membered chelate ring are possible, and interconversion of these conformations could be slow due to the presence of the methyl groups on the Me2dach ligand. Our molecular mechanics calculations showed three structures that were relatively low in energy and free of steric clashes.
The presence of one methyl group on each N is sufficient to prevent coordination of a second N-AcMet (Figure 4). However, the rate of coordination of the first methionine does not appear to be affected, as Pt(dach)(ox) and Pt(Me2dach)(ox) reacted with N-AcMet at similar rates (Figure 6). By contrast, [Pt(Me4en)(D2O)2]2+ reacted with N-AcMet approximately 16 times slower than did [Pt(en)(D2O)2]2+.[11] Thus, whereas ligands such as Me4en and Me5dien would be expected to reduce the reactivity of methionine relative to other protein or DNA targets, the Me2dach ligand would not. The Me2dach ligand thus is unique in that the additional methyl groups affect coordination of the second coordination site but not of the first coordination site.
Supplementary Material
Highlights.
Formed Pt(Me2dach)(oxalate), where Me2dach = N,N′-dimethyl-1,2-diaminocyclohexane
Stereochemistry determined to be (S,R,R,S) for Pt(Me2dach)(ox)
Pt(dach)(ox) and Pt(Me2dach)(ox) react with N-acetylmethionine at similar rates
Pt(dach)(ox) can form [Pt(dach)(N-AcMet-S)2] or [Pt(dach)(N-AcMet-S,N)]
Pt(Me2dach)(ox) can form only [Pt(Me2dach)(N-AcMet-S,O)]+
Acknowledgments
This research was supported by awards from the NIH (1R15 GM074663-01) and Western Kentucky University (Research and Creative Activities Program RCAP 11-8042) to KMW. KMW and DCJ were also supported by a Faculty Undergraduate Student Engagement grant from Western Kentucky University (12-SF145). JDH was supported as part of a Research Experience for Undergraduates award from the National Science Foundation (DBI-1004665).
Abbreviations
- dach
1,2-diaminocyclohexane
- Me2dach
N,N′-dimethyl-1,2-diaminocyclohexane
- N-AcMet
N-acetylmethionine
- ox
oxalate
- Me4en
N,N,N′,N′-tetramethylethylenediamine
- Et2en
N,N-diethylethylenediamine
- Me5dien
N,N,N′,N′,N″-pentamethyldiethylenetriamine
Appendix A. Supplementary Material
Stereo renderings of minimum energy molecular mechanics structures of [Pt(dach)(9-EtG)2], [Pt(dach)(Met)2], [Pt(Me2dach)(9-EtG)2], [Pt(Me2dach)(Met)2], and the three lowest energy structures of [Pt(Me2dach)(Met-S,O)].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kartalou M, Essigmann JM. Mutat Res. 2001;478:23–43. doi: 10.1016/s0027-5107(01)00141-5. [DOI] [PubMed] [Google Scholar]
- 2.Arnér ESJ, Nakamura H, Sasada T, Yodoi J, Holmgren A, Spyrou G. Free Radical Biol Med. 2001;31:1170–1178. doi: 10.1016/s0891-5849(01)00698-0. [DOI] [PubMed] [Google Scholar]
- 3.Larson CA, Adams PL, Blair BG, Safaei R, Howell S. Mol Pharmacol. 2010 doi: 10.1124/mol.110.064766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Djuran MI, Lempers ELM, Reedijk J. Inorg Chem. 1991;30:2648–2652. [Google Scholar]
- 5.Dutcã LM, Ko KS, Pohl NL, Kostić NM. Inorg Chem. 2005;44:5141–5146. doi: 10.1021/ic050137w. [DOI] [PubMed] [Google Scholar]
- 6.Milović NM, Dutcã LM, Kostić NM. Chem-Eur J. 2003;9:5097–5106. doi: 10.1002/chem.200304772. [DOI] [PubMed] [Google Scholar]
- 7.Hong J, Jiao Y, He WJ, Guo ZJ, Yu Z, Zhang JF, Zhu LG. Inorg Chem. 2010;49:8148–8154. doi: 10.1021/ic101191m. [DOI] [PubMed] [Google Scholar]
- 8.van Zutphen S, Kraus M, Driessen C, van der Marel GA, Overkleeft HS, Reedijk J. J Inorg Biochem. 2005;99:1384–1389. doi: 10.1016/j.jinorgbio.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 9.Witte AB, Anestal K, Jerremalm E, Ehrsson H, Arner ES. Free Radical Biol Med. 2005;39:696–703. doi: 10.1016/j.freeradbiomed.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 10.Sandlin RD, Starling MP, Williams KM. J Inorg Biochem. 2010;104:214–216. doi: 10.1016/j.jinorgbio.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sandlin RD, Whelan CJ, Bradley MS, Williams KM. Inorg Chim Acta. 2012;391:135–140. doi: 10.1016/j.ica.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams KM, Rowan C, Mitchell J. Inorg Chem. 2004;43:1190–1196. doi: 10.1021/ic035212m. [DOI] [PubMed] [Google Scholar]
- 13.Williams KM, Chapman DJ, Massey SR, Haare C. J Inorg Biochem. 2005;99:2119–2126. doi: 10.1016/j.jinorgbio.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 14.Summa N, Schiessl W, Puchta R, Hommes NvE, van Eldik R. Inorg Chem. 2006;45:2948–2959. doi: 10.1021/ic051955r. [DOI] [PubMed] [Google Scholar]
- 15.Ano SO, Intini FP, Natile G, Marzilli LG. J Am Chem Soc. 1998;120:12017–12022. [Google Scholar]
- 16.Kiser D, Intini FP, Xu Y, Natile G, Marzilli LG. Inorg Chem. 1994;33:4149–4158. [Google Scholar]
- 17.Barnham KJ, Guo ZJ, Sadler PJ. J Chem Soc, Dalton Trans. 1996:2867–2876. [Google Scholar]
- 18.Yao S, Plastaras JP, Marzilli LG. Inorg Chem. 1994;33:6061–6077. [Google Scholar]
- 19.Fröhling CDW, Sheldrick WS. J Chem Soc, Dalton Trans. 1997:4411–4420. [Google Scholar]
- 20.Siebert AFM, Sheldrick WS. J Chem Soc, Dalton Trans. 1997:385–393. [Google Scholar]
- 21.Christoforou AM, Marzilli PA, Marzilli LG. Inorg Chem. 2006;45:6771–6781. doi: 10.1021/ic0606375. [DOI] [PubMed] [Google Scholar]
- 22.Park GY, Wilson JJ, Song Y, Lippard SJ. Proc Natl Acad Sci USA. 2012;109:11987–11992. doi: 10.1073/pnas.1207670109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strickmann DB, Küng A, Keppler BK. Electrophoresis. 2002;23:74–80. doi: 10.1002/1522-2683(200201)23:1<74::AID-ELPS74>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 24.Küng A, Strickmann DB, Galanski M, Keppler BK. J Inorg Biochem. 2001;86:691–698. doi: 10.1016/s0162-0134(01)00225-2. [DOI] [PubMed] [Google Scholar]
- 25.Heudi O, Mercier-Jobard S, Cailleux A, Allain P. Biopharm Drug Dispos. 1999;20:107–116. doi: 10.1002/(sici)1099-081x(199903)20:2<107::aid-bdd161>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 26.Sun Y, Yin R, Gou S, Zhaojian J Inorg Biochem. 2012;112:68–76. doi: 10.1016/j.jinorgbio.2012.03.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.