

Abstract
The cysteinyl leukotrienes (cys-LTs), leukotriene C4 (LTC4) and its metabolites, LTD4 and LTE4, are proinflammatory lipid mediators in asthma and other inflammatory diseases. They are generated through the 5-lipoxygenase/LTC4 synthase (LTC4S) pathway and act via at least two distinct G protein-coupled receptors. The inhibition of human LTC4S will make a simple way to treat the cys-LT relevant inflammatory diseases. Here, we show that compounds having 5-(5-methylene-4-oxo-4,5-dihydrothiazol-2-ylamino) isophthalic acid moiety suppress LTC4 synthesis, glutathione conjugation to the precursor LTA4, in both an enzyme assay and a whole-cell assay. Hierarchical in silico screenings of 6 million compounds provided 300,000 dataset for docking, and after energy minimization based on the crystal structure of LTC4S, 111 compounds were selected as candidates for a competitive inhibitor to glutathione. One of those compounds showed significant inhibitory activity, and subsequently, its derivative 5-((Z)-5-((E)-2-methyl-3-phenylallylidene)-4-oxo-4,5-dihydrothiazol-2-ylamino) isophthalic acid (compound 1) was found to be the most potent inhibitor. The enzyme assay showed the IC50 was 1.9 µM and the corresponding 95% confidence interval was from 1.7 to 2.2 µM. The whole-cell assay showed that compound 1 was cell permeable and inhibited LTC4 synthesis in a concentration dependent manner.
Keywords: enzyme inhibitor, membrane protein, leukotriene, inflammation, in silico screening
Leukotrienes (LTs) are arachidonic acid metabolites obtained through the 5-lipoxygenase (5-LO) pathway (1). The 5-LO product, LTA4, is converted to the leukocyte chemoattractant LTB4 and leukotriene C4 (LTC4) by LTA4 hydrolase and LTC4 synthase (LTC4S), respectively (Fig. 1), in hematopoietic cells. LTC4 and its extracellular enzymatic metabolites LTD4 and LTE4, collectively called the cysteinyl leukotrienes (cys-LTs), play a role in smooth muscle constriction (2–4) and inflammation (5–8). The cys-LTs act through at least two distinct G protein-coupled receptors, cysteinyl leukotriene type 1 receptor (CysLT1R) and cysteinyl leukotriene type 2 receptor (CysLT2R) (9). The cys-LTs are particularly implicated in the pathophysiology of asthma, because treatment with CysLT1R antagonists or 5-LO inhibitors is efficacious to control asthma attacks (10–12), indicating the importance of cys-LT functions via the CysLT1R. In contrast, evidence of a role of CysLT2R in inflammatory diseases has been limited by the lack of a specific receptor antagonist. Studies with CysLT2R-deficient mice suggest that the CysLT2R also has proinflammatory functions, such as increasing vascular permeability in myocardial ischaemia-reperfusion injury (13) and passive cutaneous anaphylaxis (8), as well as promoting bleomycin-induced pulmonary fibrosis (8). Thus, cys-LTs participate in a wide range of inflammatory diseases as the studies on cys-LT receptors have been carried out. Therefore, the inhibition of LTC4S could provide an alternative and simple way to treat cys-LT relevant diseases.
Fig. 1.

5-LO pathway. The enzymes in the 5-LO pathway are shown in squares. In addition to these enzymes, there is 5-LO activating protein having no enzyme activity but presenting arachidonic acid to 5-LO.
LTC4S is a membrane protein embedded in the nuclear membrane that is the enzyme responsible for cys-LT biosynthesis (14, 15). LTC4S catalyses the conjugation of LTA4 with GSH to form LTC4, and the LTC4 is successively converted into LTE4 via LTD4 by extracellular hydrolytic enzymes (16). The crystal structure of the homo-trimer of LTC4S depicts the architecture of the active site formed at the space between two adjacent monomers in the homo-trimer as the biological unit which functions in the conjugation reaction (17–19). The active site is composed of two neighbouring substrate-binding sites, each with a distinct physicochemical character, the bent hydrophilic GSH and the extended hydrophobic LTA4 sites, respectively. In the hydrophilic GSH-binding site, the architecture consists of nine polar amino acid residues that allow the site-specific binding of GSH in the unique U-shaped conformation, the two terminal carboxyl groups of which reside in close vicinity with the inter carboxyl carbon distance of ∼3.9 Å. The hydrophobic LTA4-binding site is the crevice formed at the interface of the two hydrophobic transmembrane α-helix bundles neighbouring in the homo-trimer of LTC4S. These features of the active sites consisting of the neighbouring hydrophilic and hydrophobic regions are well suited to the binding of GSH and LTA4. The characteristic active site with the neighbouring hydrophilic and hydrophobic cavities is a good target to design inhibiting agents, because the hydrophilic pocket contributes to the site-specific binding through polar interaction, while the hydrophobic space enables the binding of inhibitors of strong affinity through hydrophobic interaction.
Here, we report that compounds with the common chemical structure of 5-(5-methylene-4-oxo-4,5-dihydrothiazol-2-ylamino) isophthalic acid from an in silico screening focused on the GSH-binding site exerted an inhibitory effect on LTC4S. Compound 1, 5-((Z)-5-((E)-2-methyl-3-phenylallylidene)-4-oxo-4,5-dihydrothiazol-2-ylamino) isophthalic acid (RTM100), was found to be the most potent inhibitor of LTC4S, acting in a concentration-dependent and reversible manner, and competed with GSH as one of the substrates. The IC50 was 1.9 µM for purified human LTC4S, and the 95% confidence interval (CI) of the IC50 was from 1.7 to 2.2 µM. The compound could be a potential lead for the future development of an LTC4S inhibitor.
Materials and Methods
In silico screening
A set of hierarchical in silico screenings of a compound database was applied to identify potent inhibitors of LTC4S. These hierarchical screenings were comprised of three types of filters: a query-based approach, molecular docking and a molecular mechanics/Poisson-Boltzmann surface area (MM/PB-SA) calculation.
For the first filter, the 2006.3 release of MDL (Accelrys) Screening Compounds Directory (SCD) was used as the compound database. This database contains ∼6 million compounds, and three-dimensional conformations were generated, ionized and energy minimized using LigPrep (Schrödinger Inc.), assuming a pH of 7.0. The SCD database was subjected to Lipinski’s ‘rule of five’ (20), and as a result, a filtered database containing ∼300,000 compounds was generated.
The second filter used was molecular docking. All the molecular docking experiments were carried out using GOLD version 3.2 and GLIDE version 4.5. The crystal structure of LTC4S [PDB entry: 2PNO(17)] was used. All the bound crystal water molecules, ligands and other organic compounds except for a GSH molecule between the first two chains were removed from the homo-trimer of LTC4S. Hydrogen atoms were added, and energy minimization of the hydrogen atoms was performed using the Molecular Operating Environment (MOE) programme (Chemical Computing Group Inc.). The molecular docking binding site was defined as the centre of the GSH molecule. In the GOLD docking, the standard default settings for the genetic algorithm parameters were used and the binding site radius was 12 Å. The docking run was performed using the GoldScore function. In the GLIDE docking, the standard precision mode was used, and the binding site was defined by a 12 Å × 12 Å × 12 Å box centred on the GSH molecule. First, the top-scoring 10,000 compounds from the first molecular docking screen using GOLD were selected from the filtered database above, then the top-scoring 1,000 compounds from the second molecular docking screen with GLIDE were selected. Finally, the docking for the top-scoring 1,000 compounds was generated by redocking using GOLD, because the third filter in this set of hierarchical in silico screenings was optimized for the docking obtained from GOLD.
As a post-molecular docking filter, the MM/PB-SA calculation of an energy-minimized complex structure was adopted. In the process, the complex conformations of the 1,000 compounds obtained from the second filter were energy-minimized using a MMs force field (hereafter we call this MM calculations). The binding affinities were calculated by the MM/PB-SA method using the coordinate sets of complexes obtained from the MM calculations. The in silico screening using MM/PB-SA method in the process was performed using the specialized computer ‘MDGRAPE-3’ system for the molecular dynamics simulations (21, 22). The procedures of the computational compound screening using the MM/PB-SA method were described in detail in a previous paper (23). The detailed information of structure-based screening containing molecular docking and MM/PB-SA calculation is shown in Supplementary Fig. S1 and Supplementary Table S1.
Preparation of the compound solution
All compounds were purchased from the Namiki Shoji Company Ltd. One hundred two of 111 compounds from the in silico screening were available for use. Ninety-four compounds could be dissolved at 10 mM in dimethyl sulfoxide (DMSO). We applied a unique RTM ID with a sequential number for each of the 94 compounds. The unique RTM IDs for the 94 compounds ranged from RTM001 to RTM094. The unique RTM IDs for the compounds utilized in the following experiment were assigned sequentially starting from RTM095. The purity of the compounds 1–14 determined by reverse phase high-performance liquid chromatography (RP-HPLC) analysis was higher than 95%. The RP-HPLC analysis for the purity test was performed with a SYSTEM GOLD 126 solvent module, a 168 detector, a 508 autosampler (Beckman-Coulter) and a Mightysil RP-18 GP (4.6 mm × 100 mm, 5 µm). Instead of Mightysil RP-18GP, Mightysil RP-8 GP (4.6 mm × 75 mm, 5 µm) was used for compound 13. Solvent A was distilled water, the pH of which was adjusted to 3.0 by trifluoroacetic acid, and solvent B was acetonitrile. The gradient was 10% (5% for compound 13) to 95% B over 8 min and a 5 min hold. The peak area was measured at the wavelength at which the peak exhibited the maximum absorption between 190 and 600 nm.
Protein expression and purification
The proteins for the enzyme assay were prepared in the same method as described in our previous report on the enzyme mechanism of LTC4S including kinetic analysis and high resolution crystal structure analysis (19). LTC4S from a Superose-12 column equilibrated with a solution of 20 mM MES-NaOH (pH 6.5), 0.1 M NaCl, 0.04% (w/v) dodecyl-β-d-maltoside (DDM), 1 mM DTT, 10% (v/v) glycerol and 5 mM GSH, was concentrated to ∼5 mg/ml, then stored at −80°C. Concentrations of the purified enzymes were determined based on UV absorption at 280 nm, and the milligram extinction coefficient 1.57 mg−1 • cm−1 was used. The purified samples were confirmed to comprise a single band using SDS-polyacrylamide gel electrophoresis.
Enzyme inhibition assay
The enzyme inhibition assay at a fixed concentration of compound was performed as follows. The test solution with 100 µM compound was composed of 2 µl of 10 mM compound solved in DMSO and 196 µl of the enzyme solution [20 ng LTC4S, 10 mM GSH, 50 mM Bis-Tris propane (pH 7.0), 10 mM MgCl2, 0.015% (w/v) DDM]. The test solution was incubated for 1 min on ice. Then, 2 µl of 2 mM LTA4 methyl ester (LTA4-Me) was added to the test solution to start the enzyme catalysis. The enzyme concentration was 5.7 nM in the enzyme catalysis. After 30 s incubation, 608 µl of the stop solution [methanol:acetic acid (75:1 by volume) containing prostaglandin B2 (PGB2) as the internal standard for RP-HPLC analysis] was added to the test solution. One hundred microlitre of the 808 µl solution was applied to the RP-HPLC analysis for the quantification of LTC4 methyl ester (LTC4-Me) as the product. The measurements were replicated twice (n = 2).
For the assay performed at different compound concentrations, the compound solution with an appropriate concentration instead of the 10 mM compound solution was added. The typical range of compound concentration was 0.1–200 µM. The concentrations of compound 1 were 100, 50, 5, 2.5, 1.0, 0.5, 0.1 and 0.01 µM. All measurements for compounds 1 and 2 were replicated three times (n = 3). All measurements for compounds 3–6 were replicated twice (n = 2). The IC50 and Hill slope were calculated by means of the nonlinear regression analysis of the concentration–activity relationship using Prism 5 (GraphPad Software, Inc.). The model used was 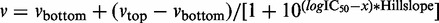 , where x is the logarithm of the inhibitor concentration.
, where x is the logarithm of the inhibitor concentration.
The LTA4-Me purchased from Cayman Chemical was dried under a N2 stream and then solved by ethanol with 3% (v/v) triethylamine, and the ethanol solution was used for the assay. The concentration of LTA4-Me was determined by UV absorbance (the molar extinction coefficient, 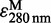 = 49,000 as shown in the product insert).
= 49,000 as shown in the product insert).
Reversibility of inhibition
Whether the compounds suppressed the enzyme activity of LTC4S in a reversible manner was tested as follows. The test solution was prepared as described earlier, with 1 and 10 mM compound solutions for compound 1 and compound 2, respectively. The test solution was incubated for 5 min on ice. The test solutions were diluted by the test solutions without both the enzyme and the compound, or without the enzyme only. Then, 2 µl of LTA4-Me was added to the diluted test solution to start the enzyme catalysis. After 8 min incubation, the enzyme catalysis was terminated by adding the stop solution. The quantification of LTC4-Me was performed using RP-HPLC analysis.
Kinetic analysis of the inhibition
The enzyme kinetic analysis was performed to assess whether compound 1 competes with GSH or LTA4-Me. In the assay with varied concentrations of GSH, GSH concentrations from 0.4 to 20 mM were used instead of the enzyme solution with the fixed concentration of GSH. For each GSH concentration, the test solutions with the compound concentrations of 2.5, 1.0, 0.5, 0.25 and 0 µM (DMSO vehicle) were measured. The test solution was incubated on ice for 1 min after adding the compound solution to the enzyme solution, and then LTA4-Me was mixed so as to be 20 µM LTA4-Me. After 2 min incubation, the enzyme catalysis was terminated by adding the stop solution. The quantification of LTC4-Me was performed using RP-HPLC analysis. The enzyme kinetic parameters were determined by means of nonlinear regression analysis of the data using Prism 5 (GraphPad Software, Inc.) with the competitive inhibition model.
In the assay using varied concentrations of LTA4-Me, the test solutions at the compound concentrations of 2.5, 1.0, 0.5 and 0 µM (DMSO vehicle) were prepared by mixing 196 µl of the enzyme solution and 2 µl of the compound solution at an appropriate concentration, and then the test solutions were incubated for 1 min on ice before the start of enzyme catalysis by the addition LTA4-Me. For each compound concentration, the enzyme activities at concentrations of LTA4-Me from 20 to 1.0 µM were measured. The enzyme catalysis was terminated by adding the stop solution after 2 min. The quantification of LTC4-Me was performed with RP-HPLC analysis. The enzyme kinetic parameters were determined by means of the nonlinear regression analysis of the data above using Prism 5 (GraphPad Software, Inc.) along with the noncompetitive inhibition model.
Reverse phase HPLC analysis
The quantification of LTC4-Me was performed using RP-HPLC with a SYSTEM GOLD 126 solvent module, a 168 detector, a 508 autosampler (Beckman-Coulter) and a YMC-Pack PolymerC18 (4.6 mm × 250 mm, S-6 µm) (19). The column was equilibrated with solvent A at a flow rate of 1 ml/min. A mixture of 80 ml methanol, 120 ml acetonitrile and 1.6 ml acetic acid was diluted to 1 l using water, and then the pH of the solution was adjusted to pH 6.0 by small aliquots of ethanolamine to prepare solvent A. Solvent B in the RP-HPLC analysis was 100% methanol. The mobile phase for the assay with LTA4-Me was 61% solvent B, and it was maintained for 13 min after injection of the sample. Then, solvent B was increased to 100% without delay and maintained at this level for 7 min. Subsequently, solvent B was returned to that of the first mobile phase without delay, and then kept for 10 min. The quantification of LTC4-Me was performed based on the ratio between the integrated areas of PGB2 as the internal standard and LTC4-Me. In this quantification, the molar extinction coefficients are 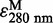 = 40,000 and 28,000 for LTC4-Me and PGB2, respectively (as described in the product insert of Cayman Chemical). The retention times of PGB2 and LTC4-Me in the mobile phase of 61% solvent B were 6.5 and 9.7 min, respectively.
= 40,000 and 28,000 for LTC4-Me and PGB2, respectively (as described in the product insert of Cayman Chemical). The retention times of PGB2 and LTC4-Me in the mobile phase of 61% solvent B were 6.5 and 9.7 min, respectively.
Effect of compounds on the 5-lipoxygenase pathway
The effect of compound 1 on the 5-LO pathway was assessed using bone marrow-derived mast cells (BMMCs) from BALB/c mice. BMMCs were established with mouse interleukin-3, as described (24). Two million BMMCs were incubated with various concentrations of compound 1 for 15 min at 37°C. Two micromolar of calcium ionophore A23187 was added and further incubated for 15 min. The reaction was terminated by adding PGB2/methanol. LTC4, LTB4, 6-trans-LTB4 as the decay products of LTA4, and 5-hydroxy-eicosatetraenoic acid (5-HETE) as the decay product of 5-hydroperoxy-eicosatetraenoic acid (5-HPETE), were measured by RP-HPLC, as described (24).
In addition to the whole-cell assay above, an assay using cell lysate was also performed. The lysates from 2 million BMMCs were prepared by sonication in Hanks’ buffer containing 1 mM CaCl2, 1 mM MgCl2 and 0.1% bovine serum albumin. The lysates were incubated with various concentrations of compound 1. GSH (10 mM) and LTA4-Me (20 µM) were then added to initiate the reaction. After incubation for 10 min at room temperature, the reaction was terminated by adding PGB2/methanol. Samples were analysed for LTC4-Me by RP-HPLC.
Statistical analysis
Statistical data were analysed with one-way ANOVA. Values of P < 0.05 were considered significant.
Results
In silico screening
Hierarchical computational screenings of the compound database were performed to search for potent inhibitors of LTC4S. The screenings consisted of both ligand-based and structure-based screenings. Based on these in silico screenings of large compound libraries, we selected 111 compounds for evaluation by enzyme assay.
The in silico screening focused on the GSH-binding site of LTC4S identified (Z)-5-(5-(4-ethylbenzylidene)-4-oxo-4,5-dihydrothiazol-2-ylamido) isophthalic acid (RTM085) (compound 2 hereafter). As described below, compound 2 at the concentration of 100 µM suppressed more than 90% of the enzyme activity in comparison with the enzyme activity of the DMSO vehicle (Fig. 2). Compound 2 in the in silico screening was predicted to bind at the GSH-binding site of LTC4S in a manner such that the isophthalic acid moiety projects deeply inside the GSH-binding site in the docking model (Fig. 3). Each of the two carboxyl groups of the isophthalic acid moiety interacted with both two neighbouring monomers which form the active site of the homo-trimer of LTC4S in the model, and the amino acid residues interacting with the two carboxyl groups were Ser23, Pro37 and Gln53 from one monomer and the amino acid residues of Arg51, Glu58, Tyr59 and Tyr97 from the other monomer. Furthermore, the carbonyl group at the thiazol moiety interacted with Tyr109. The ethylbenzyl group extended not towards the LTA4-binding site comprised of the space surrounded by the side chain of Trp116, and the α helices I and IV, but towards the weakly polarized shallow concave which was composed of the C-terminal part of the α-helix I and the base of the loop overlapping the GSH-binding site in the docking region (Fig. 3). In the shallow concave, the almost atoms existing in the distance of 5 Å from the ethylbenzyl group were the atoms of peptide backbone of Ile27, Arg30, Arg31, Arg34, Val35 and Ser36, and in addition to those, the hydrophobic alkyl chains of the guanidium side chains of Arg30, Arg31 and Arg34, and the hydroxyl side chain of Ser36 existed, resulting in the weakly polarized surface.
Fig. 2.

Residual enzyme activity in the presence of 100 µM compound. The open square at the left edge is the uninhibited enzyme activity in the DMSO vehicle (n = 18). Closed circles show the residual enzyme activities with the standard error at a 100 µM compound concentration (n = 3). Compound 2 is the rightmost point indicated by three asterisks. The asterisks indicate significantly decreased enzyme activities in comparison with the vehicle; ***P < 0.001, **P < 0.01, *P < 0.05.
Fig. 3.

The in silico docking model of compound 2 in the active site of the homo-trimer of LTC4S. Compound 2 and the amino acid residues surrounding compound 2 are represented by stick models with cyan and green carbons, respectively. The polar interactions between compound 2 and LTC4S are shown by the dashed lines. The gray and light purple ribbons are the neighbouring LTC4S monomers in the homo-trimer of LTC4S as the functional molecular unit. The roman numerals are the sequential number of α helices from the N-terminal.
Screening by enzyme assay
We performed the enzyme assay to assess an inhibitory effect of each compound obtained from the in silico screening at 100 µM in comparison with DMSO as a vehicle control (Fig. 2). Compounds having a similar retention time as LTC4-Me or PGB2 as the internal standard in the RP-HPLC analysis were excluded in the enzyme assay to avoid uncertainty by overlapping with the product or internal standard, resulting in 90 tested compounds. In comparison to the uninhibited LTC4S activity with the vehicle, the average % inhibition over the collection of the 90 tested compounds at 100 µM was 12 ± 23%. Seven of the 90 compounds reduced the enzyme activity of LTC4S to more than half of the vehicle. The efficacious compound was compound 2 with the % inhibition of 91.3 ± 1.6 (the rightmost point indicated with three asterisks in Fig. 2).
Based on the docking model from the in silico screening, the isophthalic acid and 4-oxo-thiazol moieties appeared to be important for the binding mode of compound 2, we performed an enzyme assay to assess the inhibitory effect of 13 additional compounds containing the 5-(5-methylene-4-oxo-4,5-dihydrothiazol-2-ylamino) isophthalic acid substructure (Fig. 4). Approximately half of the compounds reduced the enzyme activity more than 90% at 100 µM as compared with the DMSO vehicle, compounds, 1: 96.9 ± 0.4%; 2: 93.8 ± 1.2%; 3: 93.2 ± 0.1%; 4: 92.8 ± 0.5%; 5: 91.2 ± 0.2%; 6: 90.8 ± 0.3%. The average % inhibition over the 14 compounds at 100 µM, including compound 2, was 75 ± 20%, which was clearly higher than the % inhibition over the first selected 90 compounds from the in silico screening.
Fig. 4.

The molecular structure of the tested compounds with % inhibition in comparison with the DMSO vehicle, IC50, 95%CI regarding IC50 and absolute figure of Hill slope.
The IC50s of the six compounds were determined by the nonlinear regression with the variable Hill slope model. The IC50s and the corresponding 95% CIs, which are shown in the parentheses following IC50s, were 1.9 µM (1.7–2.2 µM) for compound 1, 15.7 µM (13.5–18.3 µM) for compound 2, 8.0 µM (5.0– 13.0 µM) for compound 3, 15.5 µM (13.6–17.7 µM) for compound 4, 14.5 µM (13.0–16.2 µM) for compound 5 and 12.6 µM (6.8–23.3 µM) for compound 6 (Fig. 4; Supplementary Fig. S2).
Reversibility of the inhibitory effects of the compounds on the LTC4S activity was examined by 10-fold dilution of the concentration of two representative compounds, that is, compound 1 with the lowest IC50, and compound 2 with the largest absolute figure of Hill slope (Fig. 5). In comparison with the DMSO vehicle, the residual enzyme activity of LTC4S recovered from 7.6 ± 0.5% to 48.3 ± 1.0% by the 10 times dilution from 10 to 1.0 µM of compound 1, and from 3.1 ± 0.5% to 35.5 ± 2.7% by the 10 times dilution from 100 to 10 µM of compound 2. These results show that compounds 1 and 2 inhibit the LTC4S activity reversibly.
Fig. 5.
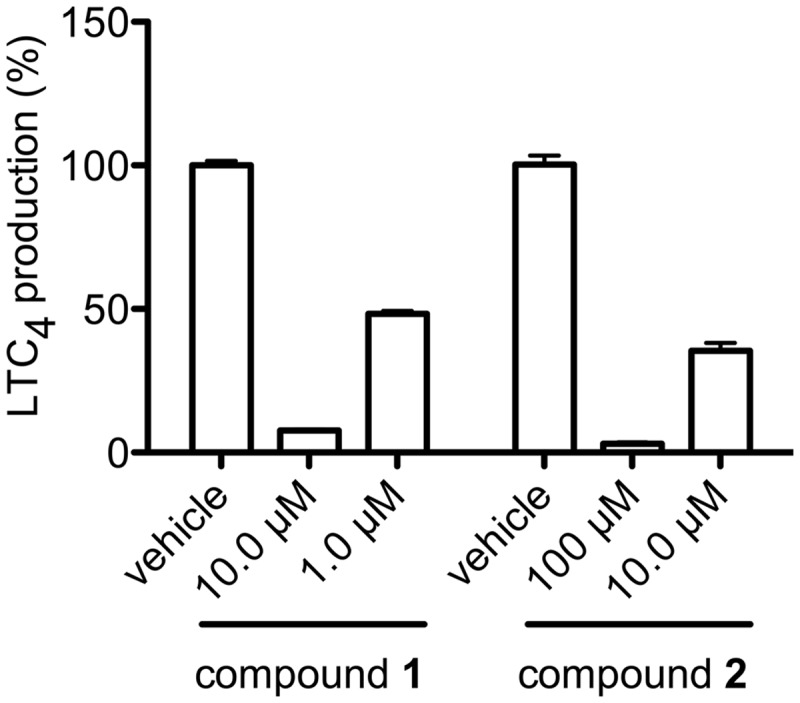
Reversible inhibition of compounds 1 and 2. The enzyme activities with 10 µM of compound 1 and 100 µM of compound 2 recovered from 7.6 ± 0.5 to 48.3 ± 1.0% and from 3.1 ± 0.5 to 35.5 ± 2.7% by 10 times dilution, respectively.
Analysis of inhibition kinetics
Compound 1, which had the smallest IC50, was subjected to enzyme kinetics analysis. Double reciprocal plots showed that compound 1 inhibited the LTC4S activities in a competitive manner against GSH and in a noncompetitive manner against LTA4-Me (Fig. 6). The Ki values of the data on GSH and LTA4-Me were 12.5 ± 2.1 and 0.55 ± 0.03 µM in the nonlinear analyses, respectively. The estimated Km for GSH and LTA4-Me were 0.27 ± 0.05 mM and 5.0 ± 0.3 µM, respectively. These Km values are comparable with those in our previous report (19).
Fig. 6.

Double reciprocal representations of enzyme kinetics. Enzyme kinetic analyses with a varying GSH concentration (A) and with a varying LTA4-Me concentration (B) were performed to determine the inhibition mechanism of compound 1, respectively. In panel A, the concentrations of compound 1 were 2.5 µM (circle), 1.0 µM (square), 0.5 µM (triangle), 0.25 µM (inversed triangle) and 0 µM (diamond). In panel B, the concentrations of compound 1 were 2.5 µM (circle), 1.0 µM (square), 0.5 µM (triangle) and 0 µM (diamond). Compound 1 was concluded to be a competitive inhibitor to GSH and a noncompetitive inhibitor to LTA4-Me.
The effect of the compound 1 on LTC4S embedded in the cell membrane
The effect of compound 1 on the enzyme activity of LTC4S was assessed using the cell lysate of 2 million BMMCs from BALB/c mice (Fig. 7), because the biological membrane, in which the membrane proteins are embedded, sometimes affects the function of these membrane proteins. Compound 1 could inhibit the LTC4S activities in the cell membrane in a concentration-dependent manner. The IC50 was 4.9 µM, and the corresponding 95% CI was from 3.3 to 7.1 µM.
Fig. 7.

The concentration-dependent inhibition of LTC4S activity by compound 1 using mouse BMMC lysates. Values show the % generation of LTC4-Me in the presence of various concentrations of compound 1 in comparison with the DMSO vehicle. Results are means ± SD from three experiments.
The whole-cell assay
The effect of compound 1 on the biosynthesis of the 5-LO pathway products was examined by whole-cell assay using mouse BMMCs after activation with the calcium ionophore, A23187. Compound 1 inhibited the biosyntheses of LTC4 and 5-HETE, the stable decomposition product of 5-HPETE, in a concentration-dependent manner. The extrapolated EC50 values of LTC4 and 5-HETE biosyntheses, which were calculated by the nonlinear regression analyses of the data without the bottom plateau region as the maximally inhibited response, were 64.7 and 114.9 µM, respectively (Fig. 8), and the corresponding 95% CIs were from 28.2 to 148.2 µM and from 43.0 to 306.9 µM, respectively. Compound 1 also inhibited the biosynthesis of LTB4 and 6-trans-LTB4, as the stable decomposition product of LTA4, although their generations were more than 50% of the controls even at 500 µM of compound 1.
Fig. 8.

Effects of compound 1 on the 5-LO pathway in mouse BMMCs. The panels A, B, C and D show the % production of LTC4, LTB4, 6-trans-LTB4 and 5-HETE, respectively, in the presence of various concentrations of compound 1 in comparison with the DMSO vehicle. Results are means ± SE from five experiments.
Discussion
In this study, in silico screening using the crystal structure coordinate of LTC4S was performed to identify candidate inhibitors that bind the GSH-binding site of LTC4S. The GSH-binding site is a good target for site-specific inhibitor molecules. Because there are many polar amino acid residues interacting with the characteristically bent form of the bound GSH. Thus, inhibitors can have polar interactions as the essential determinant of the site-specific binding in an exothermic manner. Furthermore, near the hydrophilic GSH-binding site, there is the hydrophobic pocket as the LTA4-binding site, which can accommodate the hydrophobic moiety of the candidates to strengthen the inhibitor binding. The LTA4-binding site was determined to be the pocket surrounding the transmembrane α helices I, II, IV, and the side chain of Trp116, as shown in Fig. 3 (17–19).
The most potent compound was compound 1, 5-((Z)-5-((E)-2-methyl-3-phenylallylidene)-4-oxo-4,5-dihydrothiazol-2-ylamino) isophthalic acid. Compound 1 was found among the derivatives of the common backbone structure, 5-(5-methylene-4-oxo-4,5-dihydrothiazol-2-ylamino) isophthalic acid in compound 2. The IC50 for the purified human LTC4S was 1.9 µM, and the corresponding 95% CI was from 1.7 to 2.2 µM, and the Ki values from the experiments with varying concentrations of GSH and LTA4-Me were 12.5 ± 2.1 and 0.55 ± 0.03 µM, respectively. The enzyme kinetics analysis showed that compound 1 inhibited LTC4S competitively for GSH and noncompetitively for LTA4-Me. The competitive inhibition for GSH indicates that compound 1 binds at the GSH-binding site.
The isophthalate and dihydrothiazol moiety contribute potent inhibitory activity of compound 1 and 2. The 5-(5-methylene-4-oxo-4,5-dihydrothiazol-2-ylamino) isophthalic acid moiety of compound 2 was shown to contribute to the binding of compound 2 at the GSH-binding site through the polar interactions, and the carbonyl group of the dihydrothiazol moiety interacting with Tyr109 determine the direction of the following ethylbenzyl moiety (Fig. 3). Compound 1 possesses the 5-(5-methylene-4-oxo-4,5-dihydrothiazol-2-ylamino) isophthalic acid moiety, and the moiety is shared with compound 2. Therefore, compound 1 would bind the GSH-binding site of LTC4S in a manner such that the isophthalic acid moiety resides at the deeper side of the GSH-binding site, and the (E)-2-methyl-3-phenylallylidene moiety extends towards the region including the C-terminal of the α-helix I and the base of the loop between the α helices I and II. As described earlier, the surface of the region was weakly polarized surface. The weakly polarized feature looks like to be better to accept the phenylallylidene moiety having the weakly polarized surface due to the π electrons.
Compounds 1 and 2 suppressed the enzyme activity of LTC4S as a reversible inhibitor (Fig. 5). This reversible suppression shows that the compounds do not impose denaturation or aggregation of the homo-trimer of LTC4S. However, there may be some structural changes that are implied by the compound-dependent Hill slope variation. The crystal structure of the homo-trimer of LTC4S bound GSH showed that LTC4S forms three equivalent active sites between interfaces of homo-trimer (17, 19). The binding of GSH as the natural substrate stabilizes the homo-trimer of LTC4S (25). It indicates some structural changes occur by the binding of GSH. Compound 1 is a competitive inhibitor for GSH. Therefore, the biding of inhibitor would induce some structural changes of the homo-trimer of LTC4S.
Substituent at the position of R1 in Fig. 4 giving each compound its own structural characteristic affected Hill slope of each compound. As discussed earlier, the compounds would share the common binding mode of the substituent where the substituent interacts with the loop between the α helices I and II. There are three GSH-binding sites in the homo-trimer of LTC4S, and the neighbouring two GSH-binding sites share an α-helix II. It infers that α-helix II following the loop interacting with the substituent R1 may be a mediator transmitting the substituent-dependent structural change of the loop, resulting in the compound-dependent Hill slope variation, although the detailed mechanism is remained to be elucidated.
The whole-cell assay with BMMCs showed that compound 1 is cell permeable and suppresses the 5-LO pathway (Fig. 8). The proteins relevant to cys-LT biosynthesis in the 5-LO pathway of the arachidonic acid cascade are 5-LO, 5-LO activating protein and LTC4S. 5-LO produces LTA4 by the oxygenation of arachidonic acid at the start of the 5-LO pathway (Fig. 1). Ten micromolar of compound 1 suppressed the production of LTC4 and 6-trans-LTB4 as the stable decay product of LTA4 in comparison with a lower concentration of compound 1. In contrast, the production of LTB4 and 5-HETE as the stable decay product of 5-HPETE at 10 µM of compound 1 was maintained at almost the same level to those of the lower concentrations. The maintenance of the LTB4 and 5-HETE productions shows that cell viability was maintained up to 10 µM of compound 1 at least. Therefore, until the concentration of 10 µM of compound 1, any reduction in LTC4 and 6-trans-LTB4 would be independent of an impact on cell viability.
As described earlier, the production of 6-trans-LTB4 is suppressed at a substantially lower concentration of compound 1 than 5-HETE. It suggests that compound 1 could be an inhibitor, which affects the dehydratase activity of 5-LO as the second step in the two-step catalysis of 5-LO. In this two-step catalysis, 5-LO first produces 5-HPETE by the oxygenation of arachidonic acid. 5-HETE is then derived from the 5-HPETE from the first oxygenase activity of 5-LO. Therefore, the 5-HETE production resistant to compound 1 means that the inhibitory effect of compound 1 on the oxygenase activity of 5-LO is limited. In the second step, 5-LO converts 5-HPETE to LTA4 by the dehydratase activity of 5-LO. As the 6-trans-LTB4 detected in the assay was originated from the LTA4 obtained from the second step dehydratase activity of 5-LO, the reduction of 6-trans-LTB4 production would reflect the LTA4 production due to the inhibition of the second step dehydratase activity of 5-LO rather than the first step oxygenase activity of 5-LO. Compound 1 might act as a dual inhibitor with effects on both LTC4S and 5-LO, as indomethacin, originally developed as a cyclooxygenase inhibitor, is a broad-spectrum drug affecting various proteins (26–32).
Supplementary Data
Supplementary Data are available at JB Online.
Funding
This work was supported by a National Institutes of Health grant HL90630 in part (to Y.K.).
Conflict of Interest
None declared.
Supplementary Material
Acknowledgements
We thank Dr K. Frank Austen for his valuable suggestions and support for this study. We are grateful for Dr N. Fujii’s suggestion and encouragement. We thank RIKEN Advanced Center for Computing and Communication for an allocation of computational resources on the RIKEN Integrated Cluster of Clusters (RICC) facility.
Glossary
Abbreviations
- BMMC
bone marrow-derived mast cell
- CI
confidence interval
- cys-LT
cysteinyl leukotriene
- CysLT1R
cysteinyl leukotriene type 1 receptor
- CysLT2R
cysteinyl leukotriene type 2 receptor
- DDM
dodecyl-β-d-maltoside
- DMSO
dimethyl sulfoxide
- 5-HETE
5-hydroxy-eicosatetraenoic acid
- 5-HPETE
5-hydroperoxy-eicosatetraenoic acid
- 5-LO
5-lipoxygenase
- LT
leukotriene
- LTA4-Me
leukotriene A4 methyl ester
- LTC4
leukotriene C4
- LTC4-Me
leukotriene C4 methyl ester
- LTC4S
leukotriene C4 synthase
- MM
molecular mechanics
- MM/PB-SA
molecular mechanics/Poisson-Boltzmann and surface area
- PGB2
prostaglandin B2
- RP-HPLC
reverse phase high-performance liquid chromatography
- SCD
Screening Compounds Directory
References
- 1.Peters-Golden M, Henderson WR., Jr. Leukotrienes. N. Engl. J. Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 2.Soter NA, Lewis RA, Corey EJ, Austen KF. Local effects of synthetic leukotrienes (LTC4, LTD4, LTE4, and LTB4) in human skin. J. Invest. Dermatol. 1983;80:115–119. doi: 10.1111/1523-1747.ep12531738. [DOI] [PubMed] [Google Scholar]
- 3.Dahlén SE, Hedqvist P, Hammarström S, Samuelsson B. Leukotrienes are potent constrictors of human bronchi. Nature. 1980;288:484–486. doi: 10.1038/288484a0. [DOI] [PubMed] [Google Scholar]
- 4.Dahlén SE, Björk J, Hedqvist P, Arfors KE, Hammarström S, Lindgren JÅ, Samuelsson B. Leukotrienes promote plasma leakage and leukocyte adhesion in postcapillary venules: in vivo effects with relevance to the acute inflammatory response. Proc. Natl. Acad. Sci. USA. 1981;78:3887–3891. doi: 10.1073/pnas.78.6.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paruchuri S, Tashimo H, Feng C, Maekawa A, Xing W, Jiang Y, Kanaoka Y, Conley P, Boyce JA. Leukotriene E4 induced pulmonary inflammation is mediated by the P2Y12 receptor. J. Exp. Med. 2009;206:2543–2555. doi: 10.1084/jem.20091240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maekawa A, Xing W, Austen KF, Kanaoka Y. GPR17 regulates immune pulmonary inflammation induced by house dust mites. J. Immunol. 2010;185:1846–1854. doi: 10.4049/jimmunol.1001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim DC, Hsu FI, Barrett NA, Friend DS, Grenningloh R, Ho IC, Al-Garawi A, Lora JM, Lam BK, Austen KF, Kanaoka Y. Cysteinyl leukotrienes regulate Th2 cell-dependent pulmonary inflammation. J. Immunol. 2006;176:4440–4448. doi: 10.4049/jimmunol.176.7.4440. [DOI] [PubMed] [Google Scholar]
- 8.Beller TC, Maekawa A, Friend DS, Austen KF, Kanaoka Y. Targeted gene disruption reveals the role of the cysteinyl leukotriene 2 receptor in increased vascular permeability and in bleomycin-induced pulmonary fibrosis in mice. J. Biol. Chem. 2004;279:46129–46134. doi: 10.1074/jbc.M407057200. [DOI] [PubMed] [Google Scholar]
- 9.Austen KF, Maekawa A, Kanaoka Y, Boyce JA. The leukotriene E4 puzzle: finding the missing pieces and revealing the pathobiologic implications. J. Allergy Clin. Immunol. 2009;124:406–414. doi: 10.1016/j.jaci.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Byrne PM, Gauvreau GM, Murphy DM. Efficacy of leukotriene receptor antagonists and synthesis inhibitors in asthma. J. Allergy Clin. Immunol. 2009;124:397–403. doi: 10.1016/j.jaci.2009.05.029. [DOI] [PubMed] [Google Scholar]
- 11.Diamant Z, Grootendorst DC, Veselic-Charvat M, Timmers MC, De Smet M, Leff JA, Seidenberg BC, Zwinderman AH, Peszek I, Sterk PJ. The effect of montelukast (MK-0476), a cysteinyl leukotriene receptor antagonist, on allergen-induced airway responses and sputum cell counts in asthma. Clin. Exp. Allergy. 1999;29:42–51. doi: 10.1046/j.1365-2222.1999.00447.x. [DOI] [PubMed] [Google Scholar]
- 12.Fish JE, Kemp JP, Lockey RF, Glass M, Hanby LA, Bonuccelli CM. Zafirlukast for symptomatic mild-to-moderate asthma: a 13-week multicenter study. The Zafirlukast Trialists Group. Clin. Ther. 1997;19:675–690. doi: 10.1016/s0149-2918(97)80092-6. [DOI] [PubMed] [Google Scholar]
- 13.Jiang W, Hall SR, Moos MP, Cao RY, Ishii S, Ogunyankin KO, Melo LG, Funk CD. Endothelial cysteinyl leukotriene 2 receptor expression mediates myocardial ischemia reperfusion injury. Am. J. Pathol. 2008;172:592–602. doi: 10.2353/ajpath.2008.070834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lam BK, Penrose JF, Freeman GJ, Austen KF. Expression cloning of a cDNA for human leukotriene C4 synthase, an integral membrane protein conjugating reduced glutathione to leukotriene A4. Proc. Natl Acad. Sci. USA. 1994;91:7663–7667. doi: 10.1073/pnas.91.16.7663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Welsch DJ, Creely DP, Hauser SD, Mathis KJ, Krivi GG, Isakson PC. Molecular cloning and expression of human leukotriene C4 synthase. Proc. Natl Acad. Sci. USA. 1994;91:9745–9749. doi: 10.1073/pnas.91.21.9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samuelsson B, Dahlén SE, Lindgren JÅ, Rouzer CA, Serhan CN. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science. 1987;237:1171–1176. doi: 10.1126/science.2820055. [DOI] [PubMed] [Google Scholar]
- 17.Ago H, Kanaoka Y, Irikura D, Lam BK, Shimamura T, Austen KF, Miyano M. Crystal structure of a human membrane protein involved in cysteinyl leukotriene biosynthesis. Nature. 2007;448:609–612. doi: 10.1038/nature05936. [DOI] [PubMed] [Google Scholar]
- 18.Martinez Molina D, Wetterholm A, Kohl A, McCarthy AA, Niegowski D, Ohlson E, Hammarberg T, Eshaghi S, Haeggström JZ, Nordlund P. Structural basis for synthesis of inflammatory mediators by human leukotriene C4 synthase. Nature. 2007;448:613–616. doi: 10.1038/nature06009. [DOI] [PubMed] [Google Scholar]
- 19.Saino H, Ukita Y, Ago H, Irikura D, Nisawa A, Ueno G, Yamamoto M, Kanaoka Y, Lam BK, Austen KF, Miyano M. The catalytic architecture of leukotriene C4 synthase with two arginine residues. J. Biol. Chem. 2011;286:16392–16401. doi: 10.1074/jbc.M110.150177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliver. Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 21.Narumi T, Ohno Y, Okimoto N, Koishi T, Suenaga A, Futatsugi N, Yanai R, Himeno R, Fujikawa S, Ikei M, Taiji M. SC'06 Proceedings of the 2006 ACM/IEEE Conference on Supercomputing. 2006. A 55 Tflops simulation of amyloid-forming peptides from Yeast Prion Sup35 with the special-purpose computer System MD-GRAPE3. In. Article No. 49 in CD-ROM. [Google Scholar]
- 22.Taiji M. Hot Chips 16. 2004. MDGRAPE-3 chip: a 165 Gflops application specific LSI for molecular dynamics simulations. IEEE Computer Society in CD-ROM. [Google Scholar]
- 23.Okimoto N, Futatsugi N, Fuji H, Suenaga A, Morimoto G, Yanai R, Ohno Y, Narumi T, Taiji M. High-performance drug discovery: computational screening by combining docking and molecular dynamics simulations. PLoS Comput. Biol. 2009;5:e1000528. doi: 10.1371/journal.pcbi.1000528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanaoka Y, Maekawa A, Penrose JF, Austen KF, Lam BK. Attenuated zymosan-induced peritoneal vascular permeability and IgE-dependent passive cutaneous anaphylaxis in mice lacking leukotriene C4 synthase. J. Biol. Chem. 2001;276:22608–22613. doi: 10.1074/jbc.M103562200. [DOI] [PubMed] [Google Scholar]
- 25.Izumi T, Honda Z, Ohishi N, Kitamura S, Tsuchida S, Sato K, Shimizu T, Seyama Y. Solubilization and partial purification of leukotriene C4 synthase from guinea-pig lung: a microsomal enzyme with high specificity towards 5,6-epoxide leukotriene A4. Biochim. Biophys. Acta. 1988;959:305–315. [PubMed] [Google Scholar]
- 26.Baek SJ, Kim KS, Nixon JB, Wilson LC, Eling TE. Cyclooxygenase inhibitors regulate the expression of a TGF-β superfamily member that has proapoptotic and antitumorigenic activities. Mol. Pharmacol. 2001;59:901–908. [PubMed] [Google Scholar]
- 27.Clish CB, Sun YP, Serhan CN. Identification of dual cyclooxygenase-eicosanoid oxidoreductase inhibitors: NSAIDs that inhibit PG-LX reductase/LTB4 dehydrogenase. Biochem. Biophys. Res. Commun. 2001;288:868–874. doi: 10.1006/bbrc.2001.5841. [DOI] [PubMed] [Google Scholar]
- 28.Hirai H, Tanaka K, Takano S, Ichimasa M, Nakamura M, Nagata K. Cutting edge: agonistic effect of indomethacin on a prostaglandin D2 receptor, CRTH2. J. Immunol. 2002;168:981–985. doi: 10.4049/jimmunol.168.3.981. [DOI] [PubMed] [Google Scholar]
- 29.Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors α and γ are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem. 1997;272:3406–3410. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- 30.Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Butler T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. A subset of NSAIDs lower amyloidogenic Aβ 42 independently of cyclooxygenase activity. Nature. 2001;414:212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- 31.Yamada T, Komoto J, Watanabe K, Ohmiya Y, Takusagawa F. Crystal structure and possible catalytic mechanism of microsomal prostaglandin E synthase type 2 (mPGES-2) J. Mol. Biol. 2005;348:1163–1176. doi: 10.1016/j.jmb.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 32.Hori T, Ishijima J, Yokomizo T, Ago H, Shimizu T, Miyano M. Crystal structure of anti-configuration of indomethacin and leukotriene B4 12-hydroxydehydrogenase/15-oxo-prostaglandin 13-reductase complex reveals the structural basis of broad spectrum indomethacin efficacy. J. Biochem. Tokyo. 2006;140:457–466. doi: 10.1093/jb/mvj176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.