

Abstract
Double-strand breaks in DNA can be repaired by homologous recombination including break-induced replication. In this reaction, the end of a broken DNA invades an intact chromosome and primes DNA replication resulting in the synthesis of an intact chromosome. Break-induced replication has also been suggested to cause different types of genome rearrangements.
The ability of cells to repair double-strand breaks (DSBs) is critical for cell viability. Failure to correct such damage can result in cell cycle arrest, cell death and, if repaired incorrectly, the loss of genetic information or the accumulation of mutations. DSBs can arise in cells by exposure to ionizing radiation1 and other types of DNA-damaging agents or through mechanical stress. In addition, some cellular processes, like DNA replication, can generate DSBs. These could arise by the action of nucleases at unprotected sites where replication forks stall, as a consequence of DNA polymerases (Pols) replicating across nicked chromosomes, as well as by the active processing of stalled replication forks by specific enzyme systems2-4. DSBs can also occur naturally and play important roles in processes such as meiosis5, mating-type switching6 and mammalian V(D)J recombination7.
Cells have developed a number of mechanisms to repair such lesions. In Saccharomyces cerevisiae, most of the repair of DSBs is carried out by mechanisms that promote homologous recombination. This requires the existence in the cell of sequences that are homologous to those affected by the DSB. Non-homologous end-joining also rejoins DSBs in S. cerevisiae albeit less efficiently than homologous recombination; non-homologous end-joining appears to be of greater importance in mammalian cells8.
General models of recombination
Several recent reviews have discussed recombination and the repair of DSBs in detail4,9-14. Given the increasing evidence that translocations and other types of genome rearrangements arise from inappropriate repair of DSBs, we will focus on the repair of DSBs by a homologous recombination pathway that involves extensive DNA synthesis. Such a process, termed break-induced replication (BIR), has been suggested based on the observations by Voelkel-Meiman and Roeder15,16. In this study, activation of the transcription enhancer element HOT1 creates a recombination hotspot suggested to act by generating DSBs that are processed resulting in strains with distal markers converted to those present in the homologous chromosome. The resulting gene conversion, which could extend over regions as long as 75 kb, was proposed to arise by replication of a primer structure generated by the centromere-containing fragment invading the homologous chromosome in a process analogous to that described for bacteriophage T4 (Ref. 17). Subsequent studies have provided greater mechanistic detail of this type of repair18,19. Additional evidence for a BIR mechanism is suggested by Morrow et al.20 who describe a process by which a chromosome fragment acquires all sequences of an endogenous targeted chromosome. This event can be explained as unscheduled DNA replication initiated by the free end of the chromosomal fragment.
The BIR model for the repair of DSBs incorporates many features of the DSB repair model proposed by Szostak et al.21 (Fig. 1a). In both cases the ends of the broken DNA molecule are degraded by exonucleases creating 3′ overhangs that invade the homologous template. The result is the formation of a D-loop in which the annealed invading end serves as a primer that is extended by the DNA synthesis machinery. This propagates the D-loop, which, in the DSB repair model, will allow pairing of the second 3′ single-strand terminal end of the DSB, which then primes DNA synthesis generating two symmetrical Holliday junctions. In this case, resolution can occur with or without crossing-over, and the resulting DNA molecules will contain replicated regions comprising both a newly synthesized DNA strand and a complementary template DNA strand, the result of which is essentially semiconservative DNA replication. Alternatively, the BIR model (Fig. 1b) involves extensive leading-strand synthesis, which occurs within the D-loop. This occurs as branch migration follows the replication fork assembled on the invading strand. Coordinated lagging-strand synthesis then converts the single strand produced by leading-strand synthesis to a double strand. The final result is conservative DNA synthesis, where the recipient chromosome has both strands newly synthesized. This model is equivalent to recombination-dependent replication proposed for E. coli and bacteriophage T4 (Refs 17,22). One interesting implication of this model is that copying of the template DNA could reach the end of the homologous chromosome, thus allowing the recipient chromosome to acquire telomere sequences23.
Figure 1.

Two models for the repair of double-strand breaks (DSBs). (a) In this model, proposed by Szostak et al.21, the ends of the DSB (1) are digested by 5′→3′ nucleases (2). The resulting 3′ overhang invades another DNA molecule at a homologous region (3) generating a primer end that is extended by a DNA polymerase (4). The strand displaced by the propagation of the D-loop can pair with sequences from the other end of the break (3), forming a primer for repair synthesis (4), which is extended exclusively by leading-strand synthesis. Two Holliday junctions are formed (5) that can be resolved independently, yielding products without an associated crossover (6) of the flanking regions (if both Holliday junctions are resolved by cleaving in the same manner) or with (7) an associated crossover (when both Holliday junctions are resolved in an opposite manner). (b) In the break-induced replication (BIR) model, the primer generated by the invading 3′ end (3) directs leading-strand synthesis (4). Lagging-strand synthesis is initiated on the leading strand producing a double-stranded DNA molecule with identical information to that of the template chromosome (5,6).
Biochemistry of recombination
A number of genes have been suggested to encode proteins that participate in the repair of DSBs by homologous recombination. Many of these are the so-called RAD genes that were identified as the genes defective in yeast mutants that were sensitive to X rays. Many of these genes are members of the RAD50 epistasis group and include RAD50, RAD51, RAD52, RAD54, RAD55, RAD57, MRE11 and XRS2 (Refs 9-11). The biochemical properties of the gene products of these recombination genes are presently the subject of extensive analysis. Rad52p has been shown to possess ssDNA-binding activity, to interact with another ssDNA-binding protein, replication protein A (RPA), and to promote single-strand annealing24-26 (Fig. 2). Strand exchange activity has been demonstrated in vitro for the RecA homolog Rad51p (Refs 27-29), which forms nucleoprotein filaments with either ssDNA or dsDNA. Strand exchange by Rad51p is facilitated by its association with Rad52p, which might help overcome the inhibitory effect of RPA under some reaction conditions (Fig. 2). A similar Rad51p accessory role has been assigned to the Rad55p–Rad57p complex30 (Fig. 2). The role of Rad54p is still unclear, although its sequence shows homology to DNA helicases and displays DNA-dependent ATPase activity in vitro31. Multiple interactions have been described between the above mentioned proteins. This has led to the proposed existence of a ‘recombinosome’ that could function early in recombination32. The composition of this recombinosome might vary depending on the type of repair required, because certain factors are not essential for each repair mechanism. For example, Rad51p is not entirely essential for BIR (Ref. 33).
Figure 2.
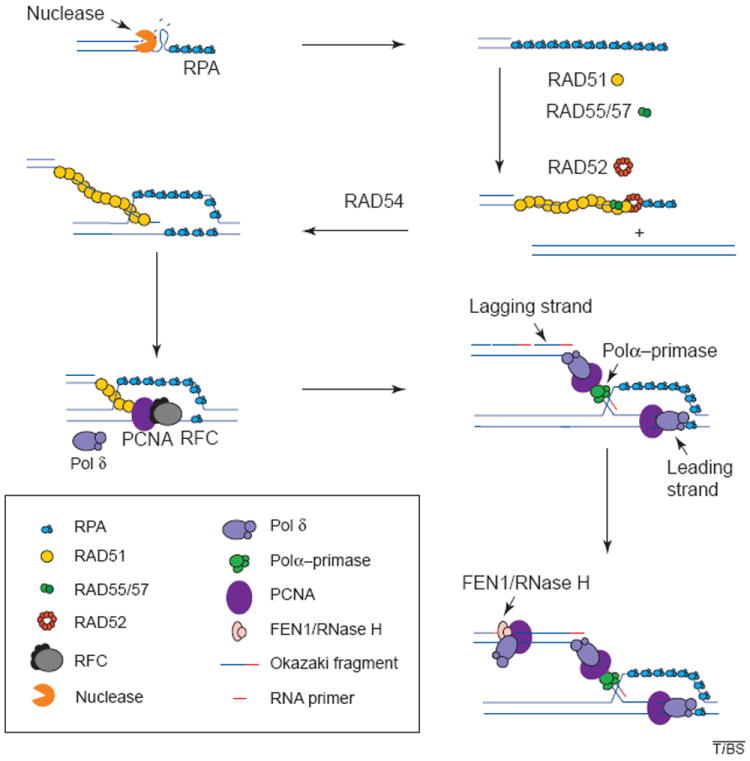
Recombination and associated DNA synthesis. The resection of the ends of the double strand creates a 3′ single-stranded DNA molecule. The binding of RPA to the single strand protects the DNA from degradation and disrupts any secondary structure. RAD51 is then loaded onto the DNA strand with the aid of RAD52, RAD55 and RAD57, and in the presence of RAD54, the RAD51 filament carries out strand exchange. The primer generated is recognized by the polymerase (Pol) δ accessory factor RFC, which loads PCNA onto the primer end generating a complex that is recognized by Pol δ or ε, which will carry out synthesis of the leading strand. Lagging-strand synthesis initiates when RNA primers are synthesized on the leading strand by the primase activity of the Pol-α–primase complex and are immediately extended by the Pol α into short DNAs. The Pol δ (or ε) system is then assembled on these DNA primers, replacing Pol α. The primers are then extended to yield Okazaki fragments. These accumulate on the lagging strand until they mature by the combined action of FEN1 and RNase H nucleases, which remove the RNA moiety of the fragment, leaving a gap that is then filled in by DNA Pol. DNA ligase seals the resulting nick yielding an intact double-stranded DNA molecule.
Biochemistry of replication
Less is known about the steps that follow strand invasion. However, certain predictions can be made based on the proposed models (Fig. 1). The Szostak model21 only calls for continuous DNA synthesis, because strand invasion is carried out by two 3′ ends that are extended until the double Holliday junction is formed. This type of synthesis is carried out by the Pol δ or ε systems that assemble on the DNA primer, which, in homologous recombination, is generated by strand invasion. Assembly of the Pol δ or ε systems begins by recognition and binding of the primer end by the multi-subunit protein complex, replication factor C (RFC), which loads the processivity factor PCNA onto the primer end in an ATP-dependent reaction34,35 (Fig. 2). The PCNA–primer complex is efficiently recognized by Pol δ or ε, which subsequently extends the primer in a stable processive manner36-38 (Fig. 2). There is also genetic evidence supporting the role of lagging-strand synthesis in the repair of DSBs (Ref. 39). However, DSB repair by BIR requires both leading- and lagging-strand synthesis, essentially as it occurs in recombination-dependent replication in E. coli and bacteriophage T4 (Refs 18,19,22). In this case, RNA priming is essential for lagging-strand synthesis. Priming in eukaryotes is carried out by the primase activity of the RNA-primase–Pol-α complex40,41 and is essential for lagging-strand synthesis, as well as for the initiation of leading-strand synthesis in normal DNA replication. Once the RNA primer is synthesized, it is extended by the Pol α generating a short DNA primer onto which the Pol δ system is assembled, similar to the leading-strand synthesis step (Fig. 2). The loaded Pol δ or ε now extends the primer until it reaches the 5′ end of the next Okazaki fragment. The switch from Pol α to Pol δ is referred to as polymerase switching42. RNA primers are removed by the concerted action of RNase H and FEN1 and the gap created is filled in by Pol δ or ε (Ref. 37). Finally, the resulting nick is then sealed by DNA ligase.
Holmes and Haber39 analysed the effect of conditional mutations in replication genes on the HO-induced mating-type switch in yeast. Repair of the DSB was inhibited by mutations affecting DNA Pol α, Rad27p (the yeast homolog of the mammalian FEN1 nuclease) and DNA primase, all components of the lagging-strand synthesis machinery. In addition, mutations affecting PCNA, RFC Pol δ and Pol ε greatly inhibited the repair of the DSB generated by the HO endonuclease. These results support the notion that both leading- and lagging-strand synthesis are necessary for the repair of DSBs. These protein requirements are likely to apply to BIR, although this has not been examined directly.
It would be interesting to examine how the DNA synthesis machinery engages the primer structure that is thought to be part, at some point, of a nucleoprotein complex containing recombination factors. No direct interaction between recombination and replication proteins have been reported, except for that implied by the coprecipitation of Rad52p by a glutathione S-transferase (GST)–RPA2 fusion protein43. Whether they have inhibitory or stimulatory effects on each other’s activities can now be assessed in vitro.
Consequence of mutagenic repair of double-strand breaks
The correct repair of DSBs by BIR can occur because genetic information exists in a redundant manner. In diploid cells, the homologous chromosome can serve as a template for repair. However, this does not apply to haploid cells, which must use other homologous sequences. In cells that have undergone replication, the repair of the DSBs can be achieved using the genetic information on the sister chromatid. These events have been observed when breaks occur in regions containing tandem repeats. The recombinants show a variable number of repeats, a phenomenon known as unequal sister-chromatid exchange44,45. One way to explain this is if the end of the broken chromosome anneals to a different repeat in the sister chromatid resulting in either loss or gain of one or more repeats11.
The analysis of recombination products that arise when controlled DSBs are created in haploid cells has yielded valuable information on how these cells cope with damage repair in the absence of a homolog. Bosco and Haber23 observed that when a break is induced in a haploid cell at a cleavage site for the HO endonuclease engineered between two selectable markers, LEU2 and URA3 at the left arm of chromosome III, 95% of the cells died. Among the surviving cells 60% were still LEU+ URA+; these could have arisen by non-homologous end-joining, where the ends are just rejoined, or by repairing the broken site by gene conversion (Fig. 1a) with Mata or HMRa, which are homologous to the 72-bp sequence at the break site. However, 35% of the survivors were LEU− URA+ and almost all of them (92%) had an identical restriction pattern. Sequence analysis showed that they had acquired part of the right arm of chromosome III, which was copied, initiating from HMRa to the end of the telomere, resulting in a chromosome with identical ends. This process is called intrachromosomal translocation (Fig. 3). The homology requirement for this mutagenic repair of DSBs is indicated by a dependency on RAD52 (Ref. 23).
Figure 3.

Mutagenic repair of double-strand breaks (DSBs). Broken chromosomes are repaired by break-induced replication. If tandem repeats (indicated as arrows) are present at the breakpoint and repair is initiated out of register, it can lead to deletions (1) of the sequences between the repeats. When the distal part of the chromosome is lost, the centromerecontaining fragment (with the centromere depicted as a solid black circle) can be repaired using another chromosome as the template leading to non-reciprocal translocations (2) where the broken chromosome acquires sequences from another chromosome (shown as gray shaded boxes) and in some cases it can be repaired using intrachromosomal regions (3). In all cases, the broken chromosome can capture telomere sequences (depicted as colored regions at the end of each chromosome) by BIR copying to the end of the template.
In another type of study, Chen et al.46,47 identified genes that, when mutated, resulted in increased rates of chromosomal rearrangements. In one assay system, they inserted the URA3 gene on chromosome V, which encodes a decarboxylase required for the synthesis of uracil, distal to the CAN1 gene (between CAN1 and the telomere), which encodes an arginine permease. Loss of function of these genes results in cells resistant to both canavanine, a toxic arginine analog, and 5-FOA, which is converted into a toxic uracil analog by the carboxylase. The inactivation of the genes was the result of three types of rearrangements: (1) interstitial deletions, where a sequence between two very short direct repeats is removed, (2) non-reciprocal translocations, where one end of chromosome V is replaced by a region from another chromosome and (3) deletion of an end of chromosome V accompanied by de novo addition of a new telomere. The deletion and translocation breakpoints observed involved either no homology or in the order of 5-bp microhomologies. The mutator genes identified included RFA1, RFA2 and RFA3 (encoding for the subunits of RPA), and RAD27, RAD52, RAD50, MRE11 and XRS2, encoding proteins required for replication, recombination and repair. Chen et al. postulated that the chromosomal rearrangements seen in these mutator mutants resulted from DSBs that were repaired in a mutagenic fashion by BIR involving sequences having little or no homology (Fig. 3). The increased rates of rearrangements seen in these mutants were suggested to be due to either increased rates of chromosome breakage, decreased repair of DSBs by normal homologous recombination or failure to suppress mutagenic repair pathways normally. Much remains to be learned about the mechanism of these mutagenic repair events.
The above examples have suggested that DSBs can result in chromosomal rearrangements when repaired by BIR in a mutagenic fashion. In addition, studies in bacteria have indicated that stalled replication forks are actively converted to DSBs as part of the replication fork restart process2,3,22 and, although not yet demonstrated, it seems likely that a similar mechanism of replication fork restart might occur in eukaryotes. These types of damage/processing events are likely candidates for activation of the G1/G2-phase damage and S-phase checkpoints48. It is tempting to speculate that one function of these checkpoints is to suppress chromosomal rearrangements resulting from mutagenic BIR, even in the case of replication fork restart.
Acknowledgments
The authors would like to thank Neelam Amin, Ruchira Das Gupta and Abhijit Datta for comments on the manuscript. H.F-R. has been supported by a fellowship from The Jane Coffin-Childs Memorial Fund for Medical Research. This work was also supported by NIH grants GM26017 and GM50006 to R.D.K.
References
- 1.Lett JT. Damage to DNA and chromatin structure from ionizing radiations, and the radiation sensitivities of mammalian cells. Prog Nucleic Acid Res Mol Biol. 1990;39:305–352. doi: 10.1016/s0079-6603(08)60630-3. [DOI] [PubMed] [Google Scholar]
- 2.Michel B, et al. DNA double-strand breaks caused by replication arrest. EMBO J. 1997;15:430–438. doi: 10.1093/emboj/16.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seigneur M, et al. RuvAB acts at arrested replication forks. Cell. 1998;30:419–430. doi: 10.1016/s0092-8674(00)81772-9. [DOI] [PubMed] [Google Scholar]
- 4.Kuzminov A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev. 1999;63:751–813. doi: 10.1128/mmbr.63.4.751-813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haber JE. Meiosis: avoiding inappropriate relationships. Curr Biol. 1998;19:832–835. doi: 10.1016/s0960-9822(07)00524-6. [DOI] [PubMed] [Google Scholar]
- 6.Haber JE. Mating-type gene switching in Saccharomyces cerevisiae. Annu Rev Genet. 1998;32:561–599. doi: 10.1146/annurev.genet.32.1.561. [DOI] [PubMed] [Google Scholar]
- 7.Gellert M. A new view of V(D)J recombination. Genes Cells. 1996;1:269–275. doi: 10.1046/j.1365-2443.1996.22023.x. [DOI] [PubMed] [Google Scholar]
- 8.Critchlow SE, Jackson SP. DNA end-joining: from yeast to man. Trends Biochem Sci. 1998;23:394–398. doi: 10.1016/s0968-0004(98)01284-5. [DOI] [PubMed] [Google Scholar]
- 9.Nickoloff JA, Hoekstra MF. DNA repair in prokaryotes and lower eukaryotes. Humana Press Inc.; Totowa, NJ: 1998. DNA damage and repair (Vol.1) [Google Scholar]
- 10.Thacker J. The role of homologous recombination processes in the repair of severe forms of DNA damage in mammalian cells. Biochimie. 1999;81:77–85. doi: 10.1016/s0300-9084(99)80041-8. [DOI] [PubMed] [Google Scholar]
- 11.Pâques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osman F, Subramani S. Double-strand break-induced recombination in eukaryotes. Prog Nucleic Acid Res Mol Biol. 1998;58:263–299. doi: 10.1016/s0079-6603(08)60039-2. [DOI] [PubMed] [Google Scholar]
- 13.Shinohara A, Ogawa T. Homologous recombination and the roles of double-strand breaks. Trends Biochem Sci. 1995;20:387–391. doi: 10.1016/s0968-0004(00)89085-4. [DOI] [PubMed] [Google Scholar]
- 14.Haber JE. DNA recombination: the replication connection. Trends Biochem Sci. 1999;24:271–275. doi: 10.1016/s0968-0004(99)01413-9. [DOI] [PubMed] [Google Scholar]
- 15.Voelkel-Meiman K, Roeder GS. A chromosome containing HOT1 preferentially receives information during mitotic interchromosomal gene conversion. Genetics. 1990;124:561–572. doi: 10.1093/genetics/124.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voelkel-Meiman K, Roeder GS. Gene conversion tracts stimulated by HOT1-promoted transcription are long and continuous. Genetics. 1990;126:851–867. doi: 10.1093/genetics/126.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mosig G. The essential role of recombination in phage T4 growth. Annu Rev Genet. 1987;21:347–371. doi: 10.1146/annurev.ge.21.120187.002023. [DOI] [PubMed] [Google Scholar]
- 18.Luder A, Mosig G. Two alternative mechanisms for initiation of DNA replication forks in bacteriophage T4: priming by RNA polymerase and by recombination. Proc Natl Acad Sci U S A. 1982;79:1101–1105. doi: 10.1073/pnas.79.4.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Formosa T, Alberts B. Purification and characterization of the T4 bacteriophage uvsX protein. J Biol Chem. 1986;261:6107–6118. [PubMed] [Google Scholar]
- 20.Morrow DM, et al. ‘Break copy’ duplication: a model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics. 1997;147:371–382. doi: 10.1093/genetics/147.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szostak JW, et al. The double-strand-break repair model for recombination. Cell. 1983;33:25–35. doi: 10.1016/0092-8674(83)90331-8. [DOI] [PubMed] [Google Scholar]
- 22.Kogoma T. Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol Mol Biol Rev. 1997;61:212–238. doi: 10.1128/mmbr.61.2.212-238.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bosco G, Haber JE. Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics. 1998;150:1037–1947. doi: 10.1093/genetics/150.3.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Firmenich AA, et al. A novel allele of Saccharomyces cerevisiae RFA1 that is deficient in recombination and repair and suppressible by RAD52. Mol Cell Biol. 1995;15:1620–1631. doi: 10.1128/mcb.15.3.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mortensen UH, et al. DNA strand annealing is promoted by the yeast Rad52 protein. Proc Natl Acad Sci U S A. 1996;93:19729–19734. doi: 10.1073/pnas.93.20.10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hays SL, et al. Studies of the interaction between Rad52 protein and the yeast single-stranded DNA binding protein RPA. Mol Cell Biol. 1998;18:4400–4406. doi: 10.1128/mcb.18.7.4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sung P. Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J Biol Chem. 1997;272:28194–28197. doi: 10.1074/jbc.272.45.28194. [DOI] [PubMed] [Google Scholar]
- 28.Shinohara A, Ogawa T. Stimulation by Rad52 of yeast Rad51-mediated recombination. Nature. 1998;391:404–407. doi: 10.1038/34943. [DOI] [PubMed] [Google Scholar]
- 29.New JH, et al. Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature. 1998;391:407–410. doi: 10.1038/34950. [DOI] [PubMed] [Google Scholar]
- 30.Sung P. Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein A to promote DNA strand exchange by Rad51 recombinase. Genes Dev. 1997;11:1111–1121. doi: 10.1101/gad.11.9.1111. [DOI] [PubMed] [Google Scholar]
- 31.Petukhova G, et al. Catalysis of homologous DNA pairing by yeast Rad51 and Rad54 proteins. Nature. 1998;393:91–94. doi: 10.1038/30037. [DOI] [PubMed] [Google Scholar]
- 32.Hays SL, et al. Complex formation in yeast double-strand break repair: participation of Rad51, Rad52, Rad55, and Rad57 proteins. Proc Natl Acad Sci U S A. 1995;92:6925–6929. doi: 10.1073/pnas.92.15.6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malkova A, et al. Double-strand break repair in the absence of RAD51 in yeast: a possible role for break-induced DNA replication. Proc Natl Acad Sci U S A. 1996;93:7131–7136. doi: 10.1073/pnas.93.14.7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee SH, et al. Studies on the activator 1 protein complex, an accessory factor for proliferating cell nuclear antigen-dependent DNA polymerase delta. J Biol Chem. 1991;266:594–602. [PubMed] [Google Scholar]
- 35.Tsurimoto T, Stillman B. Replication factors required for SV40 DNA replication in vitro. I. DNA structure-specific recognition of a primer-template junction by eukaryotic DNA polymerases and their accessory proteins. J Biol Chem. 1991;266:1950–1960. [PubMed] [Google Scholar]
- 36.Hurwitz J, et al. The in vitro replication of DNA containing the SV40 origin. J Biol Chem. 1990;265:18043–18046. [PubMed] [Google Scholar]
- 37.Waga S, Stillman B. Anatomy of a DNA replication fork revealed by reconstitution of SV40 DNA replication in vitro. Nature. 1994;369:207–212. doi: 10.1038/369207a0. [DOI] [PubMed] [Google Scholar]
- 38.Lee SH, Hurwitz J. Mechanism of elongation of primed DNA by DNA polymerase delta, proliferating cell nuclear antigen, and activator 1. Proc Natl Acad Sci U S A. 1990;87:5672–5676. doi: 10.1073/pnas.87.15.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holmes AM, Haber JE. Double-strand break repair in yeast requires both leading and lagging strand DNA polymerases. Cell. 1999;96:415–424. doi: 10.1016/s0092-8674(00)80554-1. [DOI] [PubMed] [Google Scholar]
- 40.Murakami Y, et al. Role of DNA polymerase alpha and DNA primase in simian virus 40 DNA replication in vitro. Proc Natl Acad Sci U S A. 1986;83:2869–2873. doi: 10.1073/pnas.83.9.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Francesconi S, et al. Mutations in conserved yeast DNA primase domains impair DNA replication in vivo. Proc Natl Acad Sci U S A. 1991;88:3877–3881. doi: 10.1073/pnas.88.9.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsurimoto T, Stillman B. Replication factors required for SV40 DNA replication in vitro. II. Switching of DNA polymerase alpha and delta during initiation of leading and lagging strand synthesis. J Biol Chem. 1991;266:1961–1968. [PubMed] [Google Scholar]
- 43.Shinohara A, et al. Rad52 forms ring structures and co-operates with RPA in single-strand DNA annealing. Genes Cells. 1998;3:145–156. doi: 10.1046/j.1365-2443.1998.00176.x. [DOI] [PubMed] [Google Scholar]
- 44.Petes T. Unequal meiotic recombination within tandem arrays of yeast ribosomal DNA genes. Cell. 1980;19:765–774. doi: 10.1016/s0092-8674(80)80052-3. [DOI] [PubMed] [Google Scholar]
- 45.Szostak JW, Wu R. Unequal crossing over in the ribosomal DNA of Saccharomyces cerevisiae. Nature. 1980;284:426–430. doi: 10.1038/284426a0. [DOI] [PubMed] [Google Scholar]
- 46.Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nature Genetics. 1999;23:81–85. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 47.Chen C, et al. Chromosomal rearrangements occur in S. cerevisiae rfa1 mutator mutants due to mutagenic lesions processed by double-strand-break repair. Mol Cell. 1998;2:9–22. doi: 10.1016/s1097-2765(00)80109-4. [DOI] [PubMed] [Google Scholar]
- 48.Weinert T. DNA damage and checkpoint pathways: molecular anatomy and interactions with repair. Cell. 1998;4:555–558. doi: 10.1016/s0092-8674(00)81597-4. [DOI] [PubMed] [Google Scholar]