

Abstract
Trypanosoma brucei cyclic nucleotide phosphodiesterase B1 (TbrPDEB1) and TbrPDEB2 have recently been validated as new therapeutic targets for human African Trypanosomiasis by both genetic and pharmacological means. In this study we report the crystal structure of the catalytic domain of the unliganded TbrPDEB1 and its use for the in silico screening for new TbrPDEB1 inhibitors with novel scaffolds. The TbrPDEB1 crystal structure shows the characteristic folds of human PDE enzymes, but also contains the parasite-specific P-pocket found in the structures of Leishmania major PDEB1 and Trypanosoma cruzi PDEC. The unliganded TbrPDEB1 X-ray structure was subjected to a structure-based in silico screening approach that combines molecular docking simulations with a protein-ligand interaction fingerprint (IFP) scoring method. This approach identified, six novel TbrPDEB1 inhibitors with IC50 values of 10–80 μM, which may be further optimized as potential selective TbrPDEB inhibitors.
INTRODUCTION
Human African Trypanomiasis (HAT), also known as African sleeping sickness, is a deadly infectious disease caused by the kinetoplastid Trypanosoma brucei (Tbr). The Trypanosoma brucei genome encodes five cyclic nucleotide phosphodiesterases (PDEs), of which TbrPDEB1 and TbrPDEB2 were recently validated as potential new drug targets for the treatment of HAT.1–4 Both TbrPDEB enzymes selectively catalyze the hydrolysis of cAMP to AMP. In a dual knock-down RNAi study, Seebeck and colleagues reported that simultaneous RNA knockdown of both TbrPDEB1 and TbrPDEB2 results in impaired division of trypanosomes and eventual death of the parasites.5 These studies have subsequently been confirmed by pharmacological targeting of TbrPDEB1 and TbrPDEB2,1–2, 6 suggesting that drug repurposing efforts and/or tapping into the wealth of knowledge around cyclic nucleotide PDEs (e.g.150 published crystal structures and over 3000 published submicromolar PDE inhibitors)7–8 might be an effective way to find new HAT treatments. Initial drug profiling and preliminary medicinal chemistry suggests that the human PDE inhibitors could be used as interesting starting scaffolds for the discovery of TbrPDEB inhibitors.1–2, 9 Using a computational design and fragment merging approach, we recently reported pyrazolinones VUF118512 (1, Figure 1) and VUF135242 (2, Figure 1) as TbrPDEB1 inhibitors. The hPDE4 inhibitor PPS540196 (3, Figure 1) was discovered in a high throughput screening of a proprietary library of 400,000 compounds by Nycomed Pharma. This PDE inhibitor is currently the most potent TbrPDEB1 inhibitor, and shows substantial in vitro trypanocidal activity. Three SAR studies starting from known hPDE inhibitors have resulted in the discovery of TbrPDEB1 inhibitors, among which piclamilast1 (4, Figure 1) was the most successful.1, 10–11 The TbrPDEB1 inhibitor, 1-(3-(4-hydroxybutoxy)-4-methoxyphenyl)-3-methylbutan-1-one9 (5, Figure 1) was originally discovered as an inhibitor of Leishmania major PDEB1 (LmjPDEB1) through structure-based design, but also appears to inhibit TbrPDEB1 to some extent.
Figure 1.
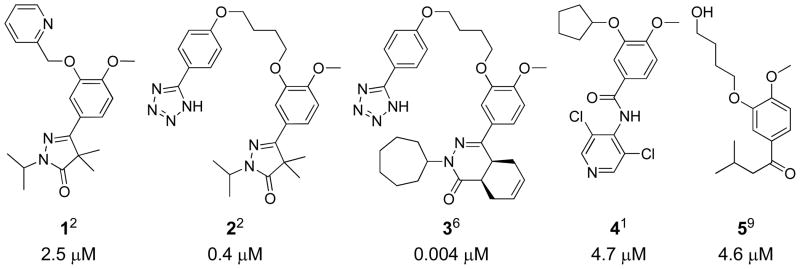
Previously reported TbrPDEB1 inhibitors, 1, 2, 3, 4 and 5, showing the IC50 values of the compounds against TbrPDEB1 in μM.
While human PDE inhibitors may provide important starting points for the discovery of novel TbrPDEB1 inhibitors, it has proven challenging to achieve parasite-selective PDE inhibition. This lack of selectivity could be a major hurdle in the development of TbrPDEB1 inhibitors as HAT drugs. To resolve this issue, we have initiated a structural biology and structure-based design program to guide the discovery of selective TbrPDEB1 inhibitors.
In this study we present for the first time a crystal structure (4I15) of the unliganded catalytic domain of the TbrPDEB1 enzyme. A parasite-specific pocket (P-pocket), first observed in the LmjPDEB1 crystal structure (2R8Q)12 and subsequently seen in TcrPDEC structures (3V93 and 3V94)4, is also present in the new TbrPDEB1 crystal structure. The high resolution crystal structure of the catalytic domain of TbrPDEB1 has been employed in a structure-based virtual screen, aiming at the identification of new TbrPDEB1 inhibitors. Virtual screening remains underutilized in the search for PDE inhibitors as shown by the fact that only three prospective structure-based virtual screening studies have been reported to date.13–15 One of these was performed using a homology model of Trypanosoma cruzi PDEC (TcrPDEC).13 In the present study we report the use of the newly resolved X-ray structure of the TbrPDEB1 catalytic domain in a customized virtual screening campaign, which lead to the identification of new TbrPDEB1 inhibitors.
RESULTS AND DISCUSSION
Unliganded TbrPDEB1 crystal structure
The full length TbrPDEB1 enzyme contains two GAF domains (residues D234 - E554) and a catalytic domain (residues V586 – R908).3 The GAF domains have been shown to bind cAMP, but only the catalytic domain is able to hydrolyse cAMP to AMP.16 Inhibition of the isolated catalytic domain and the full length enzyme by recently identified TbrPDEB1 inhibitors occurs at similar inhibitor concentrations.2 The catalytic domain (residues 576–918) of TbrPDEB1, expressed and purified from Escherichia coli, was used in our studies.
Following crystallization, the TbrPDEB1 structure with residues 586–918 could be resolved by X-ray diffraction at a resolution of 1.65 Å (Table 1). The catalytic domain comprises 16 α-helices, 7 310-helices and no β-strands (Figure 2A). Two divalent metal atoms are present which have been designated zinc and magnesium in accordance with common PDE metal ligation without further verification. The metal ions each form an octahedral geometry; zinc coordinates H673, H709, D710, D822 and two water molecules, while magnesium coordinates D710 and five water molecules. The TbrPDEB1 X-ray structure shows the presence of the parasite-specific P-pocket, which is formed with the invariant Q874 on H15 and the M-loop (Figure 2C) on opposing sides, and is adjacent to the substrate binding pocket.
Table 1.
Statistics on diffraction data and structural refinement.
| Data collection | |
| Space group | C2 (C121) |
| Unit cell (a, b, c, Å) | 115.01, 115.34, 68.51, 90, 108.1, 90 |
| Resolution (Å) | 30.00 – 1.65 |
| Unique reflections | 92,150 |
| Fold of redundancy | 6.8 |
| Completeness (%) | 90.9 (54.8a) |
| Average I/δ | 13.2 (2.7a) |
| Rmerge | 0.059 (0.285a) |
| Structure refinement | |
| R-factor | 0.203 |
| R-free | 0.223 |
| Reflections | 87,881(8781b) |
| RMS deviation for bond (Å) | 0.005 |
| Angle (degree) | 1.06 |
| Average B-factor (Å2) | |
| Protein(# of atoms) | 31.8 (5272) |
| Waters(# of atoms) | 36.7 (404) |
| Zn(# of atoms) | 26.8 (2) |
| Mg(# of atoms) | 26.0 (2) |
| Ramachandran plot statistics (%) as defined in CCP4 | |
| Most favored regions | 91.7 |
| Additionally allowed regions | 7.7 |
| Generously allowed regions | 0.7 |
| Disallowed regions | 0.0 |
For the resolution shell of 1.71–1.65 Å.
Reflections omitted for calculation of R-free.
Figure 2.

(A) An overview of the X-ray crystal structure of TbrPDEB1, helices are colored yellow and labeled where visible, 310-helices are shown in fuchsia, and loops are shown in purple. The metal ions magnesium (lime) and zinc (gray) are shown. (B) A superposition of crystal structures of TbrPDEB1 (yellow), LmjPDEB1-IBMX (2R8Q in blue)12, TcrPDEC-WYQ16 (3V94 in green)4, and a crystal structure of hPDE4B-piclamilast (1XM4 in magenta).17 Close-ups of the region between Q874 on H15 and the M-loop are shown in: (C) TbrPDEB1; (D) LmjPDEB1; (E) TcrPDEC; and (F) hPDE4B.
The structural superposition of TbrPDEB1 with LmjPDEB1 (2R8Q),12 TcrPDEC (3V94)4 and hPDE4B (1XM4)17 yielded RMSDs of 0.86 Å, 1.78 Å and 1.61 Å respectively for comparable Cα atoms (Figure 2B), indicating high overall structural similarity. Taking a closer look at the region between the Q874 and the M-loop it is clear that the P-pockets of TbrPDEB1 and LmjPDEB1 overlap well. However, the P-pocket of TcrPDEC is displaced by about 5 Å, and hPDE4B does not have a P-pocket12 (Figure 2C–F). These structural differences can be explained by sequence differences in H14, H15, and the M-loop. A key difference seems to be residue T841 (L539 TcrPDEC) and T841 (M411 hPDE4) in H14. The conformation of the M-loop also plays a role in the appearance of the P-pocket. This conformation is influenced by the length of the M-loop which is two residues shorter in TcrPDEC and one in hPDE4, when compared to the TbrPDEB1 and LmjPDEB1 M-loops.
The secondary structure elements of TbrPDEB1 and LmjPDEB1 are also almost identical, whereas those of TcrPDEC and hPDE4B deviate significantly (Figure 3). Comparing the residues of the substrate binding pocket, the only difference between TbrPDEB1 and LmjPDEB1 is to I823 (V836 LmjPDEB1) and superposing the pocket Cα atoms results in an RMSD of 0.40 Å. In the case of TcrPDEC, the residues that are different are; N825 (A524 TcrPDEC), V826 (S525), S833 (A532), A837 (L536), V840 (I538), T841 (L539), Y845 (A543), M861 (G559) and W911 (Y606), the RMSD of the pocket residues is 1.03 Å. For hPDE4B, the substrate binding pocket residues that are different are; I823 (L393 hPDE4B), V826 (P396), S833 (Y403), A837 (T407), V840 (I410), T841 (M411) and W911 (Y480), comparing the pocket residues results in a lower RMSD of 0.74 Å. These data are in line with the percentage of residue identity between the catalytic domains of TbrPDEB1 and LmjPDEB1 (66%), TbrPDEB1 and TcrPDEC (21%), and TbrPDEB1 and hPDE4B (27%). Moreover, the full length TcrPDEC does not contain GAF domains and has been shown to have a dual specificity for cAMP and cGMP, further indicating its evolutionary distance from TbrPDEB1.4, 13, 18 The high sequence identity of TbrPDEB1 and LmjPDEB1 on the other hand, suggests that a single compound may well inhibit both parasite PDEs and might be useful against both kinetoplastids.
Figure 3.

Sequence alignment between TbrPDEB1, LmjPDEB1, TcrPDEC and hPDE4B. The bars indicate secondary structure features, α-helices are shown in yellow and 310-helices are shown in fuchsia. Residues of the substrate binding pocket are shown in red. The helix numbers of TbrPDEB1 are indicated above the sequence, as are the H-loop and the M-loop.
The crystallographic asymmetric unit contains two molecules of TbrPDEB1 that form a dimer in the crystal lattice. Molecule B is involved in further crystal lattice contacts which close off part of the active site from bulk solvent. However, as only one of the two chains is involved in formation of these crystallographic lattice contacts, we suggest that this is likely to represent a crystallographic artifact without biological relevance. Superposition of the Cα atoms of the A and B chains of the asymmetric unit with MOE (Molecular Operating Environment version 2011.10)19 resulted in an RMSD of 0.49 Å. The only significant rotamer deviation seen in the pocket residues is an 86° rotation of V840. The overall similarity between the A and B chains indicates that the observed lattice contacts have little influence on the conformation of the protein.
The substrate binding pocket of the unliganded structure contains two well defined water networks, one surrounding the invariant glutamine (Q874) and the other surrounding the metal ions (Figure 4). Analysis of published PDE crystal structures containing inhibitors revealed that in all cases several of the water molecules surrounding Q874 are displaced, most often to allow the formation of hydrogen bonds between Q874 and the bound ligand. The water molecules surrounding the metal ions are coordinated tightly and only three inhibitors; an2898 in hPDE4B (3O0J), rolipram in hPDE4B2B (1RO6) and zardaverine in hPDE4D (1XOR), and the substrate products AMP and GMP displace some of them. A further feature of PDE inhibitor binding is the occupation of the hydrophobic clamp, the space between F877 and V840, which is invariably occupied by an aromatic or highly conjugated ring system. In the TbrPDEB1 crystal structure the B-factor of the M-loop residues is high in comparison to the other residues in the structure, reflecting the flexibility of this loop. The conformation of the M-loop is stabilized by the π-stacking of F862 against F844 (Figure 4), as shown by F862 having a lower B-factor than other residues in the M-loop. The parasite-specific P-pocket forms a pore through the protein between Q874 and Y845. Although inhibitors have not been shown to occupy this region in crystal structures, in silico modeling suggests that the occupation of this region may result in selective TbrPDEB1 inhibitors.2
Figure 4.

The substrate binding pocket of TbrPDEB1 (chain A) with the carbon atoms colored by B-factor, the range is shown on a color bar. Two water networks (red spheres) are shown with hydrogen bonds (gray dashes), one of which surrounds the invariant glutamine (Q874) and a second, which surrounds the metal ions. Pocket residues are shown as sticks and labeled where visible. The P-pocket is shown as a gray surface.
Structure-based virtual screening
We have performed a prospective structure-based virtual screening study, evaluating both the new TbrPDEB1 crystal structure and the customized virtual screening method, for the computer-aided discovery of novel TbrPDEB1 inhibitors. We used a protocol that combines a docking scoring function (PLANTS20) with a protein-ligand interaction fingerprint (IFP21) scoring method to rank molecular docking poses of 385,000 commercially available molecules (Figure 5). The IFP scoring method determines ligand binding mode similarity using interaction types (negatively and positively charged, H-bond acceptor and donor, aromatic face-to-edge and face-to-face, and hydrophobic) encoded as bits for each pocket residue (Figure 6). A Tanimoto coefficient measuring IFP similarity to a reference ligand pose (derived from a computationally predicted protein-ligand binding mode in this case) is used to score the docking poses of a large database of compounds, in an approach similar to that used to successfully discover several novel histamine H1 receptor ligands with a high virtual screening hit rate of 73%.22 The in silico predicted binding modes of compounds 1 and 2 in the unliganded TbrPDEB1 structure (Figure 6A–B) were used to define reference IFPs. These were used to select PLANTS docking poses in the prospective structure-based virtual screening study for novel TbrPDEB1 inhibitors.
Figure 5.

Overview of the compound selection process. (I) A selection of unique structures retrieved from a select set of 14 suppliers in the ZINC library. (II) Structures that pass a set of physical chemical filters and contain a substructure moiety able to interact with Q874 and the hydrophobic clamp. (III) Structures that have at least one docking pose that scored −90 or lower in PLANTS.22 With the GLN Filter only docking poses that form hydrogen bonds with both Q874 side chain amide hydrogen atoms were selected. The IFP1 score is the Tanimoto score between the IFP of each docked pose and the IFP of the putative binding mode of 1, a cut-off of 0.75 was applied.22 (B) The IFP2 score was calculated using the putative binding mode of 2 as the reference and those docked poses scoring 0.75 and above were kept. Compounds from routes A, B, and C were then ranked by PLANTS scores before being filtered for diversity. The top 50 compounds from routes A, B, C and D were selected and 9, 4, 8 and 8 compounds were selected respectively, according to diversity, availability and visual inspection of the docking poses. Of the compounds selected using routes A, B, C and D; 3, 2, 1 and 0 compounds, respectively, were active.
Figure 6.

Panels A–D show the TbrPDEB1 backbone as a gold ribbon, key pocket residues and ligands as sticks, metal atoms as spheres, and the P-pocket as a gray transparent surface. The modeled binding poses of compounds 1 (green, panel A) and 2 (red, panel B) are similar to those proposed previously for close analogues,2 and are used as references for the calculation Tanimoto scores based on IFP similarity.21–22 Compounds 16 (magenta) and 17 (cyan) (panel C) show a similar binding mode to compound 1, while compound 19 (panel D) shows a greater similarity to the binding mode of compound 2. Bit strings are used in IFPs to indicate the absence (0) or presence (1) of an interaction type between the ligand and each residue, as shown in panel E for compounds 2, 4, 16, 17 and 18. A sequence of seven bits is used for each residue and the position of each bit determines the interaction type (see legend). For the sake of clarity, only the bit strings of V840, T841, F844, Y845, Q874 and F877 are shown, of the 42 pocket residues.
Protein-ligand interactions described in IFPs are encoded as seven bits per residue as follows: 1. Apolar; 2. Aromatic face-to-face; 3. Aromatic edge-to-face; 4. H-bond donor (protein) – H-bond acceptor (ligand); 5. H-bond donor (ligand) – H-bond acceptor (protein); 6. Ionic interaction positive (protein) – negative (ligand); 7. Ionic interaction positive (ligand) – negative (protein). An additional H-bond bit was included for GLN874 (GLN Filter): * H-bond formation with both polar hydrogen atoms of the Q874 side-chain.
Figure 5 gives an overview of the virtual screening steps (I–III) and the hit selection routes (A–D). Consecutive virtual filters of increasing complexity are applied throughout the virtual screening process. These facilitate the selection of a small number of compounds from a large compound collection, while at the same time allowing the identification of novel chemotypes. Starting with 7.5 million unique compounds from 14 suppliers in the ZINC database23 (step I), physicochemical property filters and substructure filters were applied, reducing the number to 385,000 molecules (Step II). The physicochemical filters were defined using the properties of 3192 known PDE inhibitors with IC50 values below 10 μM, retrieved from the ChEMBL database as a reference.8 The total polar surface area (TPSA) and LogP filter values were adjusted to select compounds more likely to penetrate the blood brain barrier.24 The substructure filter (Supporting Information: Figure S2) was defined following the analysis of 132 PDE crystal structures. These indicated that the presence of an aromatic or conjugated ring system, which contains (or is connected to) a hydrogen bond acceptor moiety, is found in all co-crystalized ligands. This substructure ensures binding of the inhibitors to the hydrophobic clamp (between F877 and V840 in TbrPDEB1) while forming an essential hydrogen bond (present in 92% of the 132 PDE crystal structures) with an invariant glutamine (Q874 in TbrPDEB1).
The 385,000 molecules were docked into the TbrPDEB1 crystal structure using PLANTS protein-ligand docking software (step III).20 Of these, 64,000 generated at least one docked pose that was scored −90 or lower by PLANTS, and were taken into the four hit selection routes (A–D, Figure 5). Using the same docking protocol for the 47 published TbrPDEB1 inhibitors yielded docking poses with PLANTS scores below −90 for 16 inhibitors, of which 13 are expected to address the P-pocket2 (Table S5 in Supporting Information). In selection routes A and B, the IFPs generated from the docked poses were compared to those of the reference ligands 1 (IFP1, Figure 5) and 2 (IFP2, Figure 5), respectively. The putative binding modes of 1 (Figure 6A) and 2 (Figure 6B) were used to calculate the reference IFPs, IFP1 and IFP2, respectively. The binding modes of 1 and 2 were explored through molecular docking and were similar to those of close analogues proposed previously.2 The tridentate interaction between the putative binding mode of 1 and Q874 is a feature that has also been proposed for compound 5.9 In order to retrieve hits able to form at least two hydrogen bonds with Q874, a filter (GLN Filter) was applied in route A. This retrieved docking poses in which ligands formed hydrogen bonds with each side chain amide hydrogen atom of Q874. Reference compound 2 was predicted to penetrate deeply into the P-pocket, forming a hydrogen bond between the tetrazole ring and Y845. Compounds in route C were selected according to their ranking by PLANTS. Route D included ligands that scored well in PLANTS and appeared to fit the pocket well, but were outside the top 50 of each route. All routes involved steps to maintain structural diversity from the other compounds being selected for testing (MACCS similarity filter applied) and also to ensure that hits would be novel from the 47 published TbrPDEB1 inhibitors (Table S1 and Table S5 in Supporting Information, ECFP424 and ROCS23 similarity scoring used).
From the docking results, 29 compounds were selected according to the selection routes A–D shown in Figure 5. Of the 29 purchased compounds, 6 were found to inhibit TbrPDEB1 with IC50 values below 100 μM, of which 3 had IC50 values of around 10 μM (Table 2), resulting in an overall hit rate of 21%. While the number of compounds tested is low, these results support the use of IFP similarity scores (routes A–B) to improve hit selection compared with the use of docking scores (route C) or visual inspection (route D) alone. Routes A and B together yielded 5 experimentally confirmed hits (38% of the 13 selected compounds), while 1 new TbrPDEB1 ligand was discovered via route C (12.5% of the 8 selected compounds), and D did not give any hit at all (of the 8 selected compounds). Interestingly, routes C–D result in a higher hit rate of experimentally confirmed hPDE4 inhibitors than routes A–B (Supporting Information Table S1). This indicates that the interactions specific to TbrPDEB1, implemented in the IFP similarity scoring procedure, are essential for the identification of hits with TbrPDEB1 activity.
Table 2.
TbrPDEB1 inhibitors identified in the virtual screening.
| cpd | Virtual screening hits | TbrPDEB1 IC50 in μMa | IFP1 (rank)c | IFP2 (rank)d | PLANTS (rank)e | ROCS (Closest scaffold)f | ECFP4 (Closest scaffold)g | hPDE4B IC50 in μMb |
|---|---|---|---|---|---|---|---|---|
| 16 |
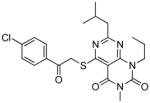
|
10 | 0.80 | 0.63 | −102.8 | 1.11 (15) | 0.18 (6 - NEU222) | 26 |
| 17 |
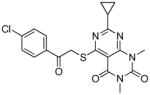
|
0.79 (33) | 0.61 | −94.7 | 1.21 (15) | 0.18 (6) | ||
| 18 |
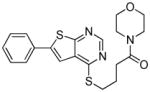
|
12 | 0.75 (44) | 0.70 | −93.8 | 1.07 (15) | 0.20 (2/4 - 8d) | 14 |
| 19 |
|
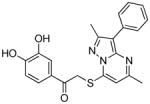
13 | 0.53 | 0.75 (13) | −101.3 | 1.02 (12) | 0.21 (5) | 5 |
| 20 |
|
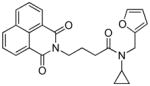
41 | 0.65 | 0.75 (6) | −103.6 | 0.95 (15) | 0.16 (2/4 - 16b) | 16 |
| 21 |
|
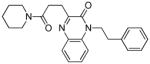
54 | 0.75 (24) | 0.63 | −95.3 | 1.10 (6 - NEU227) | 0.24 (2/4 - 8d) | 15 |
| 22 |
|
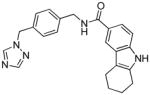
75 | 0.45 | 0.55 | −109.1 (9) | 0.924 (2/4 - 13b) | 0.21 (6 - NEU432) | 68 |
The structures of the inhibitors are given with their IC50 concentrations for full-length TbrPDEB1 and hPDE4B. It should be noted that the virtual screening hits did not reach 100% inhibition at the highest concentrations measured. Curves were assumed to plateau at 100% inhibition and fitted according to a one site competitive binding model to derive IC50 values. The measured curves and their fits are provided in Figure S3 of the Supporting Information.
The IFP1, IFP2 and PLANTS scores of the docking poses used to select the compounds are shown, with the scores printed in bold according to whether the compound was selected using routes A, B or C respectively. The ranking in selection routes A–C is also given for the score used to select the compound.
ROCS and ECFP4 score structural similarity between compounds. A maximum similarity cutoff of 1.4/2 was set for ROCS and 0.4/1 for ECFP4. All hits were found to be dissimilar to known TbrPDEB1 inhibitors. The closest published TbrPDEB1 inhibitor as identified through ROCS or ECFP4 is shown in brackets.
Reference compound 1 was predicted to form a tridentate interaction with Q874 (Figure 6A). This interaction profile aids the selection of compounds which fill the entrance of the P-pocket and strengthens the interactions with Q874. The virtual screening hit 17, selected using route A, was found to be out of stock at its supplier and the close analogue 16 was chosen to replace it (Figure 6C). The compounds adopt the same binding mode in docking studies, and thus the discovery of the active hit can be attributed to the success of the virtual screening method. Compound 19, selected using route B, forms a hydrogen bond with Y845 seen in the binding mode of 2 (Figure 6B, D). Protein-ligand interactions between the docked poses and a select group of pocket residues are described in bit string form in Figure 6E. The IFP Tanimoto scores compare all interactions between the reference ligand and the protein to those between the docked poses of the screened compounds and the protein. The entire pocket is used for the comparison to allow subtle interaction motifs to be picked up which may be missed during visual inspection. Although numbers are too low to for conformation, the higher hit rate of routes A and B suggests that the combined use of these IFP Tanimoto scores and the PLANTS docking scores aids hit selection.
Occupation of the P-pocket was implemented as a selectivity driver in the virtual screening and the corresponding selection routes A–B indeed yielded more TbrPDEB1 hits than the other selection routes (Figure 5, Table 2). Nevertheless, all confirmed TbrPDEB1 hits still showed inhibition of hPDE4 with selectivity ratios of 0.4 – 4 (TbrPDEB1 IC50 over hPDE4 IC50), suggesting adaptation of the binding mode or induced fit to accommodate the ligands in hPDE4. These selectivity ratios are a clear improvement over published TbrPDEB1 inhibitors, all of which (including compounds 1 and 2) have significant selectivity against at least one of the human PDEs.1–2, 5–6, 10–11, 25 While the molecular docking simulations suggest that the virtual screening hits and compounds 1 and 2 target the P-pocket of TbrPDEB1 (Figure 6), this has yet to be verified structurally or using site directed mutagenesis.
Prospective structure-based virtual screening studies on PDE targets remain scarce, with just three reported in literature. In one study, a virtual screening for TcrPDEC inhibitors used a library of 60,000 compounds,13 and a homology model built with a hPDE4B crystal structure (1XMY) as a reference. Following docking, hits were filtered on drug like properties, rescored using consensus scoring, analyzed for hydrogen bonding and hydrophobic profiles, and visually inspected, resulting in the selection of 25 compounds for pharmacological screening, 10 of which were TcrPDEC inhibitors with low micromolar IC50 values.13 A hPDE3 study has also been reported in which 3,000 compounds, out of an initial library of 113,000, were docked, and 80 were screened for enzymatic inhibition, resulting in one low micromolar hit.14 Finally in a virtual screening on hPDE5 that began with a library of 5 million compounds, an iterative process of filtering, docking and pharmacological screening was used to retrieve 22 low micromolar and 12 submicromolar inhibitors from a set of 196 tested compounds.15 The current study provides further evidence for the utility of structure-based virtual screening in the discovery of PDE inhibitors. Even though only an unliganded TbrPDEB1 structure was available, a customized scoring method has been able to identify hits with improved selectivity as starting points for further optimization. With over 150 crystal structures now available, the time is ripe for the application of structure-based virtual screening to the discovery of novel human and parasite PDE inhibitors.
CONCLUSIONS
The crystal structure of the catalytic domain of TbrPDEB1 was resolved at a resolution of 1.65 Å. The resolved crystal structure corroborates the existence of the parasite PDE-specific P-pocket and represents a significant step forward for HAT-directed drug discovery efforts. In the present study the unliganded structure has been used to identify TbrPDEB1 inhibitors by virtual screening. Although the new hits show only moderate to low micromolar inhibition, each of them represents a novel scaffold and thereby a new potential starting point for future drug discovery efforts on TbrPDEB1. The combined use of docking scores and protein-ligand interaction fingerprints (IFPs) has allowed us to identify novel TbrPDEB1 inhibitors. It is noted that the new experimentally confirmed TbrPDEB1 hits are equipotent for TbrPDEB1 and hPDE4 in contrast to previously published TbrPDEB1 inhibitors, all of which have significant selectivity against at least one of the human PDEs. This suggests that targeting the P-pocket in structure-based studies might indeed be a way forward to the design and discovery of selective TbrPDEB1 inhibitors.
EXPERIMENTAL SECTION
Subcloning, expression, purification and crystallization of catalytic domain tbrPDEB1
The catalytic domain of TbrPDEB1 with amino acids 576–918 was amplified by PCR and subcloned into the expression vector pET28a. The resultant plasmid pET-PDEB1 was transferred into E. coli strain BL21 (Codonplus) for overexpression. The E. coli cell carrying pET-PDEB1 was grown in LB medium at 37°C to absorption of A600 = 0.7 and then 0.1 mM isopropyl β-D-thiogalactopyranoside was added to induce the overexpression at 15°C for 24 hours. Recombinant PDEB1 was passed through a Ni-NTA column (Qiagen), subjected to the thrombin cleavage, and further purified by the columns of Q-Sepharose and Sephacryl S300 (Amersham Biosciences). A typical purification yielded over 10 mg TbrPDEB1 with a purity >95% from a 2-liter cell culture.
Crystallization, structure determination and analysis of TbrPDEB1
The catalytic domain of the unliganded TbrPDEB1 (576–918) was crystallized by vapor diffusion against a well buffer of 20% PEG3350, 0.4 M Na formate, 0.2 M guanidine, 0.1 M MES pH 6.5 at 4°C. Diffraction data was collected on a beamline X29 at the Brookhaven National Laboratory and processed by program HKL2000.26 The crystal of the unliganded TbrPDEB1 has the space group C2 with cell dimensions of a = 115.0, b = 115.3, c = 68.5 Å, and β = 108.1°. The 6.8-fold redundant measurement yielded R-merge of 0.059 for 92,150 unique reflections that represent 90.9% completeness to 1.65 Å resolution. The structure of TbrPDEB1 was solved by molecular replacement program AMoRe,27 using the LmjPDEB1 structure as the initial model.12 The structure was rebuilt by the program O28 and refined by the program CNS29 to R-factor/R-free of 0.205/0.224. Secondary structure determination was performed using DSSP.30–32 Sequence alignment of the TbrPDEB1, LmjPDEB1, TcrPDEC and hPDE4B catalytic domains was performed using ClustalW 2.1.33 Structural superposition of the Cα atoms of TbrPDEB1 (4I15, chain A) with LmjPDEB1 (2R8Q, chain A), TcrPDEC (3V94, chain A) and hPDE4B (1XM4, chain B) was performed with MOE (Molecular Operating Environment version 2011.10).19 All structures were visualized using Pymol 1.5.0.3.34
Database preparation for virtual screening
The databases of 14 suppliers (Table S3 in Supporting Information) containing 7.5 million unique structures in total were downloaded from ZINC (17-06-2011).23 The structures were passed through Openeye’s filter (version 2.1.1) in smiles format.35 Those with physical chemical properties that fell within all of the following ranges were kept for further processing: heavy atoms 20 – 30, ring systems 3 – 6, rotatable bonds 2 – 8, TPSA 50 – 90, LogP −10 – 4, hydrogen bond acceptors 3 – 5, hydrogen bond donors 0 – 2, and charge −1 – +1. Note that due to a difference in filter definition, ring systems, rather than rings, were counted, leading to unintended filtering of the database (Figure S1 in Supporting Information). The structures then passed through ChemAxon’s Jcsearch (version 5.4.1.1) and those compounds with at least one of the following features were kept for further processing: aromatic rings containing at least one nitrogen atom, conjugated rings with carbonyl or imines, or phenols (Figure S2 in Supporting Information). The remaining structures were prepared for docking by converting them from isomeric SMILES to mol2 format using Molecular Networks’ Corina (version 3.46).36
Protein preparation for virtual screening
The A chain of the TbrPDEB1 crystal structure was prepared for docking using MOE (version 2011.10) by adding and minimizing the hydrogen atoms using the protonate 3D function.19 The water molecules and metal ions were removed from the structure. The pocket of protein was defined for use during the processing of the docking results and both the protein and pocket were utilized in mol2 format.
Virtual screening
Each of the filtered ZINC23 compounds was docked 25 times using the speed 2 setting of PLANTS and the chemPLP scoring function.20 After docking, the protein-ligand interaction fingerprint (IFP)21–22 of each docking pose was calculated and compared to the IFPs of the reference compounds 1 and 2, resulting in a Tanimoto similarity score. A filter was applied to the generated docking poses so that only those which scored ≤−90 were kept for further processing. In route A, a filter required docked poses to form hydrogen bonds with both hydrogen atoms of the side chain amide of Q874 before an IFP1 score cutoff of ≥0.75 was applied. In route B, an IFP2 score cutoff of ≥0.75 was applied. The results from routes A–C were ranked by docking scores and the top 50 diverse structures were selected based on MACCS clustering in MOE19 using a Tanimoto similarity cutoff of 70%. The selected compounds were then visually inspected and compared to known TbrPDEB1 inhibitors (Table S1 and Table S5 in Supporting Information, ROCS: Comboscore ≤1.4 out of 237; ECFP-4: score ≤0.4 out of 138), and checked for supplier availability or close analogue availability if the compounds were unavailable. A total of 21 compounds were selected for purchase from this set. In addition, 8 compounds that fell outside the criteria of routes A–C were selected for purchase in route D on basis of visual inspection of a larger set of docking poses. For route D, binding modes of the top 300 diverse compounds from each of the routes A–C (excluding the top 50 used in those routes) were visually inspected. Docking poses were selected based on the following criteria: i) occupation of the P-pocket,2 ii) hydrogen bonding with Q874,17 iii) occupation of the hydrophobic clamp by an aromatic moiety,17 iv) hydrogen bonding to Y845 or π-stacking to F844.2
IFP processing
The IFPs were defined using 7 protein-ligand interaction types between residues of the pocket and the ligand as described previously.21 The interaction types used were: 1. Apolar; 2. Aromatic face-to-face; 3. Aromatic edge-to-face; 4. H-bond donor (protein) – H-bond acceptor (ligand); 5. H-bond donor (ligand) – H-bond acceptor (protein); 6. Ionic interaction positive (protein) – negative (ligand); 7. Ionic interaction positive (ligand) – negative (protein). The pocket was defined by 40 residues for IFP1 calculations, Y668, H669, H673, H709, D710, H713, L716, N717, N718, S719, T783, M785, A786, G789, D822, I823, N825, V826, S833, W836, A837, V840, T841, E843, F844, L859, P860, M861, F862, N867, M868, E869, G873, Q874, G876, F877, I878, F880, V881, A882 and 42 residues for IFP2 calculations with the addition of Y845 and L870. For each docking pose, the coordinates of pocket including the rotated hydroxyl hydrogen atoms were recorded and used to define the IFP. Standard IFP scoring parameters and a Tanimoto coefficient measuring IFP similarity with the reference molecule, were used to filter and rank docking poses. The reference IFP strings are available as Supporting Information.
Compounds selected by virtual screening
The compounds selected by virtual screening were purchased from available screening collections of two vendors: Enamine (www.enamine.com) and Vitas-M (www.vitasmlab.com). The purities of all 29 purchased compounds were verified by liquid chromatography-mass spectrometry (LC-MS), all experimentally validated hits had a purity of 95% or higher (Table S2 in Supporting Information).
Inhibition assay
The scintillation proximity assay (SPA) described by De Koning et al., was followed exactly for the determination of full length PDE activities.2, 6 The assay was used to determine IC50 values of purchased compounds on TbrPDEB1 and hPDE4B using a cAMP substrate concentration of 0.5 μM. The PDE was collected by sonification of supernatants of PDE overexpressing Sf21 cells. The PDE activity of the enzyme was determined in at least duplicates by published procedures.39–40 Enzyme concentrations were set so that <20% of the cAMP was consumed during the assay. Blank values were measured in the presence of denatured protein and always resulted in <2% of the total radioactivity. For inhibitors, which did not reach 100% inhibition at the highest concentration measured, inhibition curves were fitted to derive IC50 values with the assumption that the inhibition curves plateau at 100% inhibition, since the inhibitors are expected to bind competitively to the PDE substrate binding pocket.
Supplementary Material
Acknowledgments
We thank beamline X29 at NSLS for collection of the diffraction data. This work was partially supported by NIH GM59791 to HK and was performed under the framework of the Top Institute Pharma project “Phosphodiesterase Inhibitors for Neglected Tropical Diseases” (T4-302) with partners VU University Amsterdam, University of Bern, The Royal Tropical Institute (KIT), Mercachem BV, Nycomed (a Takeda company), IOTA Pharmaceuticals Ltd., Drugs for Neglected Diseases initiative (DNDi), and TI Pharma. Netherlands Organization for Scientific Research (NWO) is acknowledged for financial support through a VENI grant (700.59.408 to C. de G.)
ABBREVIATIONS USED
- HAT
Human African trypanosomiasis
- IFP
Protein-ligand interaction fingerprint
- LmjPDEB1
Leishmania major phosphodiesterase B1
- TbrPDEA
Trypanosoma brucei phosphodiesterase A
- TbrPDEB1
Trypanosoma brucei phosphodiesterase B1
- TbrPDEB2
Trypanosoma brucei phosphodiesterase B2
- TbrPDEC
Trypanosoma brucei phosphodiesterase C
- TbrPDED
Trypanosoma brucei phosphodiesterase D
- TcrPDEC
Trypanosoma cruzi phosphodiesterase C
- SPA
scintillation proximity assay
Footnotes
Accession Code
The coordinates and structure factors of the TbrPDEB1 crystal structure described here have been deposited onto RCSB Protein Data Bank (www.pdb.org) with accession code 4I15.
Figures and Tables containing details of physical chemical filters, substructure filter and purchased compounds. Several files used to perform the virtual screening. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bland ND, Wang C, Tallman C, Gustafson AE, Wang Z, Ashton TD, Ochiana SO, McAllister G, Cotter K, Fang AP, Gechijian L, Garceau N, Gangurde R, Ortenberg R, Ondrechen MJ, Campbell RK, Pollastri MP. Pharmacological Validation of Trypanosoma brucei Phosphodiesterases B1 and B2 as Druggable Targets for African Sleeping Sickness. J Med Chem. 2011;54:8188–8194. doi: 10.1021/jm201148s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orrling KM, Jansen C, Vu XL, Balmer V, Bregy P, Shanmugham A, England P, Cos P, Maes L, Adams E, van de Bogaart E, Chatelain E, Ioset J-R, Stolpe A, Zorg S, Veerman J, Seebeck T, Sterk GJ, de Esch IJP, Leurs R. Catechol Pyrazolinones as Trypanocidals: Fragment-Based Design, Synthesis and Pharmacological Evaluation of Nanomolar Inhibitors of Trypanosomal Phosphodiesterase B1. J Med Chem. 2012 doi: 10.1021/jm301059b. [DOI] [PubMed] [Google Scholar]
- 3.Shakur Y, Koning HP, Ke H, Kambayashi J, Seebeck T. Therapeutic Potential of Phosphodiesterase Inhibitors in Parasitic Diseases Phosphodiesterases as Drug Targets. In: Francis SH, Conti M, Houslay MD, editors. Handb Exp Pharmacol. Vol. 204. Springer; Berlin Heidelberg: 2011. pp. 487–510. [DOI] [PubMed] [Google Scholar]
- 4.Wang H, Kunz S, Chen G, Seebeck T, Wan Y, Robinson H, Martinelli S, Ke H. Biological and Structural Characterization of Trypanosoma cruzi Phosphodiesterase C and Implications for Design of Parasite Selective Inhibitors. J Biol Chem. 2012;287:11788–11797. doi: 10.1074/jbc.M111.326777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oberholzer M, Marti G, Baresic M, Kunz S, Hemphill A, Seebeck T. The Trypanosoma brucei cAMP phosphodiesterases TbrPDEB1 and TbrPDEB2: flagellar enzymes that are essential for parasite virulence. FASEB J. 2007;21:720–731. doi: 10.1096/fj.06-6818com. [DOI] [PubMed] [Google Scholar]
- 6.de Koning HP, Gould MK, Sterk GJ, Tenor H, Kunz S, Luginbuehl E, Seebeck T. Pharmacological Validation of Trypanosoma brucei Phosphodiesterases as Novel Drug Targets. J Infect Dis. 2012;206:229–237. doi: 10.1093/infdis/jir857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. ( www.pdb.org) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seebeck T, Sterk GJ, Ke H. Phosphodiesterase inhibitors as a new generation of antiprotozoan drugs: exploiting the benefit of enzymes that are highly conserved between host and parasite. Future Med Chem. 2011;3:1289–1306. doi: 10.4155/fmc.11.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang C, Ashton TD, Gustafson A, Bland ND, Ochiana SO, Campbell RK, Pollastri MP. Synthesis and evaluation of human phosphodiesterases (PDE) 5 inhibitor analogs as trypanosomal PDE inhibitors. Part 1. Sildenafil analogs. Bioorg Med Chem Lett. 2012;22:2579–2581. doi: 10.1016/j.bmcl.2012.01.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ochiana SO, Gustafson A, Bland ND, Wang C, Russo MJ, Campbell RK, Pollastri MP. Synthesis and evaluation of human phosphodiesterases (PDE) 5 inhibitor analogs as trypanosomal PDE inhibitors. Part 2. Tadalafil analogs. Bioorg Med Chem Lett. 2012;22:2582–2584. doi: 10.1016/j.bmcl.2012.01.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang H, Yan Z, Geng J, Kunz S, Seebeck T, Ke H. Crystal structure of the Leishmania major phosphodiesterase LmjPDEB1 and insight into the design of the parasite-selective inhibitors. Mol Microbiol. 2007;66:1029–1038. doi: 10.1111/j.1365-2958.2007.05976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.King-Keller S, Li M, Smith A, Zheng S, Kaur G, Yang X, Wang B, Docampo R. Chemical Validation of Phosphodiesterase C as a Chemotherapeutic Target in Trypanosoma cruzi, the Etiological Agent of Chagas’ Disease. Antimicrob Agents Chemother. 2010;54:3738–3745. doi: 10.1128/AAC.00313-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim KY, Lee H, Yoo SE, Kim SH, Kang NS. Discovery of new inhibitor for PDE3 by virtual screening. Bioorg Med Chem Lett. 2011;21:1617–1620. doi: 10.1016/j.bmcl.2011.01.120. [DOI] [PubMed] [Google Scholar]
- 15.Tomori T, Hajdu I, Barna L, Lorincz Z, Cseh S, Dorman G. Combining 2D and 3D in silico methods for rapid selection of potential PDE5 inhibitors from multimillion compounds’ repositories: biological evaluation. Mol Divers. 2012;16:59–72. doi: 10.1007/s11030-011-9335-0. [DOI] [PubMed] [Google Scholar]
- 16.Kunz S, Luginbuehl E, Seebeck T. Gene Conversion Transfers the GAF-A Domain of Phosphodiesterase TbrPDEB1 to One Allele of TbrPDEB2 of Trypanosoma brucei. PLOS Neg Trop Dis. 2009;3:e455. doi: 10.1371/journal.pntd.0000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Card GL, England BP, Suzuki Y, Fong D, Powell B, Lee B, Luu C, Tabrizizad M, Gillette S, Ibrahim PN, Artis DR, Bollag G, Milburn MV, Kim SH, Schlessinger J, Zhang KYJ. Structural Basis for the Activity of Drugs that Inhibit Phosphodiesterases. Structure. 2004;12:2233–2247. doi: 10.1016/j.str.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 18.Kunz S, Oberholzer M, Seebeck T. A FYVE-containing unusual cyclic nucleotide phosphodiesterase from Trypanosoma cruzi. FEBS J. 2005;272:6412–6422. doi: 10.1111/j.1742-4658.2005.05039.x. [DOI] [PubMed] [Google Scholar]
- 19.MOE (Molecular Operating Environment), version 2011.10. Chemical Computing Group, Inc; Montreal, Canada: [Google Scholar]
- 20.Korb O, Stützle T, Exner TE. Empirical Scoring Functions for Advanced Protein-Ligand Docking with PLANTS. J Chem Inf Model. 2009;49:84–96. doi: 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- 21.Marcou G, Rognan D. Optimizing Fragment and Scaffold Docking by Use of Molecular Interaction Fingerprints. J Chem Inf Model. 2007;47:195–207. doi: 10.1021/ci600342e. [DOI] [PubMed] [Google Scholar]
- 22.de Graaf C, Kooistra AJ, Vischer HF, Katritch V, Kuijer M, Shiroishi M, Iwata S, Shimamura T, Stevens RC, de Esch IJP, Leurs R. Crystal Structure-Based Virtual Screening for Fragment-like Ligands of the Human Histamine H1 Receptor. J Med Chem. 2011;54:8195–8206. doi: 10.1021/jm2011589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irwin JJ, Shoichet BK. ZINC - A Free Database of Commercially Available Compounds for Virtual Screening. J Chem Inf Model. 2004;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2:541–553. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zoraghi R, Seebeck T. The cAMP-specific phosphodiesterase TbPDE2C is an essential enzyme in bloodstream form Trypanosoma brucei. Proc Natl Acad Sci US A. 2002;99:4343–4348. doi: 10.1073/pnas.062716599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 27.Navaza J, Saludjian P. AMoRe: an automated molecular replacement program package. Methods Enzymol. 1997;276:581–594. doi: 10.1016/S0076-6879(97)76079-8. [DOI] [PubMed] [Google Scholar]
- 28.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr Sect A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 29.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr, Sect D: Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 30.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 31.Joosten RP, te Beek TA, Krieger E, Hekkelman ML, Hooft RW, Schneider R, Sander C, Vriend G. A series of PDB related databases for everyday needs. Nucleic Acids Res. 2011;39:D411–D419. doi: 10.1093/nar/gkq1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu H. DSSP and Stride Plugin for PyMOL. TU Dresden: BIOTEC; 2011. [Google Scholar]
- 33.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2. 0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 34.The PyMOL Molecular Graphics System, version 1.5.0.3. Schrödinger, LLC; New York, NY, U.S.A: [Google Scholar]
- 35.FILTER, version 2.1.1. Openeye Scientific Software; Santa Fe, NM: [Google Scholar]
- 36.Corina, version 3.46. Molecular Networks GmbH; Erlangen, Germany: [Google Scholar]
- 37.Nicholls A, McGaughey GB, Sheridan RP, Good AC, Warren G, Mathieu M, Muchmore SW, Brown SP, Grant JA, Haigh JA, Nevins N, Jain AN, Kelley B. Molecular Shape and Medicinal Chemistry: A Perspective. J Med Chem. 2010;53:3862–3886. doi: 10.1021/jm900818s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wawer M, Bajorath Similarity-Potency Trees: A Method to Search for SAR Information in Compound Data Sets and Derive SAR Rules. J Chem Inf Model. 2010;50:1395–1409. doi: 10.1021/ci100197b. [DOI] [PubMed] [Google Scholar]
- 39.Thompson JW, Brooker G, Appleman MM. Assay of cyclic nucleotide phosphodiesterases with radioactive substrates. Methods Enzymol. 1974;38:205–212. doi: 10.1016/0076-6879(74)38033-0. [DOI] [PubMed] [Google Scholar]
- 40.Bauer AC, Schwabe U. An improved assay of cyclic 3′,5′-nucleotide phosphodiesterases with QAE-Sephadex columns. Naunyn-Schmiedeberg’s Arch Pharmacol. 1980;311:193–198. doi: 10.1007/BF00510259. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.