

Abstract
The role of soil methylotrophs in methanol exchange with the atmosphere has been widely overlooked. Methanol can be derived from plant polymers and be consumed by soil microbial communities. In the current study, methanol-utilizing methylotrophs of 14 aerated soils were examined to resolve their comparative diversities and capacities to utilize ambient concentrations of methanol. Abundances of cultivable methylotrophs ranged from 106–108 gsoilDW−1. Methanol dissimilation was measured based on conversion of supplemented 14C-methanol, and occurred at concentrations down to 0.002 μmol methanol gsoilDW−1. Tested soils exhibited specific affinities to methanol (a0s=0.01 d−1) that were similar to those of other environments suggesting that methylotrophs with similar affinities were present. Two deep-branching alphaproteobacterial genotypes of mch responded to the addition of ambient concentrations of methanol (⩽0.6 μmol methanol gsoilDW−1) in one of these soils. Methylotroph community structures were assessed by amplicon pyrosequencing of genes of mono carbon metabolism (mxaF, mch and fae). Alphaproteobacteria-affiliated genotypes were predominant in all investigated soils, and the occurrence of novel genotypes indicated a hitherto unveiled diversity of methylotrophs. Correlations between vegetation type, soil pH and methylotroph community structure suggested that plant–methylotroph interactions were determinative for soil methylotrophs.
Keywords: aerated soil, amplicon pyrosequencing, mxaF, mch, fae, Michaelis–Menten kinetics
Introduction
Aerobic methylotrophic bacteria in soils have been recognized as drivers of methane fluxes in terrestrial ecosystem (Dunfield, 2007; Trotsenko and Murrell, 2008; Conrad, 2009; Kolb, 2009a). However, their role in methanol exchange with the atmosphere has widely been overlooked, even though the annual global emission rate (5 Tmol per year) of methanol is close to that of methane (that is, 10 Tmol per year; Jacob et al., 2005), and most methylotrophs utilize methanol (Lidstrom, 2006; Chistoserdova et al., 2009; Kolb, 2009a).
Methanol is distantly the second most abundant organic compound in the atmosphere (0.1–10 p.p.b.) after methane (1800 p.p.b.), but it is chemically more reactive than methane. Tropospheric methanol reacts with nitrogen oxides to produce HOx radicals and affects the oxidizing capacity of the troposphere and ozone formation. The main source of atmospheric methanol is plant biomass (Galbally and Kirstine, 2002; Jacob et al., 2005). Methanol can be released from the methoxy groups of pectin and lignin (Donnelly and Dagley, 1980; Schink and Zeikus, 1980; Fall and Benson, 1996; Warneke et al., 1999). Estimates of global rates of methanol emission based on plant biomass production are considerably higher (26 Tmol per year) than the observed rates (Galbally and Kirstine, 2002; Jacob et al., 2005; Kolb, 2009a) suggesting that methylotrophs of terrestrial ecosystems likely consume methanol and thereby partially mitigate its emission into the atmosphere (Kolb, 2009a). However, quantitative information on this activity in terrestrial ecosystems is lacking. As methanol is plant-derived in terrestrial ecosystems, plant surfaces, such as leaves, can be considered as favored habitats of aerobic methylotrophs. It has indeed been demonstrated that leaf surfaces of various temperate plant species are inhabited by active methylotrophs, which constitute about 14–20% to the total microbial community of the phyllosphere (Holland et al., 2002; Delmotte et al., 2009; Fedorov et al., 2011; Wellner et al., 2011; Mizuno et al., 2012). However, the upper soil layers of forests and grasslands are also well supplied with oxygen, and belowground parts of plants are likely sources of methanol.
Utilization of methanol requires unique metabolic pathways that differentiate methylotrophs from other aerobic heterotrophs. Genes encoding enzymes associated with mono carbon metabolisms are therefore suitable targets for detecting methylotrophs in the environment (McDonald et al., 2008). Methylotrophs oxidize methanol sequentially to carbon dioxide. The pyrroloquinolinequinone-dependent methanol dehydrogenase (PQQ MDH) (encoded by mxaFI) catalyzes the first step of methanol dissimilation in various Gram-negative bacteria (Chistoserdova et al., 2009). To date, mxaF is the only targeted gene in environmental studies of methylotrophs that encodes for a methanol-oxidizing enzyme (McDonald et al., 2005, 2008). A homolog (mdh2) exists in Burkholderiaceae, but mdh2 is not as widely distributed within methylotrophs as mxaF (Kalyuzhnaya et al., 2008). xoxF, a similar gene, occurs in all known methylotroph genomes (Chistoserdova, 2011). Its gene product XoxF oxidizes in vitro methanol (Schmidt et al., 2010). XoxF is needed for PQQ MDH activity and is involved in the regulation of PQQ MDH in Methylobacterium extorquens AM1. Whether XoxF can also function as an alternative MDH in vivo is unknown and currently under debate (Skovran et al., 2011). In many Gram-negative methylotrophs, formaldehyde is transferred to tetramethanopterine (H4MPT) by the formaldehyde-activating enzyme (encoded by fae), and oxidized to formate by reactions that employ H4MPT-dependent enzymes (Vorholt et al., 1999). One of those enzymes, methenyl-cyclohydrolase (encoded by mch), has been successfully used to resolve methylotrophic community structure and diversity in a lake (Kalyuzhnaya et al., 2004, 2005).
Phylogenetic identities of methylotrophs in soils that utilize methanol at in situ concentrations have not been resolved. Taxa that assimilated supplemented [13C]-methanol in an aerated forest soil have been identified (Radajewski et al., 2002), but current knowledge is based on pure cultures and soil experiments (Radajewski et al., 2002; Kolb, 2009a), in which methanol was supplemented in millimolar concentrations that likely do not occur in soil. Substrate affinities (that is, half-saturation constant, Km) of methanol dehydrogenases of soil-derived methylotrophs are above 1000 nmol l−1 (Arfman et al., 1989; Hektor et al., 2002; Nojiri et al., 2006). Recent measurements of Km (Km; 9.3 nmol l−1) in marine surface water samples (Dixon et al., 2011) suggest that unknown and low methanol concentration-adapted methylotrophs utilize methanol in marine ecosystems. Such information is missing for terrestrial ecosystems, which are not only the main source of atmospheric methanol but are also a sink for it (Karl et al., 2005; Schade et al., 2011).
The objectives of the current study were (a) to determine substrate affinities for aerobic methanol dissimilation in aerated soils, (b) to identify genotypes that respond to micro molar concentrations of methanol in aerated soils, (c) to assess community structures of methylotrophs that were detected by mxaF, mch and fae pyrosequencing and cultivation in aerated forest and grassland soils, and (d) to relate site-specific environmental parameters with methylotroph community structures.
Materials and methods
Study sites, sampling and sample preparation
Top mineral soils of 14 different temperate, aerated grassland and forest soils in Germany were investigated (Table 1). The grassland soils FG, OG and HEG 6 were used in experiments in which methanol dissimilation rates and Michaelis–Menten kinetics were determined. In case of FG and OG, top mineral soil of three subsites was sampled and pooled. Stones and other large materials were removed manually. The grassland soils AEG 2, AEG 7, HEG 6, HEG 9, SEG 2 and SEG 6, and the forest soils AEW 5, AEW 8, HEW 5, HEW 12, SEW 5 and SEW 9 were a subset of sampling plots that are distributed in three regions over a transect from northeast to southwest of Germany and set up within a joint research project that assesses biodiversity and soil parameters at about 3000 sampling sites (Fischer et al., 2010). The sites used in the current study represented grasslands and forests in three German regions (that is, Schorfheide Chorin, Hainich and Schwäbische Alb) with both high and low land use intensity. On several occasions from April 2008 to October 2009 (Table 1), five soil cores were taken per site, and A horizon (that is, top mineral soil) material was manually prepared and pooled. A detailed sampling schedule can be found in Supplementary Table S1. All soil samples were transported on ice and immediately frozen at −80 °C for molecular analyses. Soils (FG, OG and HEG 6) that were used for methanol oxidation rate measurements were stored at 2 °C for further analysis.
Table 1. Description of analyzed soils, including sampling period, and assignment to conducted analyses.
| Soil | Soil typea | Location | Vegetationb (land use intensity)c | pHd | NO3−/NH4+ (μg gTG−1) | Samplede | Analysis |
|---|---|---|---|---|---|---|---|
| FG | Cambisol | N50°08′ E11°52′ | Grassland (ni) | 6.0 | — | 2010 | MeOH dis |
| OG | Histosol | N49°57′37′′ E11°35′42′′ | Grassland (ni) | 7.0 | — | 2010 | MeOH dis |
| AEG 2 | Leptosol | N48° 22′ 36.686′′ E9° 28′ 22.023′′ | Grassland (i) | 6.9 | 2.8/23.8 | Pyro Isol MPN | |
| AEG 7 | Leptosol | N48° 23′ 29.116′′ E9° 22′ 36.65′′ | Grassland (ni) | 7.6 | 1.8/23.2 | Pyro MPN | |
| AEW 5 | Cambisol | N48° 25′ 10.626′′ E9° 24′ 52.854′′ | Forest (i) | 5.6 | 1.3/24.7 | Pyro MPN | |
| AEW 8 | Cambisol | N48° 22′ 57.322′′ E9° 22′ 56.584′′ | Forest (ni) | 6.4 | 2.2/25.0 | Pyrof Isol MPN | |
| HEG 6 | Stagnosol | N51° 12′ 53.766′′ E10° 23′ 28.395′′ | Grassland (i) | 6.5 | 0.4/20.2 | MeOH dis Pyro TRFLP Isol MPN | |
| HEG 9 | Stagnosol | N51° 13′ 26.031′′ E10° 22′ 50.834′′ | Grassland (ni) | 7.0 | 0.3/19.7 | 2008 2009 | Pyro Isol MPN |
| HEW 5 | Luvisol | N51° 15′ 49.961′′ E10° 14′ 27.448′′ | Forest (i) | 5.4 | 1.0/25.0 | Pyro Isol MPN | |
| HEW 12 | Luvisol | N51° 6′ 2.477′′ E 10° 27' 18.659” | Forest (ni) | 4.8 | 0.6/20.9 | Pyro Isol MPN | |
| SEG 2 | Histosol | N53° 5′ 21.505′′ E13° 58′ 48.169′′ | Grassland (i) | 7.5 | 1.6/24.9 | Pyro | |
| SEG 6 | Histosol | N53° 6′ 12.583′′ E13° 37′ 22.2′′ | Grassland (ni) | 5.8 | 1.6/23.7 | Pyro MPN | |
| SEW 5 | Cambisol | N53° 3′ 25.321′′ E 13° 53′ 7.318′′ | Forest (i) | 4.0 | 0.6/16.4 | Pyrog,f,h Isol MPN | |
| SEW 9 | Cambisol | N53° 2′ 40.513′′ E13° 48′ 36.371′′ | Forest (ni) | 4.5 | 0.4/15.2 | Pyrog Isol MPN |
Abbreviations: Isol, isolation of pure cultures; MeOH dis, determination of methanol dissimilation based on the amount of 14CO2 produced from 14C-methanol; MPN, most probable numbers of methanol-utilizing methylotrophs (details in Supplementary Information ‘Methods'); Pyro, pyrosequencing of genes mxaF, fae and mch; TRFLP, terminal restriction fragment length polymorphism analysis with mch.
Data were taken from Fischer et al., 2010.
Grassland, temperate grassland with various grasses and weeds; Forest, mixed hardwood forests that were dominated by beech (Fagus sylvatica L.).
i, intense land use (i.e., grasslands were regularly fertilized and forests were regularly harvested); ni, non-intense land use (i.e., grasslands no fertilized and forests were not subjected to land use since at least 60 years).
pH in water; mean values from three independent measurements.
Detailed sampling scheme is given in Supplementary Table S1.
In these soils amplification of mch was not successful.
In these soils amplification of mxaF was not successful.
In these soils amplification of fae was not successful.
Methanol Oxidation Rates in Grassland soils FG, OG and HEG 6
1) Measuring Michaelis-Menten kinetics for samples of soils FG and HEG 6. One gram fresh weight of soil was suspended in sterile water adding up to a volume of 5 ml in gas-tight tubes (Bellco Glass Inc., Vineland, NJ, USA). Slurries were supplemented with 12C-methanol with six different concentrations between 0.001 and 250 μmol gsoilfreshweight −1. Traces of 14C-methanol (1.2 nmolsoilfreshweight −1) were added. Slurries were incubated at room temperature, and formed carbon dioxide (CO2) was trapped in two sequential sodium hydroxide traps over a period of 3 days. CO2 traps consisting of tubes containing sodium hydroxide (per trap tube 1 ml NaOH (1 M) were connected to the slurries with gas-tight tubing. Flushing with sterile air was used to drive 14CO2 into the traps. Hundred-microliter aliquots were measured by scintillation counting (LS 6500, Beckman Coulter GmbH, Krefeld, Germany) after overnight incubation in the scintillation cocktail (Ecolume Liquid Scintillation Cocktail, MP Biomedicals Germany GmbH, Eschwege, Germany). Sum of counts per minute from both traps, and the specific activity of 14C-methanol (Biotrend GmbH, Köln, Deutschland) were used to calculate formed 14CO2. Based on the proportion of 14CO2 to 12CO2, the total amount of formed CO2 was determined. Production of CO2 was used to estimate the dissimilated amount of methanol. All measurements were conducted in experimental duplicates. As controls, abiotic formation of 14CO2 was measured in treatments with sterile water that was supplemented with 14C-methanol, and in soil slurries with radioactive methanol that were biologically inactivated by adding 10 mℳ potassium cyanide (Watanabe et al., 1996). Maximal velocity (V max) and K m were determined based on calculated oxidation rates of 14C-methanol at different concentrations that were nonlinearly fitted (SigmaPlot, version 10.0, Systat Software GmbH, Erkrath, Germany; Segel, 1993).
2) Localization of methanol oxidation activity in soil OG. Washed roots, root-free soil from soil OG and HEG 6, and a sterilely grown grassland plant (Arabidopis thaliana, cultivation procedure in Supplementary Information ‘Methods') were supplemented with total concentrations of 14C- and 12C-methanol of 51 nmol gfreshweigt −1. The difference in produced 14CO2 between the start of the experiment and after 13 days was determined. For each sample, a biologically inactivated treatment with sodium cyanide was set up.
Terminal restriction fragment length polymorphism of mch genes in samples of 14 C-methanol-supplemented slurries
DNA was extracted from slurry samples of the experiments, in which Michaelis–Menten kinetics were determined in soils FG and HEG6, according to a previously published protocol (Stralis-Pavese et al., 2004). Sufficient amounts of DNA could not be extracted from slurry samples of FG, and OG, and amplification of genes mxaF and fae was not successful with extracts of slurries of soil HEG 6. Primer mch-2a was covalently labeled with fluorescent dye IRDye 681 (Microsynth AG, Lindau, Germany). PCR products were digested with restriction enzyme BslI (New England Biolabs GmbH, Frankfurt a.M., Germany), and separated on sequencer (NEN4300, Licor GmbH, Bad Homburg, Germany) procedure and settings were the same that were used in a previous study, in which 16S ribosomal RNA (rRNA) gene terminal restriction fragment (TRF) length polymorphism was conducted (Schellenberger et al., 2010). Retrieved TRF patterns were used to identify those TRFs that were enriched in methanol-supplemented slurries. mch was also amplified using the same primers as for TRF length polymorphism but without fluorochrome label. These amplicons were pooled, and separated by TA cloning into competent Escherichia coli cells (LGC Genomics GmbH, Berlin, Germany). Ninety-six inserts in transformed plasmid vectors were sequenced (LGC Genomics GmbH). Sequence information was used to assign TRFs to genotypes of mch (TRFCUT; Ricke et al., 2005). Briefly, mch sequences were aligned and hypothetical TRFs were determined. Measured TRFs were compared with predicted TRFs to assign TRFs to mch sequences.
Most probable numbers of methanol-utilizing methylotrophs
Viable cell numbers of methanol-utilizing, aerobic methylotrophs were determined by most probable number (MPN) technique (Alef, 1991). From soils that were sampled in April of 2008 and 2009 (Table 1, Supplementary S1 and S3), MPNs were determined in mineral media (M1; Dedysh et al., 1998) or 125 (DSMZ Methylobacterium medium, Deutsche Sammlung für Mikroorganismen und Zellkulturen GmbH, Braunschweig, Germany). Media were supplemented with 10 mℳ of methanol at pH 3.1 or 6.8, with or without additional nitrate (0.5–1.0 mℳ) and with or without vitamine solution (Atlas, 1993). Serial dilutions of soil (102 up to 109 fold) were inoculated in 10 replicates of fresh medium in 96-well microtiter plates (Sarstedt GmbH, Nümbrecht, Germany). For each plate, a row of wells were filled with sterile medium as controls. Plates were incubated at 20 °C for up to 72 days (details in Supplementary Information ‘Methods'). Wells were regularly checked for growth until no change in number of growth-positive wells was observed. Growth was detected based on turbidity measured at 660 nm (μQuant Universal Microplate Spectrophotometer, BIO-Tek Instruments GmbH, Bad Friedrichshall, Germany). Measured optical density was corrected for values at the start of incubation to account for different amounts of inoculated cells. Based on the variability of data, a change of 0.03 in OD660 was interpreted as growth. Determined MPNs (based on tables in Alef) per milliliter of inoculum were converted to gram dry weight of soil (Supplementary Table S2). These MPNs were correlated with environmental parameters in subsequent statistical analyses.
Amplicon pyrosequencing of mxaF, mch and fae
DNA that was used for barcoded amplicon pyrosequencing was extracted from 0.3 g of soil according to a published protocol (Stralis-Pavese et al., 2004). The procedure includes lysis by beat beating. Extracted DNA was dissolved in 100 μl DNAse and RNAse-free water, and stored at −80 °C for further analysis. Undiluted or one-hundred-fold diluted DNA extract was used in subsequent amplification of mxaF, fae and mch genes. The used gene-specific primers were tagged at the 5′ terminus with a six-nucleotide barcode that encoded for a specific soil. mxaF was amplified with primers 1003f (5′-GCGGCACCAACTGGGGCTGGT-3′) and 1555r (5′-CATGAABGGCTCCCARTCCAT-3′) (McDonald and Murrell, 1997; Neufeld et al., 2007), fae was amplified with primers fae1f (5′-GTCGGCGACGGCAAYGARGTCG-3′) and fae1r (5′-GTAGTTGWANTYCTGGATCTT-3′) (Kalyuzhnaya et al., 2004), and mch was amplified with primers mch-2a (5′-TGCCTCGGCTCKCAATATGCYGGBTGG-3′) and mch-3 (5′-GCGTCGTTKGTKCKBCCCAT-3′) (Vorholt et al., 1999). For amplification of mch and fae, 50 μl reactions were performed on a Primus 96 thermocycler (PeqLab Biotechnologie GmbH, Erlangen, Germany), which contained 25 μl of Master Amp PCR premix (2.5-fold; Epicentre Biotechnologies Inc., Madison, WI, USA), 0.15 μl of Taq DNA polymerase (5 U μl−1; Invitrogen GmbH, Karlsruhe, Germany), 5 μl of each primer solution (10 μℳ), 5 μl of template DNA and 9.85 μl of RNase- and DNase-free water. mch and fae were amplified according to the following temperature program, that is, intial denaturation (95 °C, 1 min) followed by a touchdown step (denaturation, 95 °C, 0.5 min; annealing, 55 down to 45 °C, 0.5 min; and elongation, 72 °C, 1.3 min), followed by 20 cycles of amplification (denaturation, 95 °C, 0.5 min; annealing, 45 °C, 0.5 min; and elongation, 72 °C, 1.3 min). The program was finished with a single elongation step (72 °C, 10 min). As mxaF could not reliably amplified with the above described reactions mix, the reaction mix 50 μl contained 10 μl of Phusion HF Buffer (fivefold; NEB), 0.5 μl of Phusion DNA polymerase (NEB), 1.5 μl of dimethyl sulphoxide (100%), 1.5 μl of MgCl2 (50 mℳ; NEB), 4 μl of didesoxy-nucleotide mix (2.5 mℳ; NEB), 3 μl of each primer solution (10 μℳ), 1 μl of template DNA, and 25.5 μl of RNase- and DNase-free water. The temperature program started with a single denaturation step (98 °C, 1 min), followed by 35 amplification cycles (denaturation, 98 °C, 0.6 min; combined annealing and elongation, 72 °C, 0.5 min). The protocol ended with a final elongation step (72 °C, 6 min).
PCR products were cut out from agarose gels, and were extracted with the Montage Gel Extraction Kit (Millipore GmbH, Schwalbach, Germany). Amplicon mixtures were treated with PreCRepair Mix (NEB) to eliminate possible PCR-blocking DNA damage that might have occurred during gel purification or storage of amplicons, and were purified by isopropanol precipitation at −20 °C overnight. Purified and dried DNA was dissolved in 20 μl of RNAse- and DNAse-free water, and amplicons of mch and fae were pooled and then pyrosequenced at the Göttingen Genomics Laboratory using the Roche GS-FLX 454 Sequencer and GS FLX Titanium series reagents according to manufacturers recommendations (Roche Diagnostics GmbH, Mannheim, Germany). In brief, an emulsion PCR was performed after ligation of A (5′-CGTATCGCCTCCCTCGCGCCATCAG-3′) and B (5′-CTATGCGCCTTGCCAGCCCGCTCAG-3′) sequencing adaptors. Subsequently, beads were transferred on a picotiter plate, and were sequenced. mxaF was independently sequenced from mch and fae as the number of reads was not sufficient when mxaF amplicons were mixed up with mch and fae PCR products.
Quality filtering and clustering of mxaF, mch and fae amplicon pyrosequencing reads
The raw reads were processed using AmpliconNoise (Quince et al., 2011). In summary, pyrosequencing flowgrams that had at least one flow with signal intensity between 0.5 and 0.7 or a cycle of four nucleotide flows (TACG) that failed to give a signal >0.5 before cycle number 400 (both indicative of noise) were discarded, and all reads were truncated at cycle number 600. Pyrosequencing noise was subsequently removed using PyroNoise (Quince et al., 2011) using the default settings, and PCR noise was removed using SeqNoise (σs=0.033 and cs=0.08) (Quince et al., 2011) after truncation at 400 bp to reduce noise further.These data were then subjected to sorting, clustering and BLAST analysis with Jaguc (Version 2.1; Nebel et al., 2011). Only reads starting at the forward primer and being longer than 200 bp were further analyzed. All reads from all soils were then clustered and identified using BLAST (Altschul et al., 1990). To get reliable BLAST search result, the following non-default parameter settings were chosen (blastn, non-default expect threshold value was 100). Genotypes of non-target genes were removed before further analysis. Sequences were affiliated to genotypes, that is, operational taxonomic units (OTUs), based on similarity cutoff values of 77%, 80% and 80% for mxaF, mch and fae, respectively, which were determined using the average neighboring method (Nebel et al., 2011). Barcode identifiers were used to determine the relative frequency of a genotype in amplicons from a certain soil after clustering. Similarity cutoff values were determined as follows. As the data basis for calculation of species-level cutoff for mxaF was sufficient (that is, 60 strains with both gene sequences were available), a comparison of pair-wise similarities of 16S rRNA genes that were related to pair-wise similarities of corresponding mxaF similarities revealed a species-level similarity cutoff of 77% for mxaF that corresponds to 97% similarity on 16S rRNA gene level (Supplementary Information ‘Methods' and Supplementary Figure S1a). There was no sufficient information of 16S rRNA sequences and affiliated mch or fae sequences. Thus, quality-filtered reads of mch and fae from all soils were clustered using decreasing cutoff values for gene similarities, based on which sequences were clustered with Jaguc. Maximally high cutoff values were chosen, at which the number of retrieved genotypes stayed constant (Supplementary Information ‘Methods' and Supplementary Figure S1b).
Reconstruction of MxaF, Mch and Fae phylograms
From each OTU, one representative amino-acid sequence was aligned with known reference sequences (MEGA5, Tamura et al., 2011), and used to reconstruct neighbor-joining trees (1000 replicated calculations, Saitou and Nei, 1987). The trees were used to provide phylogenetic assignments to novel genotypes. All presented sequences of rarified data sets were deposited at the European Bioinformatics Institute, that is, accession numbers HE970319–HE970434.
Statistical analyses of community structure (mxaF, mch and fae) and abundance (MPN) data sets
Genotypes (that is, number of OTUs) coverages were always above 98% (Supplementary Table S5). As numbers of reads per gene and soil were variable (Supplementary Table S5), data sets for each gene were rarefied using QIIME (Caparose et al., 2010). Sample size was adjusted to the soil with lowest number of reads (Supplementary Table S5). Normal distribution of the data set was tested for the 10 most abundant OTUs of each gene by the Shapiro–Wilk test (Royston, 1995) using the software OriginPro 8G SR4 (OriginLab Corporation, Northhampton, MA, USA). As most of the tested data were not normally distributed, canonical correspondence analyses (CCA) with log-transformed data were conducted. The effect of genotype and MPN data on the environmental parameters (that is, vegetation type (forest and grassland), land use intensity (managed and not managed), soil pH (Table 1), gravimetric water content (data not shown), nitrate concentration (Table 1), ammonium concentration (Table 1; personal communication I Schöning), total carbon content (personal communication I Schöning) and total nitrogen content (personal communication I Schöning)) was tested using the software Canoco (version 4.5, Microcomputer Power, Ithaca, NY, USA) (Ter Braak and Smilauer, 2002). Effect of each variable on the community structure based on mxaF, mch, fae and MPN data sets was tested by permutation test (9999 replicates, Monte–Carlo test) of the same software package. First and second axes of CCA results were visualized in ordination diagrams (CanoDraw, Microcomputer Power). As vegetation type was significantly correlated with all data sets, indicator genotypes were determined to affiliate vegetation type to defined genotypes. Indicator values for vegetation type were calculated using the software PC-ORD (version 4.01, MjM Software Design) (Dufrene and Legendre, 1997). Indicator values for defined OTUs that were between 57% and 100% (P-value <0.04; Monte–Carlo test (1000 permutations)) were regarded as to be significant. Correlation of single MPN data sets (that is, highest or average MPN values; Supplementary Table S2) with environmental parameters was tested by Spearman rank correlation (Excel-based tool XLSTAT 2012; Microsoft GmbH, Unterschleißheim, Germany).
Results
Methanol dissimilation and associated taxa in grassland soils
All experimental replicates of all three grassland soils that were tested for their capability to oxidize low concentrations (putatively ambient) of methanol to CO2 (that is, HEG 6, OG and FG) exhibited similar dissimilation rates when methanol was provided at 74 nmol methanol gsoil DW−1 (that is, concentration corresponds to 10 μℳ in slurries). Observed half-saturation constants and maximal velocities were different between the soils FG and HEG 6 (Table 2; Supplementary Figure S2). These kinetic parameters might have been affected by diffusion limitation during the incubation. Specific affinities, which are independent of diffusion limitation, were similar (Table 2). Hence, methanol-oxidizing enzymes with similar substrate affinities were active in both soils. Highest capacities of aerobic methanol dissimilation occurred in soil that was associated with roots (Figure 1a). Root material and microorganism-free plant material of a typical grassland species (A. thaliana) displayed minimal activity (Figure 1a).
Table 2. Kinetic parameters of grassland soils OG, FG and HEG 6.
| Soil | Methanol dissimilation rate at 74 nmol methanol gDW−1 (μmol gDW−1d−1)a | νmax (mM)b | Km (mM)b | a0s (d−1) |
|---|---|---|---|---|
| OG Slurry 1 | 0.001 | — | — | — |
| OG Slurry 2 | 0.001 | — | — | — |
| FG Slurry 1 | 0.005 | 0.09 (<0.01) | 1.31 (0.06) | 0.07 |
| FG Slurry 2 | 0.007 | 0.16 (0.01) | 4.0 (0.7) | 0.04 |
| HEG Slurry 1 | 0.0006 | 0.39 (0.01) | 236.3 (21.0) | 0.01 |
| HEG Slurry 2 | 0.0006 | 0.34 (0.01) | 200.9 (16.0) | 0.01 |
Abbreviations: a0s, specific affinity; Km, Michaelis–Menten constant; νmax, maximal velocity; —, not measured.
74nmol methanol per gram dry weight of soil corresponds to 10 μℳ in slurry.
In parentheses, standard errors of predicted parameters are given (a0s is the quotient of νmax divided by Km, thus no error is given). Regressions are presented in Supplementary Figure S2.
Figure 1.

(a) Percentages of mineralized supplemented 14C-methanol in soil and plant samples. CN, cyanide added (10 mℳ potassium cyanide) sterile water, abiotic control was autoclaved water. (a) and (b) Indices of experimental replicates. (b) Changes of relative frequencies of mch TRFs in slurries of soil HEG 6 after 13 days with different concentrations of methanol. Methanol dissimilation rates at 0.002, 0.6, 6.0 and 302.0 μmol methanol gsoilDW−1 ∼0.0001, 0.027, 0.041, 1.185 μmol methanol gsoilDW−1 per day. Relative frequencies of TRFs shown at t0 are means of four experimental replicates, and relative frequencies of TRFs at different concentrations after 13 days are means of TRF patterns of two experimental replicates.
The TRF 430 bp was enriched at all supplemental methanol concentrations, and TRF 180 bp was enriched at concentrations greater than 600 nmol methanol gsoilDW−1 (Figure 1b). TRFs 430 and 180 bp were assigned to two novel genotypes (Grassland Soil Clusters (GSC) 1 and 2) within the Alphaproteobacteria (Supplementary Figure S3) based on comparison of measured TRFs with hypothetic TRFs, but were only distantly related to known genotypes of mch (30.9% distance on amino-acid level to Starkeya novella DSM 506 and 28.9% to Methylocella palustris BL2 for GSC1 and 2, respectively).
Correlations between genotype composition and environmental parameters
Genotype composition was significantly affected by soil pH, vegetation type (forest and grassland) and nitrate concentrations (Table 3; Figures 2a–c). Two mxaF genotypes, one mch genotype and one fae genotype were indicator genotypes for forest, while one mxaF, three mch and two fae genotypes were indicator genotypes for grassland (Table 4). The parameter vegetation type integrates numerous different environmental parameters, such as pH, gravimetric water content, ammonium and nitrate concentration. The latter three may represent a proxy for soil water regime and inorganic nitrogen availability. However, only nitrate concentration was significantly correlated with fae gene composition (Table 3).
Table 3. Effect of genotype composition of mxaF, mch and fae, and MPN data on environmental parameters by permutation tests.
| Data type | Environmental parametera | P-value | Trace value |
|---|---|---|---|
| mxaF | Vegetation type | 0.0007 | 0.275 |
| pH | 0.0298 | 0.198 | |
| mch | Vegetation type | 0.0001 | 0.322 |
| pH | 0.0005 | 0.361 | |
| fae | Vegetation type | 0.0018 | 0.433 |
| pH | 0.0005 | 0.470 | |
| Nitrate concentration | 0.0390 | 0.341 | |
| MPN | pH | 0.0069 | 0.109 |
| Ammonium concentration | 0.0267 | 0.095 |
Abbreviation: MPN, most probable number.
Only significantly correlated environmental parameters are presented and used for subsequent statistical analyses (Figures 2 and 3). The full set of tested environmental factors is available in online supplementary information (Supplementary Table S4).
Figure 2.

Effect of soil pH and vegetation type on mxaF, mch (a, b) genotype composition, and effect of soil pH, nitrate concentration, vegetation type on genotype composition of fae (c). Soil pH and nitrate concentration were used as vectors in CCA plots as they were significantly correlated to the respective genotype composition (Table 3). No data were available for mxaF of forest soil SEW 9. Thus, the data point of SEW 9 is missing in panel a. Closed circles, data from forest soils. Open circles, data from grassland soils. pH, soil pH; [NO3−], nitrate concentration; λ, eigen value.
Table 4. Indicator genotypes for vegetation types.
| Gene | GT | Vegetation type | IV (%) | P |
|---|---|---|---|---|
| mxaF | 6 | Forest | 99.0 | 0.023 |
| 15 | Forest | 94.7 | 0.015 | |
| 2 | Grassland | 56.9 | 0.006 | |
| mch | 185 | Grassland | 100 | 0.006 |
| 184 | Forest | 90 | 0.025 | |
| 279 | Grassland | 83.3 | 0.042 | |
| 281 | Grassland | 83.3 | 0.047 | |
| fae | 1233 | Forest | 100 | 0.002 |
| 1206 | Grassland | 97.7 | 0.004 | |
| 1222 | Grassland | 83.3 | 0.014 |
Abbreviations: GT, genotype; P, value of significance; IV, indicator value.
Abundances of cultivated methylotrophs correlated with environmental parameters
Abundances of methylotrophs as determined by MPN analysis ranged from 1 × 106 cells gsoilDW−1 to 3 × 108 cells gsoilDW−1. Highest cell numbers were determined for AEG 2 (that is, (3±7) × 108 cells gsoilDW−1), and lowest for HEG 6 (that is, (1±2) × 106 cells gsoilDW−1) (Supplementary Table S2). A correlation between highest or average MPNs of soils with any of the tested parameters was not significant (that is, tested by Spearman rank correlation). However, combined data sets of abundance data (that is, per soil MPNs of nine different cultivation treatments, Supplementary Table S2) significantly correlated with soil pH and ammonium concentration, but not with vegetation type based on Monte–Carlo tests. However, vegetation type was a determinative environmental parameter for genotype community composition (Table 3), and the combined set of abundances of forest soils were largely separated from those of grassland soils (Figure 3).
Figure 3.
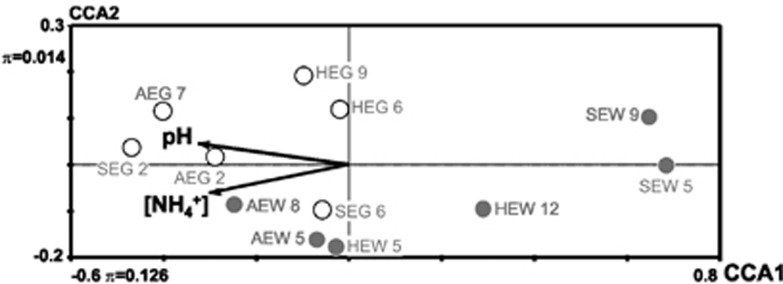
Effect of soil pH and ammonium concentration on MPNs (combined values of nine treatments) of methanol-grown methylotrophs. Soil pH and ammonium concentration were used as vectors in CCA plots as they were significantly correlated to the MPN data (Table 3). Closed circles, data from forests. Open circles, data from grassland soils. λ, eigen value.
Genotype diversity of mxaF, mch and fae
In total, 32 898, 26 503 and 17 500 reads that started with the forward primers of mxaF, mch and fae amplicons were retrieved from 11 top soils, respectively (Table 1; Supplementary Table S5). 31, 70 and 63 genotypes were detected for mxaF, mch and fae, respectively. Estimated maximal numbers of genotypes were 31, 76 and 72 for mxaF, mch and fae, respectively (Supplementary Table S5). Genotype numbers were slightly reduced after rarefaction of data sets (Supplementary Table S5). Estimated numbers of mch and fae genotypes were greater than those of mxaF (Supplementary Table S5). The numbers of mch- and fae-like genes were greater suggesting a broader diversity of detected methylotroph taxa. mch- and fae-like genes also occur in non-methylotrophs (Chistoserdova et al., 2009; Chistoserdova, 2011). Thus, the high diversity of novel mch and fae genotypes may be additionally caused by codetected non-methylotrophs.
The diversity of detected genotypes was unevenly distributed and dominated by two genotypes of mxaF, namely OTUs 2 (78% of all sequences) and 6 (13%). OTU 2 affiliated with Methylobacterium (Figure 4). Most abundant mch OTUs were 178 (46%), 201 (15%), 186 (6%) and 185 (5%). fae genotypes were more even distributed (Supplementary Table S5), which was reflected by a larger number of genotypes with a relative abundance above 1%, that is, those were 1193 (53%), 1190 (12%), 1233 (10%), 1209 (3%), 1212 (4%), 1222 (3%), 1205 (3%) and 1206 (2%).
Figure 4.

MxaF protein tree of detected genotypes. Bold genotypes, genotypes with relative abundances >1%, i.e., relative frequencies are given in parentheses. Numbers at nodes, bootstrap values from 1000 replicated trees, those >50% are shown. Scale bar represents an evolutionary distance of 5%. OTU, mxaF genoytpes of rarefied data sets.
Methylobacterium-affiliated mxaF genotypes were predominant in all soils and exhibited relative abundances of >59% in each soil (data not shown). A substantial number of low-abundant genotypes (that is, OTUs 15, 16, 17, 18, 19, 21, 22, 23, 24, 25, 26 and 27) were not closely affiliated with genotypes of known methylotrophic species. These low-abundant genotypes affiliated with genome-derived genotypes of bacterial strains, of which methylotrophy is not known to date (for example, with mxaF' of Rhizobiales) (Figure 4).
Dominant mch genotypes and mxaF genotypes affiliated with Alphaproteobacteria but with different genera, that is, mch genotypes, were distantly related with Granulibacter and Methylocella (OTU 201), and Starkeya (OTUs 178, 185 and 186) (Supplementary Figure S3). The two mch genotypes (GSC 1and 2) that responded to low methanol concentrations in soil HEG 6 (Figure 1b) were closely related on amino-acid level with the two most abundant mch genotypes (OTUs 178 and 201) (Supplementary Figure S3). Some Alphaproteobacteria-affiliated mch genoytpes were only distantly related to mch genes of known isolates (Supplementary Figure S3) suggesting that numerous unknown Alphaproteobacteria harbor mch genes.
All detected fae genotypes belonged to fae genes sensu stricto as classified in a previous study (Chistoserdova, 2011), and were mainly affiliated with those of Alpha, Beta- and Gammaproteobacteria (Supplementary Figure S4). The most abundant genotypes (OTUs 1193, 1190 and 1233, which collectively constituted 75% of all fae reads) were affiliated with members of Alphaproteobacteria (Supplementary Figure S4). Owing to the low number of known reference sequences and low similarity to known ones, it was difficult to determine if detected OTUs represented known or novel genera. The most closely related isolates for many fae OTUs were well-known methylotrophs, such as Hyphomicrobium, Methylobacterium, Methylibium and Methylotenera, but several OTUs were closely related to Burkholderia species, which are not known to be capable of methylotrophy (Kolb, 2009a).
A substantial number of novel genotypes of mch and fae were not affiliated with isolated species (Supplementary Figures S3 and S4). As the diversity of known mch- and fae-like genes is not restricted to methylotrophs (Chistoserdova, 2011), it is likely that the detected deep-branching mch and fae genotypes does not exclusively represented aerobic methylotrophs.
Discussion
Methanol-utilizing methylotrophs have been known since the nineteenth century, and have been repeatedly isolated from soils worldwide (Loew, 1892; Lidstrom, 2006; Chistoserdova et al., 2009; Kolb, 2009a). Methanol-utilizing methylotrophs can be metabolically stimulated by adding millimolar concentrations of methanol to aerated and flooded soil (Radajewski et al., 2002; Lueders et al., 2004). However, the methylotrophs responsible for in situ methanol consumption are unresolved. Information on ranges of methanol concentrations in soil and on ‘hot spots' of methanol production are not available. Concentrations down to nanomoles per gram dry weight of soil (that is, corresponds to about 0.1 μℳ in slurries) were oxidized in the current study suggesting that methylotrophs have methanol-oxidizing enzymes with micro to nanomolar half-saturation constants that could oxidize such low methanol concentrations. Based on maximal atmospheric methanol concentrations (10 p.p.b.) (Galbally and Kirstine, 2002; Jacob et al., 2005), it can be estimated that diffusion into soil yield concentrations of several nanomoles of methanol per gram dry weight of soil (that is, ∼20 nmol methanol gsoilDW−1). Thus, the observed capacity of soil microorganisms to oxidize concentrations at nanomoles methanol gsoilDW−1 may explain the capacity of aerated soils to consume atmospheric methanol (Karl et al., 2005; Schade et al., 2011). Observed minimal methanol oxidation rates were even below 20 nmol methanol gsoilDW−1. It can be speculated that unknown Alphaproteobacteria (that is, as suggested by responding mch genotypes) might have high-affinity methanol dehydrogenases that enable them to utilize atmosphere-derived methanol.
Methanol oxidation by methylotrophs in grassland soils FG, HEG 6 and OG
Two novel mch genotypes (GSC 1 and 2) responded to concentrations of methanol below 1 μmol gsoilDW−1. GSC 1 and 2 were distantly related to mch genotypes of known methylotrophs, and grouped with those of Alphaproteobacteria. Although amplification of mch failed in several samples of the kinetic experiments that were conducted with the soils FG and HEG 6, gene marker-based assessment of methylotroph community structures in the remaining 11 aerated soils revealed that Alphaproteobacteria represented the most frequently detected phylum. The mch data set suggested that GSC 1- and 2-like genotypes were frequently detected putative methanol utilizers in both temperate grasslands and forests.
Substrate affinities of active methylotrophs in grassland soils FG and HEG 6 were in the upper range of values of purified MDH (Arfman et al., 1989; Hektor et al., 2002), and higher than those of a previous environmental study that addresses ocean surface water communities (Dixon et al., 2011). Accordingly, the specific affinity values a0s were lower, and νmax were higher than the solely known values that have been obtained from the environment (a0s: 0.12 to 0.96 d−1; νmax: up to 24 nmol d−1l−1) (Dixon et al., 2011). The lowest measured Km values (3 μmol l−1) of MDH are known of the Gram-negative soil methylotroph Hyphomicrobium denitrificans (Nojiri et al., 2006), whereas Km values of MDH of other soil methylotrophs can be much higher (that is, Bacillus methanolicus, Km>200 μmol l−1) (Arfman et al., 1989; Hektor et al., 2002). H. denitrificans has a PQQ MDH (encoded by MxaFI) that has been intensively studied in the model methylotroph M. extorquens AM1 (Chistoserdova et al., 2009). PQQ MDH also occurs in many other Gram-negative methylotrophs (Nunn et al., 1989; Schmidt et al., 2010; Chistoserdova, 2011). Utilization of micro molar concentrations is known from M. extorquens AM1, and the marine methylotrophic strain HTCC2181 (Betaproteobacteria) (Giovannoni et al., 2008; Halsey et al., 2012). Strain HTCC2118 assimilates 10 μmol l−1 methanol, which is substantially above observed minimal concentrations (0.002 μmol gsoilDW−1) at which methanol dissimilation was measured in the present study. Hence, it is likely that various soil methylotrophs have the capability of dis- and assimilation of concentrations above several hundred nanomoles of methanol per gram dry weight of soil. If those can also be active at minimal concentrations observed in the current study remains speculative.
Many soil methylotrophs are facultatively methylotrophic, that is, can utilize multicarbon compounds (Loew, 1892; Lidstrom, 2006; Chistoserdova et al., 2009; Kolb, 2009a). Moreover, it has been demonstrated that methylotrophs that are restricted to methanol as carbon source exhibit increased methanol assimilation rates when they codissimilate alternative monocarbon compounds (Halsey et al., 2012). Thus, it is likely that methanol utilizers in aerated soils are not restricted to ambient methanol utilization and occupy different ecological niches in regard to the alternative substrates they can metabolize (Kolb, 2009a).
High methanol dissimilation capacity in root-associated grasslands soil
OG and HEG 6 root-associated soils exhibited the highest methanol oxidation activities, indicating that microorganisms in root-associated soil were mainly responsible for the capacity of soils to oxidize methanol. Root-free soil samples were gained by a mild washing procedure with sterile water suggesting that soil methylotrophs of grasslands are not necessarily tightly associated with the plant surface as are phyllosphere methylotrophs (for example, Delmotte et al., 2009). Roots constitute likely a source of methanol, and the minimal capacity of roots to oxidize methanol (Figure 1a) suggests that roots are not net sinks for methanol in grassland soils.
Environmental parameters that shape methylotrophic communities in aerated soils
All three gene marker data sets and the combined MPN analyses were significantly correlated with soil pH and, in case of gene data sets, also by vegetation type suggesting that the observed influences of environmental parameters are largely method independent. Vegetation type and soil pH are site parameters that are stable over several years (Fischer et al., 2010). The more dynamic site parameters (that is, water content, ammonium and nitrate concentration) did not significantly affect all of the data set patterns. This lack of general correlation does not necessarily mean that such dynamic parameters were irrelevant, but it is more likely that a seasonal data set of these parameters and methylotroph communities would have led to more conclusive results. Moreover, fae genotype composition was significantly affected by nitrate and MPN data set by ammonium concentration indicating that nitrogen availability was also a key parameter for soil methylotrophs, which is in accordance with knowledge of physiologies of methylotrophs that can assimilate dinitrogen gas or nitrate and ammonium as sources of nitrogen (Kolb, 2009a, and references therein).
Soil pH is a factor that can be correlated with soil prokaryotic community structure (Fierer and Jackson, 2006). A correlation between soil pH and aerobic methanotroph genotypes is also likely (Kolb, 2009b). Known methylotrophic species cover the full range of pH optima, that is, from alkalophilic to acidophilic species (Lidstrom, 2006; Kolb, 2009a, 2009b and references therein). Plant species composition can also be correlated with the overall community structure of soil bacteria (Berg and Smalla, 2009). Methylotroph plant interactions can be very specific (for example, root nodule formation) or can be less stringent when soil methylotrophs modulate growth of their host plants (for example, Sy et al., 2001; Fedorov et al., 2011). In the case of methane-oxidizing methylotrophs, the presence of trees correlates with certain methanotrophic genotypes (Kolb, 2009b; Degelmann et al., 2010). Thus, a growing body of evidence supports the hypothesis that community structure of soil methylotrophs in temperate grassland and forest soils is influenced by plant diversity and soil pH.
Diversity of soil methylotrophs
Alphaproteobacterial methylotrophs dominated the soil communities. Knowledge of the in situ diversity of methanol-utilizing methylotrophs in soils is limited despite the fact that many soil-derived methanol-utilizing methylotrophs have been isolated or detected by molecular analyses by employing high and likely not in situ-relevant methanol concentrations (Radajewski et al., 2002; Lueders et al., 2004; Lidstrom, 2006; Kolb, 2009a). Methanol-utilizing Burkholderiaceae, Beijerinkiaceae and Bradyrhizobiacea occur in aerated soils (Radajewski et al., 2002). These taxa as well as a larger diversity of known and unknown soil methylotrophs were detected in the current study. The gene survey revealed a large number of novel genotypes within the Proteobacteria. Actinobacteria and Flavobacteria were frequently isolated by cultivation with methanol-supplemented mineral media (Supplementary Figure S5); that these taxa were not revealed by the gene survey suggests that targeting methylotrophs by cultivation may identify additional taxa in the overall diversity of methanol-utilizing microorganisms.
The dominance of Methylobacterium in phyllosphere communities (Holland et al., 2002; Delmotte et al., 2009; Wellner et al., 2011; Mizuno et al., 2012) is coincident with the finding that Methylobacterium-affiliated genotypes were predominant in the mxaF data sets of investigated soils. Methylobacterium-affiliated mxaF genoytpes were statistically indicative for grassland vegetation. The phyllosphere is inoculated by methylotroph community of soil during plant growth (Romanovskaya et al., 2001), which is supported by the finding that Methylobacterium species were important soil methylotrophs. Various mxaF and mch genotypes affiliated with genotypes of Rhizobiales (Figure 4; Supplementary Figure S3) that are not known to be capable of methylotrophy (Lidstrom, 2006; Kolb, 2009a). Similar genotypes were also detected in a forest soil by methanol-dependent stable isotope probing (Radajewski et al., 2002).
Some fae genotypes were affiliated with aerobic members of Planctomycetales and Leptothrix (Supplementary Figure S4). However, it is not likely that these taxa are capable of aerobic methylotrophy (Lidstrom, 2006; Chistoserdova et al., 2009; Kolb, 2009a). Thus, the fae analysis likely detected some non-methylotrophs and biased the statistical analyses. mch and fae genotype analyses also suggest that hitherto unkown methylotrophic taxa were present.
A limitation of gene markers currently available for the detection of methylotrophs is that they are biased towards Gram-negative methylotrophs. mch and fae only occur in those methylotrophs that employ the H4MPT-dependent formaldehyde oxidation pathway (Kalyuzhnaya et al., 2004; Kalyuzhnaya and Chistoserdova, 2005; Chistoserdova, 2011). mxaFI genes have been found in some Actinobacteria and methylotrophy are widely distributed in this taxon (Anesti et al., 2004; Hung et al., 2011). Biochemically and evolutionary distinct methanol oxidoreductases of Gram positives cannot be detected with currently available primers (for example, those of B. methanolicus; Hektor et al., 2002; Naerdal et al., 2011). Thus, future efforts are needed to improve the assessment of Gram-positive methylotrophs.
Conclusions
This study demonstrated that atmospheric methanol concentrations can be dissimilated by methylotrophs in temperate soils. It remains speculative if methylotrophic methanol consumption in soil can explain the large gap between theoretical production and observed emission into the atmosphere. Alphaproteobacteria, especially those that are represented by mch genotypes GSC 1 and 2, may be capable of utilization of atmospheric methanol. In addition to Methylobacterium, other taxa may be important methanol consumers in temperate ecosystems. The correlation of vegetation type and the community structure of methylotrophs suggests that interactions between plants and methylotrophs are important to methylotroph diversity in aerated soil. The flux of methanol in temperate grassland and forest ecosystems is likely linked to the broad diversity of soil methylotrophs, and further resolution of ecological niches and physiological properties of such methylotrophs is warranted.
Acknowledgments
We thank the managers of the three Exploratories, Swen Renner, Sonja Gockel, Andreas Hemp, Martin Gorke and Simone Pfeiffer for their work in maintaining the plot and project infrastructure, and Markus Fischer, the late Elisabeth Kalko, Eduard Linsenmair, Dominik Hessenmöller, Jens Nieschulze, Daniel Prati, Ingo Schöning, François Buscot, Ernst-Detlef Schulze and Wolfgang Weisser for their role in setting up the Biodiversity Exploratories project. The study has been funded by the DFG Priority Program 1374 ‘Infrastructure-Biodiversity-Exploratories' (DFG Dr 310/5-1), and the University of Bayreuth. Field work permits were issued by the responsible state environmental offices of Baden-Württemberg, Thüringen and Brandenburg (according to § 72 BbgNatSchG). We thank A Mustroph (Plant Physiology; University of Bayreuth) in advising how to grow sterile A. thaliana plants, R Daniel and coworkers (Göttingen Genomics Laboratory) for pyrosequencing of mxaF, mch and fae PCR products, and Ingo Schöning for providing total carbon and total nitrogen content data, and ammonium concentrations. The New Zealand Marsden Fund provided financial support for CKL (UOW1003).
Footnotes
Supplementary Information accompanies the paper on The ISME Journal website (http://www.nature.com/ismej)
Supplementary Material
References
- Alef K. Methodenhandbuch Bodenmikrobiologie: Aktivitäten, Biomasse, Differenzierung. Ecomed Verlagsgesellschaft: Landsberg/Lech; 1991. [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Anesti V, Vohra J, Goonetilleka S, McDonald IR, Straubler B, Stackebrandt E, et al. Molecular detection and isolation of facultatively methylotrophic bacteria, including Methylobacterium podarium sp nov., from the human foot microflora. Environ Microbiol. 2004;6:820–830. doi: 10.1111/j.1462-2920.2004.00623.x. [DOI] [PubMed] [Google Scholar]
- Arfman N, Watling EM, Clement W, Vanoosterwijk RJ, Devries GE, Harder W, et al. Methanol metabolism in thermotolerant methylotrophic Bacillus strains involving a novel catabolic NAD-dependent methanol dehydrogenase as a key enzyme. Arch Microbiol. 1989;152:280–288. doi: 10.1007/BF00409664. [DOI] [PubMed] [Google Scholar]
- Atlas RM. Handbook of Microbiological Media. CRC Media: FL, USA; 1993. [Google Scholar]
- Berg G, Smalla K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol Ecol. 2009;68:1–13. doi: 10.1111/j.1574-6941.2009.00654.x. [DOI] [PubMed] [Google Scholar]
- Boden R, Thomas E, Savani P, Kelly DP, Wood AP. Novel methylotrophic bacteria isolated from the River Thames (London, UK) Environ Microbiol. 2008;10:3225–3236. doi: 10.1111/j.1462-2920.2008.01711.x. [DOI] [PubMed] [Google Scholar]
- Caparose JGKJ, Stombaugh J, Bittinger K, Bushman FD. QIIME allows analysis of high-throughput community sequencing data. Nat Met. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistoserdova L. Modularity of methylotrophy, revisited. Environ Microbiol Rep. 2011;13:2603–2622. doi: 10.1111/j.1462-2920.2011.02464.x. [DOI] [PubMed] [Google Scholar]
- Chistoserdova L, Kalyuzhnaya MG, Lidstrom ME. The expanding world of methylotrophic metabolism. Annu Rev Microbiol. 2009;63:477–499. doi: 10.1146/annurev.micro.091208.073600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad R. The global methane cycle: recent advances in understanding the microbial processes involved. Environ Microbiol Rep. 2009;1:285–292. doi: 10.1111/j.1758-2229.2009.00038.x. [DOI] [PubMed] [Google Scholar]
- Dedysh SN, Panikov NS, Liesack W, Grosskopf R, Zhou JZ, Tiedje JM. Isolation of acidophilic methane-oxidizing bacteria from northern peat wetlands. Science. 1998;282:281–284. doi: 10.1126/science.282.5387.281. [DOI] [PubMed] [Google Scholar]
- Degelmann DM, Borken W, Drake HL, Kolb S. Different atmospheric methane-oxidizing communities in European Beech and Norway Spruce soils. Appl Environ Microbiol. 2010;76:3228–3235. doi: 10.1128/AEM.02730-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmotte N, Knief C, Chaffron S, Innerebner G, Roschitzki B, Schlapbach R, et al. Community proteogenomics reveals insights into the physiology of phyllosphere bacteria. Proc Natl Acad Sci USA. 2009;106:16428–16433. doi: 10.1073/pnas.0905240106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JL, Beale R, Nightingale PD. Microbial methanol uptake in northeast Atlantic waters. ISME J. 2011;5:704–716. doi: 10.1038/ismej.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly MI, Dagley S. Production of methanol from aromatic-acids by Pseudomonas putida. J Bacteriol. 1980;142:916–924. doi: 10.1128/jb.142.3.916-924.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufrene ML, Legendre P. Species assemblage and indicator species: the need for a flexible asymmetrical approach. Ecol Monogr. 1997;67:345–366. [Google Scholar]
- Dunfield PF.2007The soil methane sinkIn: Reay DS, Hewitt CN, Smith KA, Grace J, (eds)Green House Gas Sinks CABI: Wallingford, UK; 152–170. [Google Scholar]
- Fall R, Benson AA. Leaf methanol—the simplest natural product from plants. Trend Plant Sci. 1996;1:296–301. [Google Scholar]
- Fedorov DN, Doronina NV, Trotsenko YA. Phytosymbiosis of aerobic methylobacteria: new facts and views. Microbiology. 2011;80:443–454. [PubMed] [Google Scholar]
- Fierer N, Jackson RB. The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci USA. 2006;103:626–631. doi: 10.1073/pnas.0507535103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Bossdorf O, Gockel S, Hansel F, Hemp A, Hessenmoller D, et al. Implementing large-scale and long-term functional biodiversity research: the biodiversity exploratories. Basic Appl Ecol. 2010;11:473–485. [Google Scholar]
- Galbally IE, Kirstine W. The production of methanol by flowering plants and the global cycle of methanol. J Atm Chem. 2002;43:195–229. [Google Scholar]
- Giovannoni SJ, Hayakawa DH, Tripp HJ, Stingl U, Givan SA, Cho JC, et al. The small genome of an abundant coastal ocean methylotroph. Environ Microbiol. 2008;10:1771–1782. doi: 10.1111/j.1462-2920.2008.01598.x. [DOI] [PubMed] [Google Scholar]
- Halsey KH, Carter AE, Giovannoni SJ. Synergistic metabolism of a broad range of C1 compounds in the marine methylotrophic bacterium HTCC2181. Environ Microbiol. 2012;14:630–640. doi: 10.1111/j.1462-2920.2011.02605.x. [DOI] [PubMed] [Google Scholar]
- Hektor HJ, Kloosterman H, Dijkhuizen L. Identification of a magnesium-dependent NAD(P)(H)-binding domain in the nicotinoprotein methanol dehydrogenase from Bacillus methanolicus. J Biol Chem. 2002;277:46966–46973. doi: 10.1074/jbc.M207547200. [DOI] [PubMed] [Google Scholar]
- Holland MA, Long RLG, Polacco JC. Methylobacterium spp.: Phylloplane Bacteria Involved in Cross-Talk with the Plant Host? APS Press: St. Paul, MN, USA; 2002. [Google Scholar]
- Hung WL, Wade WG, Boden R, Kelly DP, Wood AP. Facultative methylotrophs from the human oral cavity and methylotrophy in strains of Gordonia, Leifsonia, and Microbacterium. Arch Microbiol. 2011;193:407–417. doi: 10.1007/s00203-011-0689-6. [DOI] [PubMed] [Google Scholar]
- Jacob DJ, Field BD, Li QB, Blake DR, de Gouw J, Warneke C, et al. 2005Global budget of methanol: constraints from atmospheric observations J Geophys Res-Atmos 110doi: 10.1029/2004JD005172 [DOI] [Google Scholar]
- Kalyuzhnaya MG, Bowerman S, Nercessian O, Lidstrom ME, Chistoserdova L. Highly divergent genes for methanopterin-linked C1 transfer reactions in Lake Washington, assessed via metagenomic analysis and mRNA detection. Appl Environ Microbiol. 2005;71:8846–8854. doi: 10.1128/AEM.71.12.8846-8854.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyuzhnaya MG, Chistoserdova L. Community-level analysis: genes encoding methanopterin-dependent enzymes. Environ Microbiol. 2005;397:443–454. doi: 10.1016/S0076-6879(05)97027-4. [DOI] [PubMed] [Google Scholar]
- Kalyuzhnaya MG, Hristova KR, Lidstrom ME, Chistoserdova L. Characterization of a novel methanol dehydrogenase in representatives of Burkholderiales: implications for environmental detection of methylotrophy and evidence for convergent evolution. J Bacteriol. 2008;190:3817–3823. doi: 10.1128/JB.00180-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyuzhnaya MG, Lidstrom ME, Chistoserdova L. Utility of environmental primers targeting ancient enzymes: methylotroph detection in Lake Washington. Microb Ecol. 2004;48:463–472. doi: 10.1007/s00248-004-0212-6. [DOI] [PubMed] [Google Scholar]
- Karl T, Harley P, Guenther A, Rasmussen R, Baker B, Jardine K, et al. The bi-directional exchange of oxygenated VOCs between a loblolly pine (Pinus taeda) plantation and the atmosphere. Atmos Chem Phys. 2005;5:3015–3031. [Google Scholar]
- Knief C, Ramette A, Frances L, Alonso-Blanco C, Vorholt JA. Site and plant species are important determinants of the Methylobacterium community composition in the plant phyllosphere. ISME J. 2010;4:719–728. doi: 10.1038/ismej.2010.9. [DOI] [PubMed] [Google Scholar]
- Kolb S. Aerobic methanol-oxidizing bacteria in soil. FEMS Microbiol Lett. 2009a;300:1–10. doi: 10.1111/j.1574-6968.2009.01681.x. [DOI] [PubMed] [Google Scholar]
- Kolb S. The quest for atmospheric methane oxidizers in forest soils. Environ Microbiol Rep. 2009b;1:336–346. doi: 10.1111/j.1758-2229.2009.00047.x. [DOI] [PubMed] [Google Scholar]
- Lidstrom ME.2006Aerobic methylotrophic bacteriaIn: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E, (eds)The Prokaryotes3rd edn.618–634.
- Loew O. Ueber einen Bacillus, welcher Ameisensäure und Formaldehyd assimiliren kann. Centralblatt für Bakteriologie, Parasitenkunde, Infektionskrankheiten und Hygiene, Abteilung II. 1892;12:462–465. [Google Scholar]
- Lueders T, Wagner B, Claus P, Friedrich MW. Stable isotope probing of rRNA and DNA reveals a dynamic methylotroph community and trophic interactions with fungi and protozoa in oxic rice field soil. Environ Microbiol. 2004;6:60–72. doi: 10.1046/j.1462-2920.2003.00535.x. [DOI] [PubMed] [Google Scholar]
- McDonald IR, Bodrossy L, Chen Y, Murrell JC. Molecular ecology techniques for the study of aerobic methanotrophs. Appl Environ Microbiol. 2008;74:1305–1315. doi: 10.1128/AEM.02233-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald IR, Murrell JC. The methanol dehydrogenase structural gene mxaF and its use as a functional gene probe for methanotrophs and methylotrophs. Appl Environ Microbiol. 1997;63:3218–3224. doi: 10.1128/aem.63.8.3218-3224.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald IR, Radajewski S, Murrell JC. Stable isotope probing of nucleic acids in methanotrophs and methylotrophs: a review. Org Geochem. 2005;36:779–787. [Google Scholar]
- Mizuno M, Yurimoto H, Yoshida N, Iguchi H, Sakai Y. Distribution of pink-pigmented facultative methylotrophs on leaves of vegetables. Biosci Biotechnol Biochem. 2012;76:578–580. doi: 10.1271/bbb.110737. [DOI] [PubMed] [Google Scholar]
- Naerdal I, Netzer R, Ellingsen TE, Brautaset T. Analysis and manipulation of aspartate pathway genes for L-lysine overproduction from methanol by Bacillus methanolicus. Appl Environ Microbiol. 2011;77:6020–6026. doi: 10.1128/AEM.05093-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebel ME, Wild S, Holzhauser M, Huttenberger L, Reitzig R, Sperber M, et al. Jaguc—a software package for environmental diversity analyses. J Bioinformat Comput Biol. 2011;9:749–773. doi: 10.1142/s0219720011005781. [DOI] [PubMed] [Google Scholar]
- Neufeld JD, Schäfer H, Cox MJ, Boden R, McDonald IR, Murrell JC. Stable-isotope probing implicates Methylophaga spp and novel Gammaproteobacteria in marine methanol and methylamine metabolism. ISME J. 2007;1:480–491. doi: 10.1038/ismej.2007.65. [DOI] [PubMed] [Google Scholar]
- Nojiri M, Hira D, Yamaguchi K, Okajima T, Tanizawa K, Suzuki S. Crystal structures of cytochrome cL and methanol dehydrogenase from Hyphomicrobium denitrificans: structural and mechanistic insights into interactions between the two proteins. Biochemistry. 2006;45:3481–3492. doi: 10.1021/bi051877j. [DOI] [PubMed] [Google Scholar]
- Nunn DN, Day D, Anthony C. The 2nd subunit of methanol dehydrogenase of Methylobacterium extorquens AM1. Biochem J. 1989;260:857–862. doi: 10.1042/bj2600857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ. Removing noise from pyrosequenced amplicons. BMC Bioinform. 2011;12:38. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radajewski S, Webster G, Reay DS, Morris SA, Ineson P, Nedwell DB, et al. Identification of active methylotroph populations in an acidic forest soil by stable-isotope probing. Microbiology. 2002;148:2331–2342. doi: 10.1099/00221287-148-8-2331. [DOI] [PubMed] [Google Scholar]
- Ricke P, Kolb S, Braker G. Application of a newly developed ARB software-integrated tool for in silico terminal restriction fragment length polymorphism analysis reveals the dominance of a novel pmoA cluster in a forest soil. Appl Environ Microbiol. 2005;71:1671–1673. doi: 10.1128/AEM.71.3.1671-1673.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanovskaya VA, Stolyar SM, Malashenko YR, Dodatko TN. The ways of plant colonization by Methylobacterium strains and properties of these bacteria. Microbiology. 2001;70:263–269. [PubMed] [Google Scholar]
- Royston JP. Shapiro-Wilk normality test and P-value. Appl Stat. 1995;44 [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method—a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Schade GW, Solomon SJ, Dellwik E, Pilegaard K, Ladstätter-Weissenmayer A. Methanol and other VOC fluxes from a Danish beech forest during late springtime. Biogeochemistry. 2011;106:337–355. [Google Scholar]
- Schellenberger S, Kolb S, Drake HL. Metabolic responses of novel cellulolytic and saccharolytic agricultural soil Bacteria to oxygen. Environ Microbiol. 2010;12:845–861. doi: 10.1111/j.1462-2920.2009.02128.x. [DOI] [PubMed] [Google Scholar]
- Schink B, Zeikus JG. Microbial methanol formation—a major end product of pectin metabolism. Curr Microbiol. 1980;4:387–389. [Google Scholar]
- Schmidt S, Christen P, Kiefer P, Vorholt JA. Functional investigation of methanol dehydrogenase-like protein XoxF in Methylobacterium extorquens AM1. Microbiol-SGM. 2010;156:2575–2586. doi: 10.1099/mic.0.038570-0. [DOI] [PubMed] [Google Scholar]
- Segel IH. Enzyme Kinetics Behaviour and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. Wiley Classics Library: NY, USA; 1993. [Google Scholar]
- Skovran E, Palmer AD, Rountree AM, Good NM, Lidstrom ME. XoxF is required for expression of methanol dehydrogenase in Methylobacterium extorquens AM1. J Bacteriol. 2011;193:6032–6038. doi: 10.1128/JB.05367-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stralis-Pavese N, Sessitsch A, Weilharter A, Reichenauer T, Riesing J, Csontos J, et al. Optimization of diagnostic microarray for application in analysing landfill methanotroph communities under different plant covers. Environ Microbiol. 2004;6:347–363. doi: 10.1111/j.1462-2920.2004.00582.x. [DOI] [PubMed] [Google Scholar]
- Sy A, Giraud E, Jourand P, Garcia N, Willems A, de Lajudie P, et al. Methylotrophic Methylobacterium bacteria nodulate and fix nitrogen in symbiosis with legumes. J Bacteriol. 2001;183:214–220. doi: 10.1128/JB.183.1.214-220.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter Braak CJF, Smilauer P. CANOCO Reference Manual and CanoDraw for Windows User () s Guide: Software for Canonical Community Ordination (version 4.5) Micocomputer Power: Ithaca, NY, USA; 2002. [Google Scholar]
- Trotsenko YA, Murrell JC. Metabolic aspects of aerobic obligate methanotrophy. Adv Appl Microbiol. 2008;63:183–229. doi: 10.1016/S0065-2164(07)00005-6. [DOI] [PubMed] [Google Scholar]
- Vorholt JA, Chistoserdova L, Stolyar SM, Thauer RK, Lidstrom ME. Distribution of tetrahydromethanopterin-dependent enzymes in methylotrophic bacteria and phylogeny of methenyl tetrahydromethanopterin cyclohydrolases. J Bacteriol. 1999;181:5750–5757. doi: 10.1128/jb.181.18.5750-5757.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warneke C, Karl T, Judmaier H, Hansel A, Jordan A, Lindinger W, et al. Acetone, methanol, and other partially oxidized volatile organic emissions from dead plant matter by abiological processes: significance for atmospheric HOx chemistry. Glob Biogeochem Cyc. 1999;13:9–17. [Google Scholar]
- Watanabe K, Hino S, Onodera K, Kajie SI, Takahashi N. Diversity in kinetics of bacterial phenol- oxygenating activity. J Ferment Bioeng Vol. 1996;81:560–563. [Google Scholar]
- Wellner S, Lodders N, Kämpfer P. Diversity and biogeography of selected phyllosphere bacteria with special emphasis on Methylobacterium spp. Sys Appl Microbiol. 2011;34:621–630. doi: 10.1016/j.syapm.2011.08.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.