

Abstract
The human gut is colonized by a complex microbiota with multiple benefits. Although the surface-attached, mucosal microbiota has a unique composition and potential to influence human health, it remains difficult to study in vivo. Therefore, we performed an in-depth microbial characterization (human intestinal tract chip (HITChip)) of a recently developed dynamic in vitro gut model, which simulates both luminal and mucosal gut microbes (mucosal-simulator of human intestinal microbial ecosystem (M-SHIME)). Inter-individual differences among human subjects were confirmed and microbial patterns unique for each individual were preserved in vitro. Furthermore, in correspondence with in vivo studies, Bacteroidetes and Proteobacteria were enriched in the luminal content while Firmicutes rather colonized the mucin layer, with Clostridium cluster XIVa accounting for almost 60% of the mucin-adhered microbiota. Of the many acetate and/or lactate-converting butyrate producers within this cluster, Roseburia intestinalis and Eubacterium rectale most specifically colonized mucins. These 16S rRNA gene-based results were confirmed at a functional level as butyryl-CoA:acetate-CoA transferase gene sequences belonged to different species in the luminal as opposed to the mucin-adhered microbiota, with Roseburia species governing the mucosal butyrate production. Correspondingly, the simulated mucosal environment induced a shift from acetate towards butyrate. As not only inter-individual differences were preserved but also because compared with conventional models, washout of relevant mucin-adhered microbes was avoided, simulating the mucosal gut microbiota represents a breakthrough in modeling and mechanistically studying the human intestinal microbiome in health and disease. Finally, as mucosal butyrate producers produce butyrate close to the epithelium, they may enhance butyrate bioavailability, which could be useful in treating diseases, such as inflammatory bowel disease.
Keywords: bowel, Lachnospiraceae, Ruminococcaceae, Anaerostipes caccae, Faecalibacterium prausnitzii, mucus
Introduction
The human colon is colonized by a complex microbiota, mostly (>90%) consisting of Bacteroidetes and Firmicutes (Eckburg et al., 2005; Claesson et al., 2011; Walker et al., 2011). Beneficial contributions of this microbiota to human health include the breakdown of otherwise indigestible food compounds (Koropatkin et al., 2012) and regulation of host metabolism (Backhed et al., 2005). Furthermore, the importance of the intestinal mcrobiota follows from the number of diseases that have been correlated with a dysbiosed microbial composition, such as inflammatory bowel diseases (IBDs; Willing et al., 2009; Walker et al., 2011) or obesity (Turnbaugh et al., 2006). A novel focus in gut microbiology is to not only study microbes in the intestinal content but also those that colonize the mucus layer (Swidsinski et al., 2008; Van den Abbeele et al., 2011c; Belzer and de Vos, 2012). The rationale is that mucosal microbes can interact more closely with the epithelium than their luminal counterparts, which may be crucial for achieving immunomodulatory effects. Moreover, by locally excreting antimicrobials or competing with pathogens, mucosal microbes more effectively limit pathogen translocation. Besides host immune effectors (Lievin-Le Moal and Servin, 2006), microbial properties, such as mucus adhesion (Roos and Jonsson, 2002) or the ability to degrade host-derived glycans (Derrien et al., 2004), also impact the distinct surface-attached microbial composition, generally characterized by an enrichment of Firmicutes (especially Clostridium cluster XIVa) over Bacteroidetes (Eckburg et al., 2005; Frank et al., 2007; Hill et al., 2009; Shen et al., 2010; Willing et al., 2010; Hong et al., 2011; Nava et al., 2011). Moreover, the mucus layer is persistently colonized by all types of hydrogenotrophs: methanogenic archaea, sulphate-reducing bacteria and acetogenic bacteria (Nava et al., 2012).
Despite their physiological relevance, human studies are often limited to faecal samples, which do not provide information on this mucosal microbiota. For this purpose, biopsies need to be collected, but given the invasive sampling procedures, these are often only taken at the end of an experiment, preventing dynamic monitoring or detailed mechanistic studies (Zoetendal et al., 2002). By contrast, in vitro studies have the advantage that they are well-suited to perform mechanistic research. However, the current models generally only provide short-term information and often ignore the interaction between luminal and mucosal microbes. Recently, a long-term dynamic in vitro model was developed, which accounts for both the luminal and mucosal microbiota (mucosal-simulator of human intestinal microbial ecosystem (M-SHIME); Van den Abbeele et al., 2011b). The simulated mucosal environment consisted of carrier material coated with commercial pig gastric mucins. When focussing on Lactobacilli, it was found that, in correspondence with in vivo data, this in vitro mucosal environment was colonized by specific Lactobacillus species (L. mucosae and L. rhamnosus GG). However, the overall microbial community shifts remain to be elucidated.
Therefore, the aim of this study was to perform an in-depth analysis of the in vitro mucosal M-SHIME microbiota using the human intestinal tract chip (HITChip), a recently developed and widely used phylogenetic micro-array (Jonkers et al., 2009; Rajilic-Stojanovic et al., 2009; Van den Abbeele et al., 2011a). Each experiment was conducted with samples from different human subjects to account for the inter-individual variability. The aims were to determine whether the inter-individual differences among human subjects can be preserved in the model, to determine the distinct composition of the mucosal microbiota compared with the luminal one and to assess the main metabolic activities of mucosal microbes. By comparing these results with recent in vivo data and results obtained with conventional in vitro models without surface-attached bacteria, the novel M-SHIME model was validated.
Materials and methods
Chemicals and preparation of growth media
Unless stated otherwise, chemicals were obtained from Sigma (Bornem, Belgium). The experiments were conducted using sugar-depleted nutritional medium containing (in g l−1) yeast extract (3.0), peptone (1.0), commercial pig gastric mucin (4.0) and cystein (0.5). Pancreatic juice contained (in g l−1) NaHCO3 (12.5), bile salts (6.0; Difco, Bierbeek, Belgium) and pancreatin (0.9). Mucin agar was prepared by boiling dH2O containing 5% commercial pig gastric mucin and 1% agar. The pH was adjusted to 6.8 with 10 ℳ NaOH.
Dynamic in vitro gut model for the luminal and mucosal microbiota (M-SHIME)
Although the conventional luminal (L)-SHIME (registered trademark, Ghent University-Prodigest, Ghent, Belgium) only simulates luminal microbes (Van den Abbeele et al., 2010), the M-SHIME also contains a niche for surface-attached microbes (Figure 1a; Van den Abbeele et al., 2011b). Briefly, microcosms (K1-carrier, AnoxKaldnes AB, Lund, Sweden) were submerged in mucin agar and combined in a polyethylene netting (Zakkencentrale, Rotterdam, The Netherlands). At the start of each experiment, 500 ml nutritional medium and 100 mucin-covered microcosms were added to each colon unit, followed by inoculation with 40 ml of a 1:5 dilution of fresh stools of a healthy human volunteer (Possemiers et al., 2004). After an initial incubation of 18 h, 140 ml nutritional medium and 60 ml pancreatic juice were supplied to each colon compartment three times per day. The M-SHIME was at 37 °C and kept anaerobic by flushing twice per day for 15 min with N2.
Figure 1.

(a) Schematic representation of the main/first experiment: five colon compartments with a simulated mucosal environment (M-SHIME) were each inoculated with a different human faecal sample (donor A, B, C, D or E). (b) Dendrogram for the total bacterial DGGE profiles of the luminal and mucosal microbiota of these five M-SHIMEs, 3 days after inoculation with faecal samples of five human donors. The average inter-individual similarity between different donors (±s.e.m.) is shown for each type of sample: inoculum, lumen and mucin layer.
Based on earlier studies, several factors were modified to optimally study mucin-adhered microbes. (i) Long-term M-SHIME studies have demonstrated that regular replacement of microcosms, results in wash-out of mucin-adhered microbes, probably due to limited cross-contamination between old and new microcosms (unpublished results). Therefore, we performed short-term experiments (3 days), which avoids renewal of microcosms. (ii) Because such short-term studies are confronted by a strong build-up of gasses and acids due to fermentation of sugar-rich nutritional medium, we used sugar-depleted medium, which allowed a more moderate carbohydrate fermentation upon inoculation. (iii) Thirdly, instead of operating two SHIME units which each consist of an ascending, transverse and descending colon (Van den Abbeele et al., 2010), we used the six colon vessels as separate units allowing to test more conditions. (iv) Finally, the pH of each unit was maintained between 6.15 and 6.4 (∼transverse colon).
Experimental design
In the first/main experiment, the overall mucin-adhered microbiota composition was characterized. Therefore, five M-SHIME units were inoculated with stools of different human subjects: donor A (male, 26 years), B (male, 33 years), C (male, 32 years), D (male, 24 years) and E (female, 32 years; Figure 1a). Samples for short-chain fatty acid (SCFA) analysis were collected after 18, 42 and 68 h. Samples of the inocula together with mucosal and luminal samples of the final time point (68 h) were snap-frozen in liquid nitrogen for microbial characterization (denaturing gradient gel electrophoresis (DGGE), HITChip) and a clone library for the butyryl-CoA:acetate-CoA transferase gene.
To validate results of this main experiment, two additional experiments were performed. In the second experiment, the contribution of mucosal microbes to the overall microbial activity and composition was evaluated. Therefore, three M-SHIMEs (with mucosal environment) and three L-SHIMEs (without mucosal environment) were simultaneously inoculated with faecal inocula of donors A, B and D (Supplementary Figure S1A). In the third experiment, the impact of the presence of mucins in the mucosal environment on mucosal microbial activity and composition was evaluated. Therefore, microcosms were coated with mucin-containing or mucin-free agar. In this way, two colon units with a mucosal environment containing mucins (M(+)-SHME), two colon units with a mucosal environment free of mucins (M(−)-SHME) and two units without a mucosal environment (L-SHIME) were inoculated with faecal samples of donors A and D (Supplementary Figure S2A). Samples for SCFA analysis were collected after 18, 42 and 68 h. Samples of the inocula together with mucosal and luminal samples of the final time point were subjected to DGGE.
Microbial community (DGGE and HITChip) and metabolic activity analysis (SCFA)
The most prominent shifts within the microbiota were monitored via DGGE. After DNA extraction (Boon et al., 2003) and PCR (Muyzer et al., 1993), gels were run using an Ingeny PhorU apparatus (Ingeny International, Goes, The Netherlands). Pearson correlation and UPGMA (Unweighted Pair Group Method using Arithmetic Mean) clustering were used to calculate dendrograms using BioNumerics v5.10 (Applied Maths, Sint-Martens-Latem, Belgium).
The HITChip was used for deep characterization of the microbiota during the first/main experiment (Rajilic-Stojanovic et al., 2009). The array contains duplicated sets of 4800 16S rRNA oligonucleotide probes targeting 1140 intestinal phylotypes (<98% identity) and 131 genus-like groups (<90% identity). Quantification of these groups has been validated with fluorescence in situ hybridization, quantitative PCR and 454 pyrosequencing (Rajilic-Stojanovic et al., 2009; Claesson et al., 2011; van den Bogert et al., 2011). Briefly, 16S rRNA genes were amplified, in vitro transcribed, labelled with Cy3/Cy5 and hybridized to the microarray, washed and scanned. Spot intensities were extracted using Agilent Feature Extraction software v9.5 and normalised using R-based scripts (http://www.r-project.org/). Analysis were performed in a custom-designed relational database, which runs under MySQL database management system (http://www.mysql.com/) using a series of custom R-scripts as described previously (Rajilic-Stojanovic et al., 2009).
Acetate, propionate, butyrate, valerate, caproate and branched SCFA (isobutyrate, isovalerate and isocaproate) were measured as described previously (De Weirdt et al., 2010).
Clone library for butyryl-CoA:acetate CoA-transferase gene
DNA was amplified with primers BCoATscrF/BCoATscrR (2.5 μℳ) (Louis and Flint, 2007). PCR products were cut from a gel, purified (QIAquick Gel Extraction Kit, QIAGEN, Antwerp, Belgium) and cloned with a TOPO TA Cloning Kit with pCR2.1-TOPO Vector (Invitrogen, Carlsbad, CA, USA). Clones were amplified with primers M13F/M13R and sequenced (AGOWA, Berlin, Germany). Sequences (∼480 bp) were manually inspected and compared with databases at the NCBI website (http://blast.ncbi.nlm.nih.gov/blast.cgi). Further, they were aligned using the ClustalW algorithm with the ClustalW 1.6 weight matrix and a neighbour-joining tree with 2500 bootstrap iterations and the Kimura 2-parameter substitution model was constructed (MEGA5), using the 4-hydroxybutyrate CoA-transferase sequence from A.caccae L1-92 as outgroup. Sequences with <98% similarity to the 32 operational taxonomic units (OTUs) described by Louis et al. (2010) were considered as novel OTUs. Sequences have been submitted to the European Nucleotide Archive under accession numbers HE984158–HE984296.
Statistics
All data were analyzed using SPSS16 (SPSS Inc., Chicago, IL, USA). Normality and homogeneity of variances were studied with a Kolmogorov–Smirnov and Levene test, respectively. If so, an analysis of variance with Bonferroni test was performed to investigate intergroup differences, otherwise a Kruskal–Wallis with Mann–Whitney test was applied.
A singular value decomposition was performed to identify which factors most strongly determined the microbial differences measured with the HITChip. These factors consisted of three locations (faecal inocula, lumen, mucin layer) and five sources (donor A/B/C/D/E). The initial matrix X consisted of 131 (=s) rows representing abundances of 131 genus-like groups and 15 (=t) columns representing the 15 samples, being organized as follows: five inocula, five luminal M- and five mucosal M-SHIME samples, each time for donor A–E. Each column was converted to ranks and this rank-transformed matrix, XR, was decomposed in 15 terms each consisting of a singular value (λj), vector uj (length s) and vector vjt (length t): 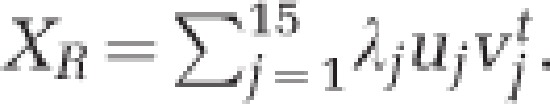 The 15 terms are ordered so that λ1⩾λ2⩾…⩾λ15. Hence, the first term is the most important in the approximation of XR, the second term the second most important, and so on. The significance of the contribution of location and source was verified by applying analysis of variance F-tests. Corresponding p-values (pL and pS) were computed based on a Monte Carlo approximation of the exact permutation null distributions.
The 15 terms are ordered so that λ1⩾λ2⩾…⩾λ15. Hence, the first term is the most important in the approximation of XR, the second term the second most important, and so on. The significance of the contribution of location and source was verified by applying analysis of variance F-tests. Corresponding p-values (pL and pS) were computed based on a Monte Carlo approximation of the exact permutation null distributions.
To assess correlation of microbial groups detected by HITChip with human inocula, mucin layer or lumen, redundancy analysis was used as implemented in Canoco for Windows 4.5. Average signal intensities for 131 genus-like groups were used as microbial data, and diagrams were plotted using the CanoDraw for Windows utility. The Monte Carlo Permutation Procedure was used to assess statistical significance of the variation in data sets in relation to sample origin.
Results
In vitro mucosal environment enhances butyrate production
SCFA were analyzed in the luminal content of M-SHIME units of five different human subjects (Figure 1a). As observed from the low standard errors, SCFA levels were similar among individuals (Table 1). Sixty-eight hours after inoculation, the acetate/propionate/butyrate ratio of the M-SHIME was 65/15/20, while for conventional models without surface-attached bacteria such as the L-SHIME, ratios of around 68/25/6 have been reported (Van den Abbeele et al., 2010). Simulating the mucosal environment thus seems to enhance butyrate levels.
Table 1. Average (±s.e.m.) absolute (mℳ) SCFA levels in the luminal content of M-SHIME units during the first 3 days (18 h, 42 h and 68 h) after inoculation with faecal samples of five different human donors (n=5).
| Time after inoculation | 0 h | 18 h | 42 h | 68 h |
|---|---|---|---|---|
| Absolute values (mℳ) | ||||
| Acetate | 7.7±1.5 | 36.5±1.4 | 31.1±1.1 | 31.3±0.5 |
| Propionate | 2.5±0.4 | 8.3±0.7 | 7.0±0.4 | 7.1±0.2 |
| Butyrate | 2.9±1.1 | 7.8±1.7 | 11.3±1.1 | 9.9±0.7 |
| Valerate | 0.6±0.1 | 1.9±1.1 | 2.4±0.9 | 1.6±0.6 |
| Caproate | 0.5±0.2 | 0.3±0.3 | 0.5±0.5 | 0.3±0.3 |
| Branched SCFA | 1.2±0.1 | 3.0±0.7 | 3.7±0.9 | 2.5±0.7 |
| Total SCFA | 15.4±3.2 | 57.9±3.7 | 56.0±3.2 | 52.9±1.7 |
| Proportional values (mol%) | ||||
| Acetate | 50.8±1.2 | 63.6±2.2 | 56.0±2.4 | 59.5±2.1 |
| Propionate | 16.6±0.8 | 14.3±0.8 | 12.6±0.4 | 13.5±0.5 |
| Butyrate | 17.1±2.4 | 13.3±2.6 | 20.1±1.2 | 18.8±1.2 |
| Valerate | 4.0±0.4 | 3.0±1.7 | 4.1±1.4 | 2.9±1.1 |
| Caproate | 2.7±0.8 | 0.4±0.4 | 0.8±0.8 | 0.6±0.6 |
| Branched SCFA | 8.8±1.7 | 5.4±1.4 | 6.5±1.3 | 4.7±1.1 |
Abbreviations: M-SHIME, mucosal-simulator of human intestinal microbial ecosystem; SCFA, short-chain fatty acid.
Compared with the conventional L-SHIME, the novel M-SHIME not only differed in the incorporation of a simulated mucosal environment but also in a shorter incubation time, sugar-depleted nutritional medium and a single-stage fermentation. To eliminate influence of the latter and straightforwardly evaluate the impact of a mucosal environment, three M-SHIME and three L-SHIME units were simultaneously inoculated with faeces of three donors (Supplementary Figure S1A). It followed that from the second day after inoculation onwards, the mucin layer induced a proportional shift from acetate (−6.3%) to butyrate (+3.9%) (Supplementary Table S1). In a third experiment, the importance of the presence of mucins in the mucosal environment was evaluated (Supplementary Figure S2A). It followed that the shift from acetate to butyrate was more profound when the microcosms were coated with mucin-containing as opposed to mucin-free agar (only −2.6% acetate and +0.9% butyrate; Supplementary Table S2).
Distinct microbial composition of human faeces, lumen and mucin layer (DGGE)
To observe the main microbial shifts upon inoculation of the M-SHIME, DGGE was performed on 16S rRNA amplicons of the total bacterial community (Figure 1b). The profiles were grouped as three separate clusters according to sample origin: human inocula, lumen and mucin layer. Based on the Pearson correlation coefficients, it followed that the mucin-adhered microbiota was very different from the luminal microbiota (only 19±5% similar). Furthermore, although similar shifts were observed for the different human donors (Supplementary Figure S3), there was significant inter-individual variability among their faecal inocula (62±3% similar), which continued to exist in the luminal (64±5% similar) and mucin-adhered (44±7% similar) M-SHIME microbiota.
Also the DGGE profiles of the second (Supplementary Figure S1B) and third (Supplementary Figure S2B) experiment revealed clustering according to sample origin (inocula, lumen and mucin layer) rather than to the donor (Supplementary Figures S4 and S5). Both experiments confirmed the distinct nature of the luminal and mucosal communities (only 14±6% similar), together with the inter-individual variability among human inocula and resulting luminal and mucosal microbiota. Both experiments also showed that the luminal microbiota of the L- and M-SHIME are highly similar (90±4% similar), suggesting that the mucosal environment does not majorly affect microbial composition in the luminal content (Supplementary Figures S1B and S2B). From the third experiment, it followed that the presence of mucins in the mucosal environment is crucial for the mucosal microbiota development. Although the microbiota that colonized the mucin-free microcosms was different from the luminal microbiota (63±1% similar), the microbiota that colonized the mucin-containing microcosms was much more distinct from the luminal microbiota (only 25±5% similar; Supplementary Figure S2B).
In vitro gut model preserves the specific inter-individual differences (HITChip)
In order to decipher the factors that influence microbial colonization in the M-SHIME, a singular value decomposition was applied on the high-resolution phylogenetic HITChip data of the five human inocula and five luminal and mucosal M-SHIME samples. The first two terms in the decomposition explained 39.6% and 28.4% of the variation in the microbial dataset, caused by the distinct microbial composition of the human inocula, lumen and mucin layer (Table 2 and Supplementary Figure S6). The next four terms also explained a significant part of the microbial changes (in total, 21.6%). Interestingly, these changes were independent of the location but attributed to the inter-individual differences among human subjects, indicating that the individual-specific microbial patterns are preserved in both the luminal and mucosal M-SHIME environment.
Table 2. The singular value decomposition of the matrix containing abundances of 131 genus-like groups (as determined with the HITChip) of the human inocula, luminal and mucosal M-SHIME samples (n=5) resulted in six terms, which significantly explained the variation in the data set caused by either the location (=human inoculum, lumen M-SHIME or mucin layer M-SHIME) or source of the sample (=human donor A, B, C, D and E).
| Term | P-value location | P-value source | % Variation explained | Cause of variation |
|---|---|---|---|---|
| 1 | 0.0002 | 0.9492 | 39.6 | Inoculum↔M-SHIME lumen/mucin layer |
| 2 | 0.0000 | 0.9931 | 28.4 | M-SHIME lumen↔M-SHIME mucin layer |
| 3 | 0.8400 | 0.0008 | 8.4 | Interindividual variability human inocula |
| 4 | 0.9858 | 0.0205 | 5.6 | Interindividual variability human inocula |
| 5 | 0.6273 | 0.0371 | 4.5 | Interindividual variability human inocula |
| 6 | 0.9637 | 0.0242 | 3.1 | Interindividual variability human inocula |
Abbreviations: HITChip, human intestinal tract chip; M-SHIME, mucosal-simulator of human intestinal microbial ecosystem.
Detailed characterization of luminal and mucosal M-SHIME microbiota (HITChip)
Not only did the HITChip data confirm the distinct microbial composition of the human inocula as opposed to the lumen and mucin layer of the M-SHIME (Supplementary Figure S7), it also provided a detailed phylogenetic characterization (Figure 2 and Supplementary Table S3). The Firmicutes phylum was significantly enriched in the mucin layer (94% of the total community) as opposed to the lumen (64%), attributed to increased levels of Bacilli (3%) and bacteria belonging to the Clostridium clusters I (10%), IV (19%), XI (2%) and especially Clostridium cluster XIVa (59%). By contrast, the mucin layer was virtually devoid of Bacteroidetes (4%) and Proteobacteria (1%), which rather colonized the luminal content (25% and 10%, respectively).
Figure 2.

The average abundance (%) of higher taxonomic groups (∼ phylum level) based on the HITChip analysis of the human faecal inocula and the resulting luminal and mucosal environment of the M-SHIME, 3 days after inoculation (n=5). An asterisks indicates a significant difference in the abundance between the lumen and mucin layer of the M-SHIME. The averages and s.e. can be found in Supplementary Table S3.
Butyrate-producing bacteria from Clostridum cluster XIVa colonize the mucin layer (HITChip)
A redundancy analysis of the HITChip data at the bacterial group level (∼131 genus-like groups) confirmed the distinct microbiota of inocula, lumen and mucin layer (P=0.02; Figure 3a). Furthermore, it specified the microbial changes as 68 genus-like groups correlated with specific locations (Figure 3b and Supplementary Table S4). Of these 68 groups, 15 specifically colonized the mucin layer (all belonging to the Firmicutes), while 29 specifically correlated to the lumen (21 belonging to the Proteobacteria or Bacteroidetes). Finally, 24 bacterial groups were equally abundant between lumen and mucin layer but differed in abundance compared with the inocula.
Figure 3.

(a) Redundancy analysis at genus-like group-level based on the HITChip data of the inoculum, lumen and mucin layer of M-SHIMEs inoculated with faecal samples of five human subjects (n=5; P=0.02). (b) Dendrogram of the same samples based on the HITChip data for 131 genus-like groups, including a heat map which shows the inter-individual distribution of microbial groups that differed among inocula, lumen and/or mucin layer.
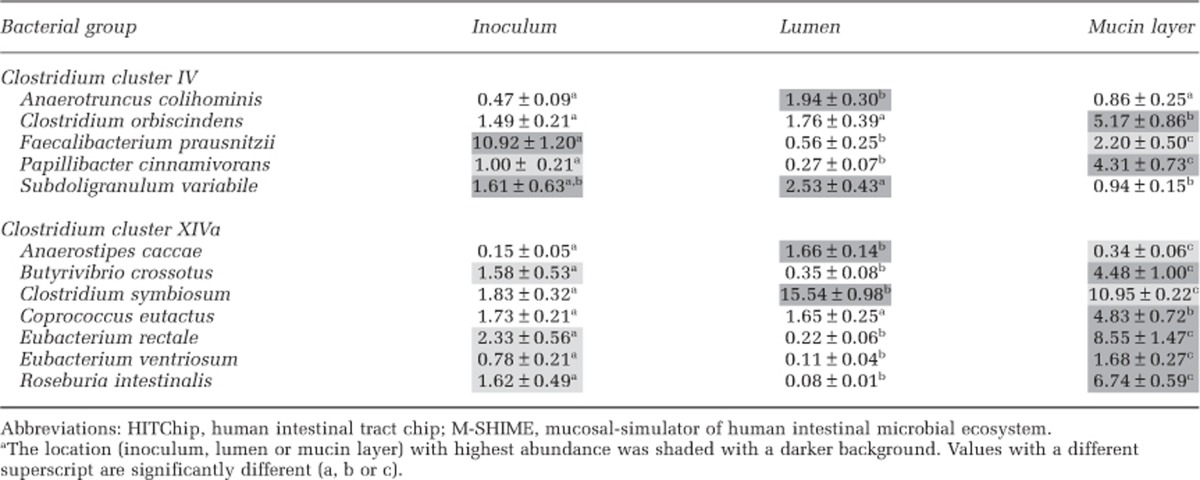
Several butyrate producers belonging to Clostridium clusters IV and XIVa, but particularly the latter, specifically colonized the mucosal M-SHIME environment (Table 3). They include (expressed as ratio of mucin-adhered/lumen) Roseburia intestinalis (84.3), Eubacterium rectale (38.9), Papillibacter cinnamivorans (16.0), Eubacterium ventriosum (15.3), Butyrivibrio crossotus (12.8), Faecalibacterium prausnitzii (3.9), Clostridium orbiscidens (2.9) and Coprococcus eutactus (2.9). By contrast, few butyrate-producing groups specifically colonized the lumen (expressed as ratio of lumen/mucin-adhered): A. caccae (4.9), Subdoligranulum variabile (2.7), Anaerotruncus colihominis (2.3) and Clostridium symbiosum (1.4).
Table 3. The abundance (%) of butyrate-producing genus-like groups belonging to Clostridium clusters IV and XIVa (<Firmicutes) that were significantly different between the luminal or mucosal M-SHIME environment, as determined with the HITChip (n=5).
Clone library for butyryl-CoA:acetate CoA-transferase gene confirms specific mucosal butyrate producers
Overall, 45% of the sequences of the butyryl-CoA:acetate CoA-transferase gene belonged to uncultured bacteria, with 16% of the sequences belonging to novel butyrate-producing species (OTU33–39). The 16S rRNA gene-derived results (HITChip) were confirmed at a functional level as the genes responsible for butyrate production were carried by different microbes in the mucin-adhered as opposed to the luminal microbiota (Figure 4). Although Roseburia species and an unknown species (OTU39) dominated the mucin-adhered microbiota (R. intestinalis, R. hominis and R. inulinvorans), the luminal butyrate producers mainly consisted of strain M62-1 and related unknown species (OTU24, 25 and 38). Five other uncultured butyrate producers, (distantly) related to F. prausnitzii, were also detected in the luminal microbiota (OTU33–37).
Figure 4.

Phylogenetic tree and the amount of butyryl-CoA:acetate-CoA transferase gene sequences (∼480 bp) identified in the luminal content and mucin layer of the M-SHIME 68 h after inoculation with human faecal samples of different donors (A, B and D; first experiment). Sequences with at least 98% sequence identity were grouped together under the same OTU number of which one sequence was chosen to build the phylogenetic tree. OTU numbers >32 indicate novel butyrate-producing species identified during the current study.
Simulating a mucosal environment avoids wash-out of surface-attached bacteria
The previously published HITChip data of a conventional L-SHIME without mucosal environment (Van den Abbeele et al., 2010), showed that this setup results in an enrichment of Bacteroidetes and Proteobacteria and wash-out of Firmicutes (Supplementary Figure S8). As the mucosal environment of the M-SHIME was specifically colonized by Firmicutes members, especially belonging to the Clostridium cluster XIVa, wash-out of these surface-attached, mucosal Firmicutes is avoided (Figure 2).
This was further confirmed at the bacterial group level, where HITChip analysis of the M-SHIME microbiota also allowed characterizing bacterial groups that rather colonized the mucin layer or lumen. Although bacteria characterized as luminal in the M-SHIME (Figure 5: area A) were maintained at high levels in the conventional L-SHIME, those characterized as mucosal in the M-SHIME (Figure 5: area B) disappeared from the conventional L-SHIME. Simulating a mucosal environment is thus crucial for many Firmicutes species in order to persist in an in vitro gut model.
Figure 5.

Average abundance (%) of genus-like groups as determined with the HITChip in a conventional L-SHIME without simulation of the mucosal environment, as reported previously (Van den Abbeele et al., 2010) (n=4), as a function of the mucin colonization of these microbes in the novel M-SHIME (n=5). Mucin colonization represents the preference of a bacterial group to colonize the in vitro mucosal environment and is expressed as the logarithm of the abundance in the mucosal environment versus the abundance in the luminal content of the M-SHIME. A value <0 indicates that the microbial group rather colonizes the lumen (area A), a value >0 indicates that this group rather colonizes the simulated mucosal environment (area B).
Discussion
We recently developed the M-SHIME, an in vitro gut model, which not only provides a niche for luminal but also for surface-attached, mucosal microbes (Van den Abbeele et al., 2011b). In this study, we confirm the inter-individual differences in microbiota composition among different human stool and mucosal samples (Zoetendal et al., 1998, 2002; Eckburg et al., 2005) and demonstrate that these microbial patterns unique to each individual are preserved in the luminal and mucosal in vitro microbiota. A microarray with high phylogenetic resolution revealed the unique composition of the in vitro mucosal microbiota in great detail.
The distinct microbial composition of the mucin layer, lumen and faeces was the most determining factor within the HITChip dataset accounting for 68% of the microbial differences, which was higher than the variation caused by the inter-individual differences among human subjects (21.6% Table 2). The Firmicutes phylum largely dominated the mucosal M-SHIME microbiota (>90%), with Clostridium cluster XIVa species accounting for 60% of the total mucosal microbiota (Figure 2). Concordant with the fact that this cluster includes many acetate- and/or lactate-converting butyrate producers, the simulation of a mucosal environment induced a shift from acetate towards butyrate, independent of the inoculum. Despite the fact that, in living animals, the continuous desquamation of mucus into the luminal content obscures the distinction between luminal and mucosal microbes, recent in vivo studies also show an enrichment of Firmicutes (especially Clostridium cluster XIVa>Lachnospiraceae family), over Bacteroidetes in biopsies compared with luminal or faecal samples, both in rodents (Hill et al., 2009; Nava et al., 2011) and humans (Eckburg et al., 2005; Frank et al., 2007; Shen et al., 2010; Wang et al., 2010; Willing et al., 2010; Hong et al., 2011). This in vivo enrichment of Firmicutes in mucus, although sometimes less strong as during our in vitro study, suggests that similar forces may drive the mucosal microbiota composition in vivo and in vitro, likely to include selection of specific groups that adhere to mucins (Leitch et al., 2007) or insoluble substrates in general (Walker et al., 2008). Furthermore, as opposed to the luminal content where the pH was maintained constant, local accumulation of acids in mucus may cause a lower pH, selecting for Firmicutes over Bacteroidetes and Proteobacteria (Duncan et al., 2009). In addition, mucins may also serve as a growth substrate for butyrate-producing Firmicutes, possibly via cross-feeding with mucin-degrading microbes that deliver partial breakdown products, acetate and/or lactate (Belzer and de Vos 2012), similar as reported for fructo-oligosaccharides (Belenguer et al., 2006; Falony et al., 2006). Independent of the underlying reason, the mucus colonization by specific butyrate-producing Firmicutes (especially Clostridium cluster XIVa) species provides novel insight in the ecology of these abundant human gut colonizers.
Although bacteria belonging to Clostridium cluster XIVa, and to a lesser extent Clostridium cluster IV, were enriched in the in vitro mucosal environment, some butyrate-producing bacterial groups belonging to these clusters had higher abundances in the luminal content (Table 3). The most specific mucosal colonizers, as identified with the HITChip, included R. intestinalis and E. rectale, while A. caccae rather colonized the lumen. In contrast to A. caccae that is non-motile, R. intestinalis and E. rectale possess flagella, which may allow for penetration into the mucus layer (Louis and Flint, 2009). Moreover, another difference is that the mucosal butyrate producers solely require acetate and no lactate for butyrate production (Louis and Flint, 2009), indicating that cross-feeding with other microbes might relate to the specific colonization of mucus.
The selective mucus colonization by specific butyrate-producing Firmicutes members may be interconnected with processes that affect the intestinal mucus as illustrated by two examples: consumption of prebiotic compounds and IBD. Firstly, prebiotics often enhance mucus production (Ten Bruggencate et al., 2004; Van den Abbeele et al., 2011a) and, according to our findings, this may stimulate the release of mucosal butyrate producers towards the lumen. This hypothesis is supported by a study in which humanized rats were treated with long-chain arabinoxylans or inulin (Van den Abbeele et al., 2011a). Long-chain arabinoxylans increased caecal mucin levels threefold, led to higher butyrate levels and higher abundances of butyrate producers, identified as mucosal butyrate producers during this study (E. rectale and R. intestinalis). Remarkably, inulin increased caecal mucin levels by sixfold and corresponded with even higher butyrate levels and higher abundances of the same mucosal butyrate producers. Besides the selective degradation of prebiotics by specific butyrate producers (Duncan et al., 2002; Schwiertz et al., 2002), stimulating mucus secretion may be a mechanism by which prebiotics increase butyrate levels. A second example is IBD, a disease in which the mucus layer becomes thinner and more discontinuous (Strugala et al., 2008). Recent in vivo (Swidsinski et al., 2005; Sokol et al., 2008; Walker et al., 2011) and in vitro (Vermeiren et al., 2012) studies revealed that this disease correlates with lower levels of mucosal butyrate producers, including Roseburia and Faecalibacterium, indicating that a damaged mucus layer may lower the ecological fitness of specific butyrate producers. In vivo validation of the preference of butyrate producers to reside in mucus together with better understanding of factors that drive these processes may allow to develop novel therapies. As mucosal butyrate producers release butyrate close to the epithelium, they may enhance butyrate bioavailability for the host, which may be particularly useful for IBD patients where transport of butyrate to colonocytes is impaired (Thibault et al., 2010).
The mucosal environment of the M-SHIME prevents wash-out of microbes that disappeared from conventional in vitro models (Figure 5). The enrichment of Firmicutes over Bacteroidetes and Protebacteria in the mucosal M-SHIME environment and the fact that this contributes to higher butyrate levels in the in vitro model further indicate that the M-SHIME is a significantly improved simulation of the intestinal microbiota compared with conventional in vitro models. The latter models are indeed confronted with lower abundances of Firmicutes, especially butyrate-producing species belonging to the Clostridium cluster XIVa and IV (Rajilić-Stojanović et al., 2010; Van den Abbeele et al., 2010), whereas Bacteroidetes and Proteobacteria are very abundant (Allison et al., 1989; Macfarlane et al., 1998; Mäkeläinen et al., 2009; Rajilić-Stojanović et al., 2010; Van den Abbeele et al., 2010), typically resulting in lower butyrate levels (Allison et al., 1989; Van den Abbeele et al., 2010). By avoiding washout of mucosal microbes, the M-SHIME allows mechanistic studies that not only focus on planktonic but also on surface-attached microbes, increasing the relevance of in vitro research.
Although meaningful results have been obtained with the M-SHIME, several adaptations may further enhance its value. Firstly, in order to obtain a large surface area, one is currently limited to use commercially available pig gastric mucins. These mucins have lower molar mass averages and contain more impurities than freshly prepared gastric mucins (Jumel et al., 1996). Moreover, the composition of these gastric mucin (mainly MUC1, MUC5AC and MUC6) differs from colonic mucins (mainly MUC2) and changes in gastrointestinal diseases (Reis et al., 1999; Corfield et al., 2000). In order to improve the mucosal simulation in the M-SHIME, more relevant mucins may be used in future experiments. Ideally, human colonic mucins would be coated on the carrier materials using an agar-independent method. Next, Akkermansia muciniphila, the only known Verrucomicrobia representative that is a marker for mucin degradation (Belzer and de Vos, 2012), did not attain high densities in the current setup. Recent studies demonstrated that Akkermansia degrades mucins in (distal) colon regions characterized by depleted nutrient levels and long residence times (Van den Abbeele et al., 2010, 2011a). As colonization of mucus by different mucolytic species may be crucial with respect to human health (Png et al., 2010), the M-SHIME may be improved by adding a distal colon region. Further, for performing long-term experiments, a setup needs to be designed in which mucin-covered microcosms can be regularly renewed without disturbing the stability of the mucosal and luminal microbiota, particularly without opening the vessel and exposing the micro-organisms to oxygen. Finally, microbes are confronted with an oxygen gradient in vivo as oxygen is continuously released from the blood towards the mucus layer. Accounting for this gradient would also add to the relevance of the M-SHIME.
In conclusion, the recently developed M-SHIME simulates not only planktonic but also surface-attached gut microbes. In correspondence with in vivo studies, the simulated mucosal environment was specifically colonized by Firmicutes members, including many butyrate producers of the Clostridium cluster XIVa. Compared with conventional in vitro models that do not account for surface-attached microbes, wash-out of the latter species was avoided resulting in a more in vivo-like microbial composition and activity, allowing for more relevant mechanistic in vitro studies to unravel the importance of mucosal gut microbes in health and disease. The M-SHIME may thus be an excellent tool to isolate novel mucosal microbes and to study factors that drive health-promoting or disease-causing microbes to reside in the mucosal microbiota from where they can closely interact with the host epithelium. The potential to isolate novel microbes was supported by the finding that 45% of the butyrate producers in the M-SHIME belonged to uncultured species. In this context, discovery of mucosal butyrate producers may lead to a novel therapy for diseases such as IBD, which are characterized by an impaired butyrate transport to the colonocytes. Further, as the microbial patterns unique to the five tested human subjects were preserved in the in vitro model, the M-SHIME may be used in future studies using larger cohorts to focus on these inter-individual differences.
Acknowledgments
PVdA is a Postdoctoral Fellow from FWO-Vlaanderen (Research Foundation of Flanders, Belgium) and RDW is a PhD student funded by the Special Research Fund (BOF) of Ghent University. This work was partially supported by a GOA (BOF12/GOA/008) project from Ghent University, an SBO project (100016) from the Agency for Innovation by Science and Technology (IWT) and an unrestricted Spinoza Award of the Netherlands Foundation for Scientific Research (NWO) and an Advanced Grant of the European Research Council (to WMdV). Finally, we thank Tim Lacoere (Ghent University) for technical assistance and Petra Louis (University of Aberdeen) for her excellent advise with respect to the construction of the butyryl-CoA:acetate-CoA transferase gene clone library and subsequent analysis.
Footnotes
Supplementary Information accompanies the paper on The ISME Journal website (http://www.nature.com/ismej)
Supplementary Material
References
- Allison C, McFarlan C, Macfarlane GT. Studies on mixed populations of human intestinal bacteria grown in single-stage and multistage continuous culture systems. Appl Environ Microbiol. 1989;55:672–678. doi: 10.1128/aem.55.3.672-678.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- Belenguer A, Duncan SH, Calder AG, Holtrop G, Louis P, Lobley GE, et al. Two routes of metabolic cross-feeding between Bifidobacterium adolescentis and butyrate-producing anaerobes from the human gut. Appl Environ Microbiol. 2006;72:3593–3599. doi: 10.1128/AEM.72.5.3593-3599.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belzer C, de Vos WM. Microbes inside—from diversity to function: the case of Akkermansia. ISME J. 2012;6 (8:1449–1458. doi: 10.1038/ismej.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon N, Top EM, Verstraete W, Siciliano SD. Bioaugmentation as a tool to protect the structure and function of an activated-sludge microbial community against a 3-chloroaniline shock load. Appl Environ Microbiol. 2003;69:1511–1520. doi: 10.1128/AEM.69.3.1511-1520.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesson MJ, Cusack Sn, O'Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci USA. 2011;108:4586–4591. doi: 10.1073/pnas.1000097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corfield AP, Myerscough N, Longman R, Sylvester P, Arul S, Pignatelli M. Mucins and mucosal protection in the gastrointestinal tract: new prospects for mucins in the pathology of gastrointestinal disease. Gut. 2000;47:589–594. doi: 10.1136/gut.47.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Weirdt R, Possemiers S, Vermeulen G, Moerdijk-Poortvliet TCW, Boschker HTS, Verstraete W, et al. Human faecal microbiota display variable patterns of glycerol metabolism. FEMS Microbiol Ecol. 2010;74:601–611. doi: 10.1111/j.1574-6941.2010.00974.x. [DOI] [PubMed] [Google Scholar]
- Derrien M, Vaughan EE, Plugge CM, de Vos WM. Akkermansia muciniphila gen. nov., sp nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol. 2004;54:1469–1476. doi: 10.1099/ijs.0.02873-0. [DOI] [PubMed] [Google Scholar]
- Duncan SH, Hold GL, Barcenilla A, Stewart CS, Flint HJ. Roseburia intestinalis sp. nov., a novel saccharolytic, butyrate-producing bacterium from human faeces. Int J Syst Evol Microbiol. 2002;52:1615–1620. doi: 10.1099/00207713-52-5-1615. [DOI] [PubMed] [Google Scholar]
- Duncan SH, Louis P, Thomson JM, Flint HJ. The role of pH in determining the species composition of the human colonic microbiota. Environ Microbiol. 2009;11:2112–2122. doi: 10.1111/j.1462-2920.2009.01931.x. [DOI] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falony G, Vlachou A, Verbrugghe K, Vuyst LD. Cross-feeding between Bifidobacterium longum BB536 and acetate-converting, butyrate-producing colon bacteria during growth on oligofructose. Appl Environ Microbiol. 2006;72:7835–7841. doi: 10.1128/AEM.01296-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DN, Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DA, Hoffmann C, Abt MC, Du Y, Kobuley D, Kirn TJ, et al. Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol. 2009;3:148–158. doi: 10.1038/mi.2009.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong P-Y, Croix JA, Greenberg E, Gaskins HR, Mackie RI. Pyrosequencing-based analysis of the mucosal microbiota in healthy individuals reveals ubiquitous bacterial groups and micro-heterogeneity. PLoS ONE. 2011;6:e25042. doi: 10.1371/journal.pone.0025042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers D, Zoetendal EG, Romberg M, Hesselink M, de Vos W, Masclee A, et al. Phylogenetic fingerprinting of the faecal microbiota in IBD-patients using the human intestinal tract (HIT) chip approach: a Twin Study. Gastroenterol. 2009;136:A21–A21. [Google Scholar]
- Jumel K, Fiebrig I, Harding SE. Rapid size distribution and purity analysis of gastric mucus glycoproteins by size exclusion chromatography multi-angle laser light scattering. Int J Biol Macromol. 1996;18:133–139. doi: 10.1016/0141-8130(95)01071-8. [DOI] [PubMed] [Google Scholar]
- Koropatkin NM, Cameron EA, Martens EC. How glycan metabolism shapes the human gut microbiota. Nat Rev Microbiol. 2012;10:323–335. doi: 10.1038/nrmicro2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch ECM, Walker AW, Duncan SH, Holtrop G, Flint HJ. Selective colonization of insoluble substrates by human faecal bacteria. Environ Microbiol. 2007;9:667–679. doi: 10.1111/j.1462-2920.2006.01186.x. [DOI] [PubMed] [Google Scholar]
- Lievin-Le Moal V, Servin AL. The front line of enteric host defense against unwelcome iIntrusion of harmful microorganisms: mucins, antimicrobial peptides, and microbiota. Clin Microbiol Rev. 2006;19:315–337. doi: 10.1128/CMR.19.2.315-337.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis P, Flint HJ. Development of a semiquantitative degenerate real-time PCR-based assay for estimation of numbers of butyryl-coenzyme A (CoA) CoA transferase genes in complex bacterial samples. Appl Environ Microbiol. 2007;73:2009–2012. doi: 10.1128/AEM.02561-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. 2009;294:1–8. doi: 10.1111/j.1574-6968.2009.01514.x. [DOI] [PubMed] [Google Scholar]
- Louis P, Young P, Holtrop G, Flint HJ. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA:acetate CoA-transferase gene. Environ Microbiol. 2010;12:304–314. doi: 10.1111/j.1462-2920.2009.02066.x. [DOI] [PubMed] [Google Scholar]
- Macfarlane GT, Macfarlane S, Gibson GR. Validation of a three-stage compound continuous culture system for investigating the effect of retention time on the ecology and metabolism of bacteria in the human colon. Microb Ecol. 1998;35:180–187. doi: 10.1007/s002489900072. [DOI] [PubMed] [Google Scholar]
- Mäkeläinen H, Hasselwander O, Rautonen N, Ouwehand AC. Panose, a new prebiotic candidate. Lett Appl Microbiol. 2009;49:666–672. doi: 10.1111/j.1472-765X.2009.02698.x. [DOI] [PubMed] [Google Scholar]
- Muyzer G, Dewaal EC, Uitterlinden AG. Profiling of complex microbial-populations by denaturing gradient gel-electrophoresis of polymerase chain reaction-amplified genes-coding for 16S ribosomal-RNA. Appl Environ Microbiol. 1993;59:695–700. doi: 10.1128/aem.59.3.695-700.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava GM, Friedrichsen HJ, Stappenbeck TS. Spatial organization of intestinal microbiota in the mouse ascending colon. ISME J. 2011;5:627–638. doi: 10.1038/ismej.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava GM, Carbonero F, Croix JA, Greenberg E, Gaskins HR. Abundance and diversity of mucosa-associated hydrogenotrophic microbes in the healthy human colon. ISME J. 2012;6:57–70. doi: 10.1038/ismej.2011.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Png CW, Linden SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, et al. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol. 2010;105:2420–2428. doi: 10.1038/ajg.2010.281. [DOI] [PubMed] [Google Scholar]
- Possemiers S, Verthe K, Uyttendaele S, Verstraete W. PCR-DGGE-based quantification of stability of the microbial community in a simulator of the human intestinal microbial ecosystem. FEMS Microbiol Ecol. 2004;49:495–507. doi: 10.1016/j.femsec.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Rajilic-Stojanovic M, Heilig H, Molenaar D, Kajander K, Surakka A, Smidt H, et al. Development and application of the human intestinal tract chip, a phylogenetic microarray: analysis of universally conserved phylotypes in the abundant microbiota of young and elderly adults. Environ Microbiol. 2009;11:1736–1751. doi: 10.1111/j.1462-2920.2009.01900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajilić-Stojanović M, Maathuis A, HGHJ Heilig, Venema K, de Vos WM, Smidt H. Evaluating the microbial diversity of an in vitro model of the human large intestine by phylogenetic microarray analysis. Microbiology. 2010;156:3270–3281. doi: 10.1099/mic.0.042044-0. [DOI] [PubMed] [Google Scholar]
- Reis CA, David L, Correa P, Carneiro F, Cd Bolós, Garcia E, et al. Intestinal metaplasia of human stomach displays distinct patterns of mucin (MUC1, MUC2, MUC5AC, and MUC6) expression. Cancer Res. 1999;59:1003–1007. [PubMed] [Google Scholar]
- Roos S, Jonsson H. A high-molecular-mass cell-surface protein from Lactobacillus reuteri 1063 adheres to mucus components. Microbiology. 2002;148:433–442. doi: 10.1099/00221287-148-2-433. [DOI] [PubMed] [Google Scholar]
- Schwiertz A, Hold GL, Duncan SH, Gruhl B, Collins MD, Lawson PA, et al. Anaerostipes caccae gen. nov., sp. nov., a new saccharolytic, acetate-utilising, butyrate-producing bacterium from human faeces. Syst Appl Microbiol. 2002;25:46–51. doi: 10.1078/0723-2020-00096. [DOI] [PubMed] [Google Scholar]
- Shen XJ, Rawls JF, Randall TA, Burcall L, Mpande C, Jenkins N, et al. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes. 2010;1:138–147. doi: 10.4161/gmic.1.3.12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux J-J, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strugala V, Dettmar PW, Pearson JP. Thickness and continuity of the adherent colonic mucus barrier in active and quiescent ulcerative colitis and Crohn's disease. Int J Clin Pract. 2008;62:762–769. doi: 10.1111/j.1742-1241.2007.01665.x. [DOI] [PubMed] [Google Scholar]
- Swidsinski A, Weber J, Loening-Baucke V, Hale LP, Lochs H. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol. 2005;43:3380–3389. doi: 10.1128/JCM.43.7.3380-3389.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swidsinski A, Loening-Baucke V, Verstraelen H, Osowska S, Doerffel Y. Biostructure of fecal microbiota in healthy subjects and patients with chronic idiopathic diarrhea. Gastroenterology. 2008;135:568–579. doi: 10.1053/j.gastro.2008.04.017. [DOI] [PubMed] [Google Scholar]
- Ten Bruggencate SJM, Bovee-Oudenhoven IMJ, Lettink-Wissink MLG, Katan MB, Van der Meer R. Dietary fructo-oligosaccharides and inulin decrease resistance of rats to salmonella: protective role of calcium. Gut. 2004;53:530–535. doi: 10.1136/gut.2003.023499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault R, Blachier F, Darcy-Vrillon B, de Coppet P, Bourreille A, Segain J-P. Butyrate utilization by the colonic mucosa in inflammatory bowel diseases: a transport deficiency. Inflamm Bowel Dis. 2010;16:684–695. doi: 10.1002/ibd.21108. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1131. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Van den Abbeele P, Grootaert C, Marzorati M, Possemiers S, Verstraete W, Gérard P, et al. Microbial community development in a dynamic gut model is reproducible, colon-region specific and selects for Bacteroidetes and Clostridium cluster IX. Appl Environ Microbiol. 2010;76:5237–5246. doi: 10.1128/AEM.00759-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Abbeele P, Gerard P, Rabot S, Bruneau A, El Aidy S, Derrien M, et al. Arabinoxylans and inulin differentially modulate the mucosal and luminal gut microbiota and mucin-degradation in humanized rats. Environ Microbiol. 2011a;13:2667–2680. doi: 10.1111/j.1462-2920.2011.02533.x. [DOI] [PubMed] [Google Scholar]
- Van den Abbeele P, Roos S, Eeckhaut V, Marzorati M, Possemiers S, Vanhoecke B, et al. Incorporation of a mucosal environment in a dynamic gut model results in a more representative colonization by lactobacilli. Microb Biotechnol. 2011b;5:106–115. doi: 10.1111/j.1751-7915.2011.00308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Abbeele P, Van de Wiele T, Verstraete W, Possemiers S. The host selects mucosal and luminal associations of coevolved gut microorganisms: a novel concept. FEMS Microbiol Rev. 2011c;35:681–704. doi: 10.1111/j.1574-6976.2011.00270.x. [DOI] [PubMed] [Google Scholar]
- van den Bogert B, de Vos WM, Zoetendal EG, Kleerebezem M. Microarray analysis and barcoded pyrosequencing provide consistent microbial profiles depending on the source of human intestinal samples. Appl Environ Microbiol. 2011;77:2071–2080. doi: 10.1128/AEM.02477-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeiren J, Van den Abbeele P, Laukens D, Vigsnæs LK, De Vos M, Boon N, et al. Decreased colonization of fecal Clostridium coccoides/Eubacterium rectale species from ulcerative colitis patients in an in vitro dynamic gut model with mucin environment. FEMS Microbiol Ecol. 2012;79:685–696. doi: 10.1111/j.1574-6941.2011.01252.x. [DOI] [PubMed] [Google Scholar]
- Walker A, Sanderson J, Churcher C, Parkes G, Hudspith B, Rayment N, et al. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011;11:7. doi: 10.1186/1471-2180-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AW, Duncan SH, Harmsen HJM, Holtrop G, Welling GW, Flint HJ. The species composition of the human intestinal microbiota differs between particle-associated and liquid phase communities. Environ Microbiol. 2008;10:3275–3283. doi: 10.1111/j.1462-2920.2008.01717.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Antonopoulos D, Zhu X, Harrell L, Hanan I, Alverdy J, et al. Laser capture microdissection and metagenomic analysis of intact mucosa-associated microbial communities of human colon. Appl Environ Microbiol. 2010;88:1333–1342. doi: 10.1007/s00253-010-2921-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willing B, Halfvarson J, Dicksved J, Rosenquist M, Järnerot G, Engstrand L, et al. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn's disease. Inflamm Bowel Dis. 2009;15:653–660. doi: 10.1002/ibd.20783. [DOI] [PubMed] [Google Scholar]
- Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, et al. 2010A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes Gastroenterology 1391844–1854.e1841. [DOI] [PubMed] [Google Scholar]
- Zoetendal EG, Akkermans ADL, De Vos WM. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl Environ Microbiol. 1998;64:3854–3859. doi: 10.1128/aem.64.10.3854-3859.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoetendal EG, von Wright A, Vilpponen-Salmela T, Ben-Amor K, Akkermans ADL, de Vos WM. Mucosa-associated bacteria in the human gastrointestinal tract are uniformly distributed along the colon and differ from the community recovered from feces. Appl Environ Microbiol. 2002;68:3401–3407. doi: 10.1128/AEM.68.7.3401-3407.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.