

Summary
Mammalian oocytes are arrested in metaphase of second meiosis (MII) until fertilization. This arrest is enforced by the cytostatic factor (CSF), which maintains the M-phase promoting factor (MPF) in a highly active state. Although the continuous synthesis and degradation of cyclin B to maintain the CSF-mediated MII arrest is well established, it is unknown whether cyclin-dependent kinase 1 (Cdk1) phosphorylations are involved in this arrest in mouse oocytes. Here, we show that a dynamic equilibrium of Cdk1 phosphorylation is required to maintain MII arrest. When the Cdc25A phosphatase is downregulated, mouse oocytes are released from MII arrest and MPF becomes inactivated. This inactivation occurs in the absence of cyclin B degradation and is dependent on Wee1B-mediated phosphorylation of Cdk1. Thus, our data demonstrate that Cdk1 activity is maintained during MII arrest not only by cyclin turnover but also by steady state phosphorylation.
Key words: Oocyte, Meiosis, Cdc25, MPF, CSF
Introduction
Female gametes must complete a specialized meiotic cell cycle to become fertilizable eggs arrested at metaphase of the second meiotic division (MII) (Jones, 2005). During fertilization, the MII arrest is released by a series of Ca2+ oscillations induced by fusion with the spermatozoon. This cell cycle blockade is essential to prevent parthenogenetic activation and the development of an embryo without paternal genome contribution.
The molecular basis of this MII block was first explored in Xenopus oocytes when it was shown by cytoplasm transfer that MII oocytes contain an activity termed cytostatic factor (CSF), which prevents cell cycle progression (Masui and Markert, 1971). This activity functions primarily to stabilize the activated state of M-phase promoting factor (MPF), a complex of cyclin-dependent kinase 1 (Cdk1) and cyclin B (CycB). To date, several pathways involving the anaphase promoting complex/cyclosome (APC/C) inhibitor Emi2 (Schmidt et al., 2005), the Mos/p90rsk kinase cascade (Gross et al., 1999; Sagata et al., 1989), or the cyclin E/Cdk2 and spindle assembly checkpoint (SAC) components (Gabrielli et al., 1993; Grimison et al., 2006) have been implicated in the establishment and maintenance of CSF in Xenopus oocytes. However, not all of the above pathways are active in maintenance of MII block in mouse oocytes. Whereas Mos/Erk and Emi2 are essential (Verlhac et al., 2000; Shoji et al., 2006), neither the downstream target of Erk, p90rsk, nor SAC is necessary or sufficient for the MII suspension (Dumont et al., 2005; Tsurumi et al., 2004). The F-box protein Emi2, ortholog of the Xenopus XErp1/Emi2, exerts its cytostatic activity by preventing Cdc20 binding and activation of APC/C, a multimeric E3 ubiquitin ligase responsible for CycB ubiquitination (Jones, 2005). Stabilization of CycB is considered the primary factor maintaining an active Cdk1 and the MII arrest (Kubiak et al., 1993). As an extension, the prevailing concept for the release from MII arrest at fertilization is that Ca2+ signals induce degradation of Emi2 and activation of the APC/C, thus initiating CycB degradation (Nixon et al., 2002; Madgwick et al., 2006) as well as inactivation of Mos/Erk pathway (Verlhac et al., 2000).
However, MPF is also regulated by the phosphorylation of two highly conserved residues in Cdk1, Thr14 and Tyr15 (Lew and Kornbluth, 1996). These inhibitory phosphorylations are catalyzed by Wee kinases, whereas dephosphorylation of these sites is mediated by the Cdc25 dual specificity phosphatases. The balance between these two activities depends on feedback regulations involving Cdk1 itself, as well the counteracting phosphatase PP2A (Glover, 2012). With the discovery of a regulatory network involving Greatwall kinase-dependent phosphorylation of the PP2A inhibitor endosulphine/Arpp19, the contribution of the Cdk1/Cdc25/Wee1 feedback to cell cycle progression has been clearly delineated (Gharbi-Ayachi et al., 2010; Mochida et al., 2010; Vigneron et al., 2009). Moreover, recent evidence in mouse oocytes indicates that the inhibitory phosphorylation of Cdk1 by Wee1 kinase plays a role in MPF inactivation during M-phase exit (Oh et al., 2011). Likewise, Cdc25C-dependent regulation of Cdk1 in CSF extracts has been documented in Xenopus oocytes (Lorca et al., 2010). Therefore, we explored the possibility that, in addition to CycB dynamics, MPF activity during MII arrest in mouse oocytes requires a balance between the activity of Wee kinases and Cdc25 phosphatases. Here we show that Cdc25A is necessary to stabilize meiotic arrest, and downregulation of this activity by different strategies causes MPF phosphorylation and inactivation and MII exit without CycB degradation.
Results and Discussion
Cdc25A is required for MII arrest in mouse oocytes
To investigate the role of Cdk1 phosphorylation in MII arrest of mouse oocytes, Cdc25 phosphatase expression was downregulated by long double-stranded RNA (dsRNA). The effectiveness and specificity of this strategy was assessed by measuring mRNA and protein levels (supplementary material Fig. S1). When dsRNAs targeting Cdc25A were injected, a large fraction of oocytes were released from the MII arrest, chromosomes decondensed, and nuclear membrane reconstituted around two pronuclei-like structures, morphological changes consistent with transition to an interphase-like state (Fig. 1A,B; supplementary material Movie 1). This phenotype was dependent on the depletion of Cdc25A activity, as it was rescued by the concomitant injection of mRNAs encoding wild-type Cdc25A, but not the catalytically inactive Cdc25A (Fig. 1C). Morphologically, Cdc25A knockdown oocytes overexpressing wild-type Cdc25A showed a well organized spindle (supplementary material Fig. S1D). Therefore, these results provide a first indication that the Cdc25A phosphatase is required to maintain MII arrest in mouse oocytes. Protein levels of Cdc25B and C could not be efficiently downregulated with this strategy, even though mRNAs coding for these phosphatases were undetectable after dsRNA injection (supplementary material Fig. S1). As an alternative approach, we attempted to use morpholino oligonucleotides (MO) targeting Cdc25B and C. However, we were unable to design specific MO because of high GC ratios in potential target sequences of Cdc25B and C. A partial knockdown of Cdc25B activity by antibody injection led to the partial release from MII arrest (supplementary material Fig. S1E) similar to that observed with dsRNA (Fig. 1A), suggesting that Cdc25B is also involved in MII arrest. Cdc25C knockdown was not informative, as little decrease in protein could be detected under these conditions. Available knockout data indicate that Cdc25C is dispensable for meiosis and mitosis (Chen et al., 2001).
Fig. 1.
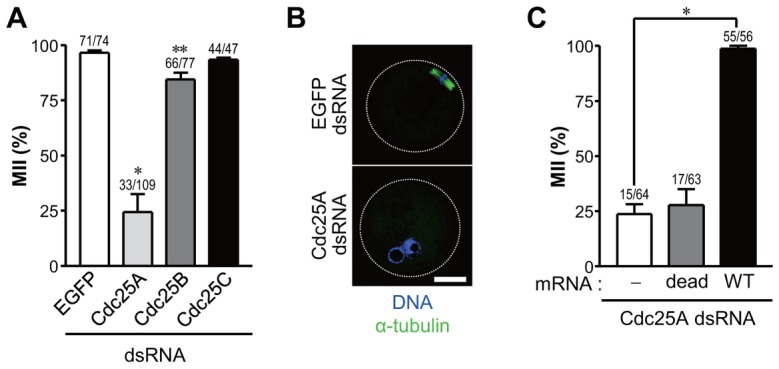
Knockdown of Cdc25 phosphatases. (A) MII oocytes were injected with long double-stranded (ds) RNAs corresponding to Cdc25A, Cdc25B or Cdc25C. EGFP–dsRNA was used as a negative control. MII oocytes were scored after 24 hours. (B) Representative images of Cdc25A knockdown oocytes harvested at 24 hours. The spindle and DNA were stained with anti-α-tubulin and DAPI, respectively. Scale bar: 20 µm. (C) MII oocytes were co-injected with Cdc25A dsRNA and mRNA encoding catalytically inactive or wild-type human Cdc25A and scored as above. Representative images are shown in supplementary material Fig. S1D. Data are means ± s.e.m. of at least three independent experiments. Numbers above the bars indicate the number of oocytes in MII and total oocyte number. *P<0.0005, **P<0.01.
MPF inactivation is mediated by Cdk1 phosphorylation rather than CycB degradation in Cdc25A-depleted oocytes
The above observation opens the possibility that the activation of MPF during the MII suspension depends on the continuous Cdk1 dephosphorylation by Cdc25A (Mailand et al., 2002). To investigate this possibility, we measured MPF and MAPK activities and found that these are significantly decreased in oocytes depleted of Cdc25A (Fig. 2A,B). We next measured CycB levels because MPF inactivation is usually associated with CycB degradation during the release from MII arrest (Jones, 2005). Surprisingly, CycB was not degraded in Cdc25A knockdown oocytes (Fig. 2C,D). Instead, CycB levels increased 12–14 hours after knockdown. Given that CycB levels increase once pronuclei are formed in fertilized eggs (Chang et al., 2004), this finding further indicates that Cdc25A knockdown oocytes released from MII arrest were in interphase. Moreover, this implies that CycB degradation is dispensable for MPF inactivation during exit from M-phase. Indeed, it has been shown that dissociation of CycB from Cdk1, not its proteolytic degradation, may be involved in MPF inactivation during M-phase exit (Chesnel et al., 2006; Nishiyama et al., 2000; Yu et al., 2006). Furthermore, Cdc25A knockdown was sufficient to override the MII arrest induced by MG132, a proteasome inhibitor (Fig. 2E), further indicating that APC/C activation is not required.
Fig. 2.

Decrease in MPF activity in Cdc25A knockdown oocytes. (A,B) Histone H1 and MBP (myelin basic protein) double kinase assay in Cdc25A knockdown oocytes. MII oocytes injected with either EGFP or Cdc25A dsRNA were cultured for 24 hours. MPF and MAPK activities were measured as 32P incorporation by histone H1 and MBP. Freshly ovulated MII oocytes were used as an additional control. (B) The ratio of 32P incorporation into H1 and MBP were quantified densitometrically and reported as means ± s.e.m. *P<0.0005 compared to EGFP dsRNA. (C,D) MII oocytes were co-injected with Cdc25A dsRNA and CycB1-GFP mRNA, and GFP levels measured every 10 minutes up to 24 hours. Representative images are shown in C. Scale bar: 60 µm. Arrowheads indicate pronuclei without GFP signal. (D) Quantification of cyclin B levels. As a control, MII oocytes injected with CycB1-GFP were parthenogenetically activated with strontium and GFP levels measured until pronuclear formation. The timing of pronuclear formation is marked by a dotted line. Data are the means ± s.e.m. using 16 oocytes from three independent experiments. (E,F) MII oocytes injected with Cdc25A dsRNA were incubated with (+) or without (−) either MG132 (E) or nocodazole (F) for 24 hours. The percentage of treated oocytes in MII was then determined. As a control, MII oocytes injected with EGFP dsRNA were activated with strontium. Numbers above the bars indicate the number of MII oocytes/total oocytes. ns, not significant.
CycB degradation is dependent on activity of APC/C in turn regulated by the SAC (Nixon et al., 2002). Therefore, we next asked whether Cdc25A knockdown overcomes the MII arrest induced by activated SAC. In agreement with the absence of CycB degradation after Cdc25A knockdown, SAC activation by nocodazole was not sufficient to prevent the release from MII arrest, indicating that Cdc25A effects are independent on APC/C-mediated proteolysis (Fig. 2F).
Because CycB is not degraded in Cdc25A knockdown oocytes, we investigated whether the decrease in MPF activity is due to a change in Cdk1 phosphorylation. Indeed, Cdk1 phosphorylation in Tyr15, which is barely detectable during MII arrest, becomes clearly evident in Cdc25A knockdown oocytes, suggesting that MPF inactivation is mostly due to accumulation of inhibitory Cdk1 phosphorylation rather than CycB degradation (Fig. 3A). Given that the inhibitory phosphorylation of Cdk1 is mediated by Wee1B kinase in mouse oocytes (Oh et al., 2011), we tested whether Wee1B downregulation rescues the phenotype induced by Cdc25A knockdown. Wee1B was downregulated by MOs as described previously (Oh et al., 2011). Strikingly, a significant number of oocytes depleted in Cdc25A and Wee1B did not exit from MII (Fig. 3B). This finding is in further support of the hypothesis that Cdc25A knockdown leads to MPF inactivation through inhibitory phosphorylations, and when the kinase responsible for this phosphorylation is downregulated, the phenotype is rescued. This scenario is also supported by an additional experiment where a constitutively active Cdk1 (Cdk1-AF) was injected and rescued the release from MII arrest induced by Cdc25A knockdown (Fig. 3C). Thus, Cdk1 phosphorylation rather than CycB degradation is the primary event downstream of Cdc25A depletion.
Fig. 3.
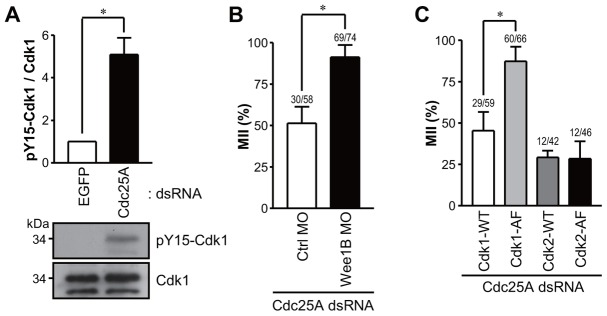
Cdk1 phosphorylation in Cdc25A knockdown oocytes. (A) Immunoblot analysis of phospho-tyrosine Cdk1 in Cdc25A knockdown oocytes. MII oocytes injected with EGFP or Cdc25A dsRNA were incubated for 24 hours and subjected to SDS-PAGE followed by immunoblotting for Cdk1 and Cdk1 phospho-tyrosine 15 (pY15-Cdk1). The pY15-Cdk1/Cdk1 ratio is reported together with an immunoblot representative of three experiments performed. (B) In vitro matured MII oocytes injected with control (Ctrl) or Wee1B MO were injected with Cdc25A dsRNA and incubated for 24 hours. (C) Cdc25A dsRNA was co-injected into MII oocytes with mRNAs encoding Cdk1-WT, Cdk1-AF, or Cdk2-WT and Cdk2-AF. The percentage of treated oocytes in MII was determined for B and C. Data are the means ± s.e.m. of at least three independent experiments. Numbers above the bars indicate the number of MII oocytes/total oocyte number. *P<0.05.
An additional possibility is that Cdk2, rather than Cdk1, is the Cdc25A target in mouse oocytes. Even though Cdk2 in complex with cyclin E is required for CSF arrest in Xenopus oocytes, and both Cdk2 and Cdk1 are substrates of Cdc25A (Gabrielli et al., 1993; Sexl et al., 1999), this is unlikely because Cdk2–cyclin E inhibits APC/C-mediated CycB degradation by regulating SAC activity (Grimison et al., 2006). Indeed, injection of a constitutively active Cdk2 (Cdk2-AF) mutant did not prevent the exit from M phase (Fig. 3C). Furthermore, we were not able to detect cyclin E in MII oocytes, and oocytes expressing Cdk2-AF mutant did not prevent Ca2+-mediated release from MII arrest (supplementary material Fig. S2). Therefore, unlike that shown in Xenopus, Cdk2 does not participate in maintaining CSF-mediated MII arrest in mouse oocytes. Consistent with this conclusion, it has been shown that SAC is not required for MII arrest in mouse oocytes (Tsurumi et al., 2004). Taken together, our data indicate that Cdc25A is required to maintain MPF activity during MII arrest, predominately by removing the inhibitory Cdk1 phosphorylation.
Recently, it has been shown that Cdc14B phosphatase inactivates Cdc25A thereby decreasing MPF activity during late mitosis (Tumurbaatar et al., 2011). Therefore, we experimentally manipulated Cdc14 activity as an alternative strategy to inactivate Cdc25A. Similar to Cdc25A knockdown, overexpression of Cdc14B induces pronuclear formation (Fig. 4A,B), whereas Cdc14A had no effect. Moreover, Cdc14B, but not Cdc14A, directly interacted with Cdc25A, and this interaction decreased the phosphatase activity of Cdc25A towards MPF but not towards MAPK (Fig. 4C–E). It should be noted that Cdc14B itself did not affect MPF or MAPK activity (Fig. 4F), indicating that the decrease in MPF activity is dependent on Cdc25A inactivation. Consistent with this finding is the observation that Cdc14B does not directly target MPF but rather regulates MPF activity by inactivating Cdc25A (Tumurbaatar et al., 2011). Thus, these data conclusively demonstrate that Cdc25A is required to maintain MPF activity during MII arrest in mouse oocytes.
Fig. 4.

Cdc14 effect on MII arrest and interaction with Cdc25A. (A) MII oocytes were injected with mRNAs coding for Cdc14A or B, and the percentage of treated oocytes in MII was scored after 24 hours. Numbers above the bars indicate the number of oocytes in the MII stage as well as total number of oocytes used. *P<0.005. (B) Representative images of Cdc14B-overexpressing oocytes. The spindle and DNA were stained with anti-α-tubulin and DAPI, respectively. Scale bar: 20 µm. (C) GST fusion proteins used in the pull-down assay (arrowhead) were separated by SDS-PAGE and stained with Coomassie Brilliant Blue. (D) GST fusion proteins were immunoprecipitated from the cell lysates overexpressing Cdc25A. Proteins recovered in the immunoprecipitation pellet were immunoblotted for Cdc25A. (E) Immunoprecipitated Cdc25As were pre-incubated with GST fusion proteins and H1/MBP double kinase assay was performed with GV oocyte lysates. (F) GST fusion proteins were subjected to H1/MBP double kinase assay with GV or MII oocytes lysates. Water was loaded as a negative control (−).
Conclusions
It is well established that CycB degradation is a key event for meiotic and mitotic exit enabling progression from metaphase to anaphase (Murray et al., 1989). Recently, however, mounting evidence suggests that MPF inactivation does not solely depend on CycB degradation during either meiosis (Chesnel et al., 2006) or mitosis (Nishiyama et al., 2000). Conversely, the inhibitory phosphorylation of Cdk1 is also necessary for MPF inactivation during M-phase exit (Chow et al., 2011; D'Angiolella et al., 2007; Oh et al., 2011; Potapova et al., 2009). Using several independent approaches, we show here that this phosphorylation participates in the MPF regulation during CSF-mediated MII arrest in mouse oocytes. MPF activity in MII is maintained by a Cdc25-mediated dephosphorylation of Cdk1 as well as by CycB levels. Disrupting this loop by downregulation of Cdc25A causes the oocyte release from MII arrest mostly through accumulation of the inhibitory phosphorylation of Cdk1. It has been shown that removal of Cdc25C from Xenopus CSF extracts also induces loss of mitotic state (Lorca et al., 2010). Given that this phosphorylation is mediated by Wee1B kinase in mouse oocytes, we propose that the balance between the activities of Wee kinases and Cdc25 phosphatases is a critical circuit required for the MII arrest in oocytes. In addition and as a complement to our findings on Wee1B regulation, it is likely that exit from the MII phase requires inactivation of Cdc25. Thus, our data demonstrate that MPF activity required for the MII arrest is maintained not only by CycB dynamics but also by Cdk1 phosphorylation.
Materials and Methods
Oocyte collection and microinjection
Oocytes were obtained, cultured and microinjected as described previously (Oh et al., 2011). The microinjected oocytes were incubated with cytochalasin B (1 µg/ml) for 24 hours to prevent second polar body extrusion. Nocodazole or MG132 was added to media at a concentration of 100 nM and 1 µM, respectively. Oocytes were observed and imaged on an inverted microscope (DMI 4000B; Leica).
Plasmids, morpholino oligonucleotides and preparation of mRNAs and double strand RNAs
The full-length cDNA coding the human Cdc25A was purchased from Origene. The catalytically inactive mutant of human Cdc25A (C431S) was generated using site-directed mutagenesis. Cdc14A and B were cloned as described previously (Schindler and Schultz, 2009b; Schindler and Schultz, 2009a). Wild-type Cdk2 (Cdk2-WT) and constitutively active Cdk2 (Cdk2-AF), GST-tagged Cdc14 phosphatases and H2B-mCherry were gifts from D. Morgan, Y. Wang and M. Anger, respectively. Capped mRNAs for microinjection were synthesized in vitro using mMESSAGE mMACHINE kit (Ambion), polyadenylated, and then purified with the MEGAclear kit (Ambion). MOs were designed as described previously (Oh et al., 2011). For double strand RNAs, PCR products were used as a template for in vitro transcription using the MEGAscript Kit (Ambion). PCR primers are reported in supplementary material Table S1.
RT-PCR
Total RNAs were extracted from oocytes (Qiagen) followed by reverse transcription using Sensiscript RT kit (Qiagen). PCR was performed using primers reported in supplementary material Table S1.
Immunoblotting and kinase assay
Oocytes were collected in phosphate-buffered saline (PBS) containing 1% polyvinylpyrrolidine (PVP) and frozen in SDS sample buffer. Western blotting was performed using antibodies against Cdc25 phosphatases (sc-65503; Santa Cruz), α-tubulin (ab7291; Abcam), Cdk1 (sc-954; Santa Cruz), Cdk1 phospho-tyrosine 15 (sc-7989; Santa Cruz), cyclin E1(sc-481; Santa Cruz), or cyclin E2 (4132; Cell Signaling). Histone H1 and myelin basic protein kinase (MBP) assays were performed as previously described (Svoboda et al., 2000). Briefly, MII oocytes injected with either EGFP dsRNA or Cdc25A dsRNA were lysed and incubated with histone H1 and MBP in the presence of [γ-32P]ATP. The intensity of spots on the autoradiogram was quantified by ImageJ software.
Protein interaction assays
GST, GST–Cdc14A, and GST–Cdc14B proteins were expressed in Escherichia coli and affinity purified. For pull-down assay, HEK293 cells were transfected with full-length Cdc25A clones and cell lysates were incubated with GST fusion proteins. The bound proteins were subjected to SDS-PAGE and immunoblotted with Cdc25A antibody. To measure the phosphatase activity of Cdc25A bound to Cdc14 proteins, immunoprecipitated Cdc25A was preincubated with GST fusion proteins, and H1/MBP kinase assay was performed with GV oocyte lysates.
Immunofluorescence imaging
Immunofluorescence was performed as described previously (Oh et al., 2011). Anti-mouse acetylated α-tubulin antibody (Sigma) was used to label the spindle. Primary antibody was detected with an Alexa-Fluor-488-conjugated goat polyclonal anti-mouse IgG (Molecular Probes) followed by incubation with DAPI for DNA staining. Immunostaining was visualized using an inverted confocal microscope (TCS SP5; Leica) with 63× objective.
Quantification and statistical analyses
Immunoreactive band or fluorescence intensity was measured using ImageJ software. Background correction and normalization were performed using Microsoft Excel software. Statistical analysis was performed using GraphPad software. Data are average of at least three independent experiments unless otherwise specified. Values were analyzed by one-way ANOVA or Student's t-test, and P<0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank David Morgan for Cdk2 plasmids, Martin Anger for H2B-mCherry plasmids, and Yisong Wang for the Cdc14-GST plasmids.
Footnotes
Author contributions
J.O. conceived and designed the study. J.O., A.S. and K.S. performed and analyzed experiments in this study. M.C. and R.S. supervised the study. J.O. wrote an initial draft of the manuscript, which all authors commented on and edited.
Funding
This work was supported by the National Institutes of Health [grant number GM08527 to M.C.]; and the Next-Generation BioGreen 21 Program, Rural Development Administration, Republic of Korea [grant number PJ009024 to J.O.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.115592/-/DC1
References
- Chang H. Y., Levasseur M., Jones K. T. (2004). Degradation of APCcdc20 and APCcdh1 substrates during the second meiotic division in mouse eggs. J. Cell Sci. 117, 6289–6296 10.1242/jcs.01567 [DOI] [PubMed] [Google Scholar]
- Chen M. S., Hurov J., White L. S., Woodford–Thomas T., Piwnica–Worms H. (2001). Absence of apparent phenotype in mice lacking Cdc25C protein phosphatase. Mol. Cell. Biol. 21, 3853–3861 10.1128/MCB.21.12.3853-3861.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesnel F., Bazile F., Pascal A., Kubiak J. Z. (2006). Cyclin B dissociation from CDK1 precedes its degradation upon MPF inactivation in mitotic extracts of Xenopus laevis embryos. Cell Cycle 5, 1687–1698 10.4161/cc.5.15.3123 [DOI] [PubMed] [Google Scholar]
- Chow J. P., Poon R. Y., Ma H. T. (2011). Inhibitory phosphorylation of cyclin-dependent kinase 1 as a compensatory mechanism for mitosis exit. Mol. Cell. Biol. 31, 1478–1491 10.1128/MCB.00891-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angiolella V., Palazzo L., Santarpia C., Costanzo V., Grieco D. (2007). Role for non-proteolytic control of M-phase-promoting factor activity at M-phase exit. PLoS ONE 2, e247 10.1371/journal.pone.0000247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont J., Umbhauer M., Rassinier P., Hanauer A., Verlhac M. H. (2005). p90Rsk is not involved in cytostatic factor arrest in mouse oocytes. J. Cell Biol. 169, 227–231 10.1083/jcb.200501027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielli B. G., Roy L. M., Maller J. L. (1993). Requirement for Cdk2 in cytostatic factor-mediated metaphase II arrest. Science 259, 1766–1769 10.1126/science.8456304 [DOI] [PubMed] [Google Scholar]
- Gharbi–Ayachi A., Labbé J-C., Burgess A., Vigneron S., Strub J-M., Brioudes E., Van–Dorsselaer A., Castro A., Lorca T. (2010). The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science 330, 1673–1677 10.1126/science.1197048 [DOI] [PubMed] [Google Scholar]
- Glover D. M. (2012). The overlooked greatwall: a new perspective on mitotic control. Open Biol. 2, 120023 10.1098/rsob.120023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimison B., Liu J., Lewellyn A. L., Maller J. L. (2006). Metaphase arrest by cyclin E-Cdk2 requires the spindle-checkpoint kinase Mps1. Curr. Biol. 16, 1968–1973 10.1016/j.cub.2006.08.055 [DOI] [PubMed] [Google Scholar]
- Gross S. D., Schwab M. S., Lewellyn A. L., Maller J. L. (1999). Induction of metaphase arrest in cleaving Xenopus embryos by the protein kinase p90Rsk. Science 286, 1365–1367 10.1126/science.286.5443.1365 [DOI] [PubMed] [Google Scholar]
- Jones K. T. (2005). Mammalian egg activation: from Ca2+ spiking to cell cycle progression. Reproduction 130, 813–823 10.1530/rep.1.00710 [DOI] [PubMed] [Google Scholar]
- Kubiak J. Z., Weber M., de Pennart H., Winston N. J., Maro B. (1993). The metaphase II arrest in mouse oocytes is controlled through microtubule-dependent destruction of cyclin B in the presence of CSF. EMBO J. 12, 3773–3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew D. J., Kornbluth S. (1996). Regulatory roles of cyclin dependent kinase phosphorylation in cell cycle control. Curr. Opin. Cell Biol. 8, 795–804 10.1016/S0955-0674(96)80080-9 [DOI] [PubMed] [Google Scholar]
- Lorca T., Bernis C., Vigneron S., Burgess A., Brioudes E., Labbé J. C., Castro A. (2010). Constant regulation of both the MPF amplification loop and the Greatwall-PP2A pathway is required for metaphase II arrest and correct entry into the first embryonic cell cycle. J. Cell Sci. 123, 2281–2291 10.1242/jcs.064527 [DOI] [PubMed] [Google Scholar]
- Madgwick S., Hansen D. V., Levasseur M., Jackson P. K., Jones K. T. (2006). Mouse Emi2 is required to enter meiosis II by reestablishing cyclin B1 during interkinesis. J. Cell Biol. 174, 791–801 10.1083/jcb.200604140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N., Podtelejnikov A. V., Groth A., Mann M., Bartek J., Lukas J. (2002). Regulation of G(2)/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J. 21, 5911–5920 10.1093/emboj/cdf567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui Y., Markert C. L. (1971). Cytoplasmic control of nuclear behavior during meiotic maturation of frog oocytes. J. Exp. Zool. 177, 129–145 10.1002/jez.1401770202 [DOI] [PubMed] [Google Scholar]
- Mochida S., Maslen S. L., Skehel M., Hunt T. (2010). Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 330, 1670–1673 10.1126/science.1195689 [DOI] [PubMed] [Google Scholar]
- Murray A. W., Solomon M. J., Kirschner M. W. (1989). The role of cyclin synthesis and degradation in the control of maturation promoting factor activity. Nature 339, 280–286 [DOI] [PubMed] [Google Scholar]
- Nishiyama A., Tachibana K., Igarashi Y., Yasuda H., Tanahashi N., Tanaka K., Ohsumi K., Kishimoto T. (2000). A nonproteolytic function of the proteasome is required for the dissociation of Cdc2 and cyclin B at the end of M phase. Genes Dev. 14, 2344–2357 10.1101/gad.823200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon V. L., Levasseur M., McDougall A., Jones K. T. (2002). Ca(2+) oscillations promote APC/C-dependent cyclin B1 degradation during metaphase arrest and completion of meiosis in fertilizing mouse eggs. Curr. Biol. 12, 746–750 10.1016/S0960-9822(02)00811-4 [DOI] [PubMed] [Google Scholar]
- Oh J. S., Susor A., Conti M. (2011). Protein tyrosine kinase Wee1B is essential for metaphase II exit in mouse oocytes. Science 332, 462–465 10.1126/science.1199211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapova T. A., Daum J. R., Byrd K. S., Gorbsky G. J. (2009). Fine tuning the cell cycle: activation of the Cdk1 inhibitory phosphorylation pathway during mitotic exit. Mol. Biol. Cell 20, 1737–1748 10.1091/mbc.E08-07-0771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagata N., Watanabe N., Vande Woude G. F., Ikawa Y. (1989). The c-mos proto-oncogene product is a cytostatic factor responsible for meiotic arrest in vertebrate eggs. Nature 342, 512–518 10.1038/342512a0 [DOI] [PubMed] [Google Scholar]
- Schindler K., Schultz R. M. (2009a). CDC14B acts through FZR1 (CDH1) to prevent meiotic maturation of mouse oocytes. Biol. Reprod. 80, 795–803 10.1095/biolreprod.108.074906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler K., Schultz R. M. (2009b). The CDC14A phosphatase regulates oocyte maturation in mouse. Cell Cycle 8, 1090–1098 10.4161/cc.8.7.8144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A., Duncan P. I., Rauh N. R., Sauer G., Fry A. M., Nigg E. A., Mayer T. U. (2005). Xenopus polo-like kinase Plx1 regulates XErp1, a novel inhibitor of APC/C activity. Genes Dev. 19, 502–513 10.1101/gad.320705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexl V., Diehl J. A., Sherr C. J., Ashmun R., Beach D., Roussel M. F. (1999). A rate limiting function of cdc25A for S phase entry inversely correlates with tyrosine dephosphorylation of Cdk2. Oncogene 18, 573–582 10.1038/sj.onc.1202362 [DOI] [PubMed] [Google Scholar]
- Shoji S., Yoshida N., Amanai M., Ohgishi M., Fukui T., Fujimoto S., Nakano Y., Kajikawa E., Perry A. C. (2006). Mammalian Emi2 mediates cytostatic arrest and transduces the signal for meiotic exit via Cdc20. EMBO J. 25, 834–845 10.1038/sj.emboj.7600953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda P., Stein P., Hayashi H., Schultz R. M. (2000). Selective reduction of dormant maternal mRNAs in mouse oocytes by RNA interference. Development 127, 4147–4156 [DOI] [PubMed] [Google Scholar]
- Tsurumi C., Hoffmann S., Geley S., Graeser R., Polanski Z. (2004). The spindle assembly checkpoint is not essential for CSF arrest of mouse oocytes. J. Cell Biol. 167, 1037–1050 10.1083/jcb.200405165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumurbaatar I., Cizmecioglu O., Hoffmann I., Grummt I., Voit R. (2011). Human Cdc14B promotes progression through mitosis by dephosphorylating Cdc25 and regulating Cdk1/cyclin B activity. PLoS ONE 6, e14711 10.1371/journal.pone.0014711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verlhac M-H., Lefebvre C., Kubiak J. Z., Umbhauer M., Rassinier P., Colledge W., Maro B. (2000). Mos activates MAP kinase in mouse oocytes through two opposite pathways. EMBO J. 19, 6065–6074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneron S., Brioudes E., Burgess A., Labbé J. C., Lorca T., Castro A. (2009). Greatwall maintains mitosis through regulation of PP2A. EMBO J. 28, 2786–2793 10.1038/emboj.2009.228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Zhao Y., Li Z., Galas S., Goldberg M. L. (2006). Greatwall kinase participates in the Cdc2 autoregulatory loop in Xenopus egg extracts. Mol. Cell 22, 83–91 10.1016/j.molcel.2006.02.022 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.