

Summary
The cell has many mechanisms for protecting the integrity of its genome. These mechanisms are often weakened or absent in many cancers, leading to high rates of chromosomal instability in tumors. Control of the cell cycle is crucial for the function of these checkpoints, and is frequently lost in cancers as well. Overexpression of Cyclin D1 in a large number of breast cancers causes overactivation of the cyclin-dependent kinases, including Cdk2. Constitutive Cdk2 activation through Cyclin D1 generates tumors in mice that are aneuploid and have many characteristics indicative of chromosomal instability. Expression of these complexes in the MCF10A cell line leads to retinoblastoma protein (Rb) hyperphosphorylation, a subsequent increase in proliferation rate, and increased expression of the spindle assembly checkpoint protein Mad2. This results in a strengthening of the spindle assembly checkpoint and renders cells more sensitive to the spindle poison paclitaxel. Constitutive Rb phosphorylation also causes a weakening of the p53-dependent tetraploidy checkpoint. Cells with overactive Cdk2 fail to arrest after mitotic slippage in the presence of paclitaxel or cytokinesis failure during treatment with cytochalasin-B, generating 8N populations. This additional increase in DNA content appears to further intensify the tetraploidy checkpoint in a step-wise manner. These polyploid cells are not viable long-term, either failing to undergo division or creating daughter cells that are unable to undergo subsequent division. This study raises intriguing questions about the treatment of tumors with overactive Cdk2.
Key words: Cyclin D1, Cdk2, Breast cancer, Centrosome, Chromosomal instability, Spindle assembly checkpoint, Mad2, Tetraploidy checkpoint
Introduction
Included in the updated Hallmarks of Cancer (Hanahan and Weinberg, 2011), unstable DNA is a common characteristic of many solid cancers. This instability can take the form of genetic instability, changes in the DNA nucleotide sequence, or chromosomal instability (CIN), changes in the number of whole chromosomes, and this instability is both a tumor initiator (Weaver et al., 2007) and a driver of tumor evolution (Nowell, 1976). The DNA content of cells can change through multiple mechanisms, either causing stochastic changes up or down or simple genome doubling. Phenomena including endoreduplication and failed cytokinesis induce an abnormal tetraploid, 4N, DNA content. Centrosome duplication can also occur concomitantly, and while a stable, true tetraploid DNA content is not sufficient to cause transformation (Fujiwara et al., 2005), centrosome amplification can lead to multipolar mitoses, lagging chromosomes, and subsequent aneuploidy (Sluder and Nordberg, 2004; Ganem et al., 2009).
Many of the mechanisms that protect the cellular genome are integrally linked to the cell cycle, and cancers with cell cycle mutations often have a high rate of chromosomal instability (Deangelis et al., 1993; Collecchi et al., 2000; Sotillo et al., 2007). Cyclin D has been found to be overexpressed in ∼50% of all breast cancers (Sweeney et al., 1998) and Nelsen et al. have shown that short-term overexpression of Cyclin D1 is capable of initiating CIN, and that the process of chromosomal changes persists even after the exogenous Cyclin D1 is no longer being produced (Nelsen et al., 2005). The molecular mechanism by which this occurs is unknown. Cyclin D1 is most commonly thought to be an activating partner of the cyclin-dependent kinases Cdk4 and Cdk6 and Cyclin D1/Cdk4 complexes have been shown to induce centrosome amplification in the presence of Ras mutations (Zeng et al., 2010) and MEK2 deficiency (Ussar and Voss, 2004). Additionally, both Cdk4 and Cdk2 are required for the Nucleophosmin hyperphosphorylation and subsequent centrosome amplification observed in p53 null cells (Adon et al., 2010).
While it may not generally be considered a canonical partner, Cyclin D1 has also been shown to form active complexes with Cdk2 (Chytil et al., 2004; Malumbres et al., 2004). Cdk2, which is normally active and bound to Cyclin E in late G1 through S phase and then with Cyclin A into late G2, is thought to play a critical role in the centrosome duplication cycle (Tokuyama et al., 2001; Chen et al., 2002). Therefore, it was unclear whether the results seen by Nelsen et al. were due to Cyclin D1 complexes with Cdk2, Cdk4, Cdk6, or any of the other cyclin-dependent kinases (Nelson et al., 2005). Further studies showed that expression of Cdk2 is required for Cyclin D1-induced CIN, hinting to the importance of Cyclin D1/Cdk2 complexes, but not eliminating the possibility that Cyclin D1 is binding to another Cdk and causing CIN in a Cdk2-dependent mechanism (Hanse et al., 2009).
We have previously published studies utilizing a Cyclin D1–Cdk2 fusion protein (D1K2), and transgenic mice expressing this protein under the control of the mammary-specific mouse mammary tumor virus (MMTV) promoter/enhancer develop spontaneous mammary tumors that are invasive, heterogeneous, and express markers associated with the basal-like subtype of breast cancer (Chytil et al., 2004; Corsino et al., 2007; Corsino et al., 2008). Here, we present analysis of the effect of D1K2 expression on the mitotic and tetraploidy checkpoints using tumor-derived cell lines and the nontransformed human mammary epithelial cell line, MCF10A, stably expressing D1K2. Our results show that D1K2 strengthens the mitotic checkpoint through upregulation of Mad2, increasing the anti-proliferative effects of paclitaxel. D1K2 expression also weakens the tetraploidy checkpoint, allowing polyploidization of cells that enter G1 as tetraploid through mitotic slippage or failed cytokinesis. Long-term survival data of these cells raise intriguing questions regarding the treatment of cancers with overactive Cdk2.
Results
MMTV-D1K2 mice develop aneuploid mammary tumors with a compromised tetraploidy checkpoint
Cdk2 activation has previously been implicated in the induction of aneuploidy and genomic instability through the processes of centrosome overduplication and endoreduplication (Hinchcliffe et al., 1999; Matsumoto et al., 1999; Stewart et al., 1999; Nishimura et al., 2005; Duensing et al., 2006; Adon et al., 2010). We previously observed aneuploidy in MMTV-D1K2 transgenic mice, suggesting that induction of tumorigenesis by Cyclin D1/Cdk2 complexes may involve these processes (Corsino et al., 2007). We examined Hematoxylin and Eosin (H&E)-stained sections of MMTV-D1K2 tumors for evidence of aberrant mitosis. As indicated in Fig. 1A, all primary MMTV-D1K2 tumors isolated exhibited mitoses that appeared abnormal. Metaphase spreads of cell lines derived from these tumors (Fig. 1B) showed a high variance in DNA content. The presence of cells with a greater than 4N DNA content indicates a weakened tetraploidy checkpoint in these tumors.
Fig. 1.

MMTV-D1K2 tumors and tumor-derived cell lines exhibit aberrant mitosis and CIN. (A) H&E-stained sections from tumor tissue generated by injecting cancer cell lines isolated from MMTV-D1K2 tumors into the mammary glands of wild-type FVB mice. Aberrant mitoses (red arrows), a normal bipolar mitotic figure (white arrow), and a bipolar mitotic figure with lagging chromosomes (black arrow) are evident. (B) Metaphase spreads of cells from the indicated MMTV-D1K2 cell lines. The number in the lower right-hand corner of each picture is the number of chromosomes in the associated spread. (C) Karyotype analysis of metaphase chromosomes by G-banding. (D) The indicated cell lines were an analyzed by immunofluorescence microscopy with antibodies to γ-Tubulin (yellow). DAPI staining is shown in blue. Yellow arrows denote cells with an abnormally high number of centrosomes (>2). Red arrows indicate cells undergoing multipolar mitosis. Rates of centrosome amplification (CA) in the indicated cell lines are shown below. (E) The indicated cell lines were treated with 1 µM paclitaxel, 1 µM nocodazole, or the 0.1% DMSO vehicle for 72 hours. Treated cells were subjected to propidium iodide staining followed by flow cytometry.
Further, karyotype analysis of the tumor-derived cell lines showed high rates of trisomy and a bi-modal population with cells concentrating around diploid or tetraploid status (Fig. 1C and data not shown). These results indicate a mechanism for genome doubling and the ability to bypass the p53-dependent tetraploidy checkpoint (Andreassen et al., 2001) in order to continue growth. This abnormal doubling would be expected to be paired with centrosome amplification, and the tumor cell lines do show high levels of centrosome amplification, ranging from 13.5 to 22.5% of individual cells showing supernumerary centrosomes marked by γ-tubulin staining (Fig. 1D). These cells also exhibit multipolar mitoses, and a hydroxyurea-based centriole reduplication assay (Balczon et al., 1995) showed a small, not statistically significant, increase in the rate of centrosome amplification in the D1K2-T1 cell line from 19.5 to 22% (data not shown). This indicates that while D1K2 tumor cell lines possibly possess a mechanism to induce minor overduplication of centrioles, which is unsurprising due to the Nucleophosmin hyperphosphorylation seen in Fig. 2A, it is likely not sufficient to account for the high rate of centrosome amplification observed. These cell lines also generate populations with 8N and 16N DNA content, as measured by flow cytometry, when grown in the presence of the spindle poisons nocodazole or paclitaxel, providing further evidence for the lack of a functional tetraploidy checkpoint.
Fig. 2.

D1K2 phosphorylates Cdk2 substrates, increases growth rate, and sensitizes cells to paclitaxel. (A) Immunoblot analysis of Cdk2 substrates in the indicated cell lines. Actin served as a loading control. (B) Growth curves of the indicated cell lines after being treated with 0.1% DMSO or 1 nM paclitaxel for 72 hours. Cells were then reseeded in six-well plates and counted in triplicate each day for 1 week. (C) [3H]Thymidine incorporation of the indicated cell lines treated with increasing concentrations of paclitaxel for 72 hours. Values are the average of three replicate samples. Error bars represent the s.d. of three replicates. *P<0.05 and **P<0.005, using the unpaired t-test.
MCF10A cells expressing D1K2 have a deregulated cell cycle and increased paclitaxel sensitivity
Since tumors derived from the mouse model presumably have undergone further mutations during tumor development, we sought to create a model in which the direct effects of D1K2 activity could be studied. We stably transduced MCF10A cells with vectors encoding a Hygromycin control vector (Hygro), D1K2, or a kinase dead version of D1K2 [D1K2(KD)]. A clonal cell line, D1K2 CL1, was derived from the polyclonal D1K2-expressing cell line in order to obtain cells expressing a homogenous amount of the transgene. Immunoblot of these cell lines (Fig. 2A) shows intracellular D1K2 activity illustrated by hyperphosphorylation of Cdk substrates, including retinoblastoma protein (Rb) and Nucleophosmin (NPM) in only those cell lines containing kinase active D1K2. Rb shows phosphorylation on sites preferred by Cdk4 (Ser249/Thr252, Ser780 and Ser795) as well as Cdk2 (Ser807/811), consistent with our previous findings (Chytil et al., 2004). Nucleophosmin is a substrate of the Cyclin E/Cdk2 complex and helps maintain control of centriole duplication (Okuda et al., 2000). It is unclear as to whether D1K2 phosphorylation of NPM leads to centrosome amplification in vivo. In the in vitro model discussed below, NPM hyperphosphorylation does not lead to abnormal centrosome numbers; however, these cells likely have redundant mechanisms to block overduplication (Ganem et al., 2009; Krzywicka-Racka and Sluder, 2011) that may be compromised during tumor development.
Rb hyperphosphorylation causes cell cycle deregulation (Sherr, 1996) and analysis of growth rates showed a dramatic increase in proliferation of the D1K2 CL1 cell line compared to the Hygro control (Fig. 2B, top panel). In addition, D1K2 expression increased the maximum confluent density of the cells, indicated by the approximately four times higher maximum cell number reached by the D1K2 CL1 cell line. Treatment of these cell lines with paclitaxel yielded interesting results. D1K2-expressing cells replated after paclitaxel washout showed growth rates equal to or less than that of the comparably treated control over the first 4 days (Fig. 2B, bottom panel). The untreated D1K2 CL1 cell line had a statistically significant increase in cell number compared to the Hygro cell line after 3 and 4 days. However, the difference in cell number of each cell line was not statistically different on days 3 and 4 after paclitaxel treatment, indicating a greater sensitivity to the growth inhibitory effects of the spindle poison. Subsequent growth, presumably after the effects of treatment had dissipated, recapitulated that seen in the untreated cells.
[3H]Thymidine incorporation in the Hygro and D1K2 CL1 cell lines after treatment with increasing concentrations of paclitaxel for 72 hours also showed a differential response between the cell lines (Fig. 2C). There was a statistically significant difference in proliferation in these cell lines, normalized to untreated controls, when grown in the presence of 1.875 or 3.75 nM paclitaxel. Interestingly, the data in Fig. 2B,C show that the difference in sensitivity to paclitaxel in the control and D1K2-expressing cells increases with time after exposure. Whereas nearly a 2 nM concentration was required to see a statistically significant difference after 72 hours of exposure in Fig. 2C, the effects of paclitaxel were seen after 72 hours of treatment and a subsequent 72-hour washout period with only 1 nM paclitaxel in Fig. 2B. Treatment with 1 µM paclitaxel for 72 hours, as required to generate tetraploid populations below, blocked nearly all proliferation (supplementary material Fig. S1).
D1K2 kinase activity strengthens the spindle assembly checkpoint
Flow cytometric analysis of the DNA content of Hygro and D1K2 CL1 cells treated with paclitaxel for 72 hours shows the appearance of a tetraploid, 8N, population in the cells expressing D1K2 but not in the control cells (Fig. 3A, left and center panels). Cells expressing the kinase dead D1K2 fail to produce this 8N population (Fig. 3A, right panel), indicating that the D1K2 kinase activity is required for the phenomenon rather than the fusion protein exerting its effects through protein/protein interactions, as has been discussed previously (Chytil et al., 2004). Similarly, co-treatment of the D1K2 CL1 cell line with the Cdk2 inhibitor CVT313 along with paclitaxel inhibited the development of this 8N population in a dose-dependent manner (Fig. 3B). Thymidine incorporation experiments showed that treating these cell lines with paclitaxel, CVT313, or a combination blocks proliferation. At the paclitaxel concentration used, a small amount of DNA synthesis remains and addition of CVT313 further decreases it, supporting the flow cytometry data (supplementary material Fig. S2A).
Fig. 3.

D1K2 kinase activity promotes polyploidy and upregulates Mad2. (A) Flow cytometry analysis of the indicated cell lines after 72 hours of treatment with 0.1% DMSO or 1 µM paclitaxel. (B) Flow cytometry analysis of the MCF10A D1K2 cell line treated with 1 µM paclitaxel and the indicated concentration of the Cdk2 inhibitor CVT313 for 72 hours. (C) Immunoblot analysis of the indicated cell lines showing Mad2, Cdk2 and D1K2 expression. (D) Flow cytometry analysis of the indicated cell lines after 72 hours of treatment with 0.1% DMSO or 1 µM paclitaxel. (E) Immunoblot analysis of the indicated cell lines at various time points after constant treatment with 1 µM paclitaxel, showing Mad2 expression levels. Flow cytometry profiles in all panels represent DNA content as measured by propidium iodide staining. Black arrowheads on the x-axes of the flow cytometry graphs indicated (left to right) the 2N, 4N and 8N peaks, respectively.
As expression of the spindle assembly checkpoint (SAC) protein Mad2 has been shown to be E2F dependent (Hernando et al., 2004) and its overexpression is capable of initiating tumorigenesis and chromosomal instability (Sotillo et al., 2007), we examined Mad2 protein levels in our cell lines. Mad2 expression was found to be increased in the D1K2 CL1 cell line compared to the Hygro and D1K2(KD) cell lines (Fig. 3C, left panel). This increased expression was confirmed to be due to D1K2 activity as partial Cdk2/D1K2 knockdown reduced Mad2 protein levels (Fig. 3C, right panel). We also observed decreased Mad2 expression in the presence of CVT313 (supplementary material Fig. S2B).
If high levels of Mad2 are responsible for a weakened SAC, leading to tetraploidy, restoration of Mad2 levels through a partial knockdown should block formation of an 8N population in the D1K2 CL1 cell line. However, we observed that Mad2 knockdown (analyzed in supplementary material Fig. S3A) cooperated with D1K2 activity to increase the formation of this population (Fig. 3D). Immunoblot analysis of cell lines treated with paclitaxel showed that, in all cases, Mad2 levels initially increased upon treatment but decreased nearly to zero upon long-term treatment (Fig. 3E). Presumably, all of the utilized cell lines treated with paclitaxel eventually suffer a failure of the SAC and undergo mitotic slippage, reentering the G1 phase of the cell cycle as 4N. Increased Mad2 expression due to D1K2 activity appears to strengthen the SAC, delaying mitotic slippage, protecting against tetraploidy, and providing a potential mechanism for the increased sensitivity to paclitaxel seen in Fig. 2. As further evidence of a strengthened SAC, the D1K2 CL1 cell line shows much greater MPM2 staining, a mitotic marker, than does the Hygro cell line after treatment with paclitaxel (supplementary material Fig. S3B).
Quantification of centrosome numbers showed amplification due to paclitaxel treatment. Whereas the untreated D1K2 CL1 cell line showed an ∼5% rate of centrosome amplification (>2 centrosomes/cell), that rate increased to 22% upon 72 hour paclitaxel treatment. Surprisingly, co-treatment with CVT313 failed to block this amplification and instead resulted in a further increase to 45% (supplementary material Fig. S4). This is presumably due to the observed decrease in Mad2 levels upon CVT313 treatment (supplementary material Fig. S2B), weakening the spindle assembly checkpoint, allowing a greater number of cells to enter G1 with two unduplicated centrosomes, which will then become four. While Cdk2 inhibition via CVT313 is sufficient to block subsequent DNA replication (Fig. 3B), it does not appear to be sufficient to block centrosome duplication. It is possible that a lower kinase activity level is required to initiate centrosome duplication compared to DNA replication. Another explanation for the results could be that Cdk4 is substituting for the inhibited Cdk2 in the centrosome duplication machinery, as has been previously seen (Adon et al., 2010), but is unable to do so in the DNA replication process.
D1K2 activity weakens the tetraploidy checkpoint
A p53-dependent tetraploidy checkpoint has been shown to arrest cells that contain a 4N DNA content in G1 phase through increased production of p21 and the subsequent blockage of Rb phosphorylation (Andreassen et al., 2001). If all of the MCF10A cell lines undergo mitotic slippage as our data suggest, the mechanism that prevents the control cells from becoming 8N must act in G1 or at the G1/S transition. Stable knockdown of p53 (analyzed in supplementary material Fig. S5) cooperated with D1K2 to increase the 8N population (Fig. 4A), providing further evidence that cells are reentering G1 and DNA replication is being inhibited by a p53-dependent checkpoint.
Fig. 4.

D1K2 weakens the tetraploidy checkpoint. (A) Flow cytometry analysis of the indicated cell lines after 72 hours of treatment with 0.1% DMSO or 1 µM paclitaxel. (B) Flow cytometry analysis of the indicated cell lines after 72 hours of treatment with 0.1% DMSO or after 24, 48 and 72 hours of treatment with 4.5 µg/ml cytochalasin-B. (C) Micrographs of the indicated cell lines after 72 hours of treatment with 0.1% DMSO or 4.5 µg/ml cytochalasin-B. The insets are enlarged images of the regions within the dashed white boxes. White arrows mark cells with more than two nuclei. (D) Flow cytometry analysis of the MCF10A D1K2 CL1 shp53 cell line after 72 hours of treatment with 0.1% DMSO or 4.5 µg/ml cytochalasin-B. Note: x-axis values differ from previous flow cytometry profiles because of the lower detector voltage required to detect the 16N peak.
To generate large populations of tetraploid cells and to examine the fidelity of the tetraploidy checkpoint in the absence of a SAC response, we utilized the cytokinesis inhibitor cytochalasin-B. Even more strikingly than is seen with paclitaxel treatment, long-term treatment with cytochalasin-B induced a large 8N population in the D1K2 CL1 cell line that was much larger than that seen in the Hygro cell line (Fig. 4B, left and center panels). Additionally, the 8N population persisted to 72 hours, whereas the small 8N population produced in the Hygro cell line after 24 and 48 hours appears to have reverted to 2N/4N or died after 72 hours. Elimination of the tetraploidy checkpoint through p53 knockdown produced results similar to that seen with D1K2 expression (Fig. 4B, right panels). Photographs of treated cell lines show that untreated cell lines are mononucleate. Upon cytokinesis failure due to cytochalasin-B treatment, the Hygro cell line remains binucleate, whereas D1K2-expressing or shp53 cell lines progress to become multinucleate (Fig. 4C). Interestingly, in the D1K2 CL1 cell line in which p53 is also knocked down, cytochalasin-B treatment not only produced a large 8N population, but also a substantial 16N population (Fig. 4D) that was not seen in any of the other cell lines. It appears that constitutive Cdk2 activation or ablation of the tetraploidy checkpoint are sufficient to allow one extra round of DNA replication, but that both conditions must be met in order to permit a subsequent duplication.
D1K2 expression prolongs Rb phosphorylation upon tetraploidy checkpoint activation
Immunoblot analysis of MCF10A cell lines treated with cytochalasin-B for 24, 48 or 72 hours shows that both Hygro and D1K2 CL1 cell lines activate the tetraploidy checkpoint upon cytokinesis failure, illustrated by an increase in p53 expression (Fig. 5A). At 24 hours, little decrease in Rb phosphorylation is shown in either cell line. After 48 hours of treatment, D1K2 CL1 exhibits higher levels of phosphorylated Rb compared to the Hygro cell line despite the D1K2 CL1 cell line having progressed further in the cell cycle and started to develop an 8N population due to its faster growth rate. At 72 hours of treatment, Rb phosphorylation has been abolished in both cell lines, presumably arresting both. It is interesting to note that the D1K2 CL1 cell line shows higher levels of p53 after checkpoint activation than does the Hygro cell line, and it appears to increase further between 48 and 72 hours. The MCF10A shp53 cell line shows no induction of p53 and maintains Rb phosphorylation through 72 hours. These results are consistent with a model in which the tetraploidy checkpoint is activated in both Hygro and D1K2 CL1 cell lines. The initial activation is adequate to arrest the Hygro cell line at 4N, whereas D1K2 expression maintains Rb phosphorylation at levels sufficient for DNA replication. This further increase in DNA content intensifies the tetraploidy checkpoint, arresting these cells at 8N after the subsequent round of failed cytokinesis.
Fig. 5.

D1K2 expression or p53 knockdown sustain Rb phosphorylation in polyploid cells. Immunoblot analysis of the indicated cell lines after 24, 48 and 72 hours of treatment with either 0.1% DMSO or 4.5 µg/ml cytochalasin-B.
Long-term outcome of tetraploid populations
In order to evaluate the long-term effects of tetraploidy, we sorted cells treated with cytochalasin-B for 72 hours using flow cytometry. 2N and 4N populations were isolated from MCF10A Hygro, D1K2 CL1 and shp53 cell lines as well as 8N populations from D1K2 CL1 and shp53. Cell viability and growth rates were observed the week following sorting by measuring Crystal Violet staining (Fig. 6A). 4N and 8N populations showed a decreased rate of proliferation compared to the 2N population. The DNA profiles of these populations were examined one week after sorting and showed a near normal 2N/4N population, with the previous polyploid population markedly absent (Fig. 6B). Additionally, D1K2 CL1 cells plated at single cell density prior to being treated with cytochalasin-B were monitored for survival (Fig. 6C). The majority of cells visually confirmed to be multinucleate after cytochalasin-B treatment died (77%) or failed to divide (19%) in the 7 days following washout. A small number of cells successfully divided once (4%) but their progeny failed to undergo subsequent divisions.
Fig. 6.

Long-term effects of polyploidy on MCF10A cell lines. (A) Growth curves of the indicated cell lines after treatment for 72 hours with 4.5 µg/ml cytochalasin-B and subsequent sorting based on DNA content. Plotted values are Crystal Violet absorbances (±s.d.) normalized to those on day 1. (B) Flow cytometry analysis of the indicated cell lines 1 week after sorting based on ploidy (2N, 4N, 8N). (C) Data obtained through the visual analysis of the fate of tetraploid D1K2 CL1 cells induced with cytochalasin-B for 72 hours.
These data are consistent with cells that are able to briefly continue to proliferate with a polyploid DNA content due to the presence of D1K2 or absence of p53 but are not viable in the long-term. The post-sorting flow cytometry showing a normal population likely reflects a 2N population initially present in the 4N and 8N populations, due to imperfect sorting, that overtakes the polyploid populations over time. This also explains the growth curve data. Indeed, further flow cytometry analysis of sorted populations detects the presence of 2N cells in the 4N and 8N populations (data not shown).
Discussion
The generation of polyploid cells, followed by subsequent multipolar mitosis and lagging chromosomes, has been suggested as a potential mechanism of aneuploidy generation in tumors, a near universal commonality in cancers. The existence of a checkpoint that is able to detect the presence of a 4N DNA content in G1 phase cells has been known for some time. A number of studies have clearly shown the ability of tetraploidy to arrest the cell cycle (Minn et al., 1996; Lanni and Jacks, 1998; Andreassen et al., 2001; Krzywicka-Racka and Sluder, 2011), but the mechanism by which it senses tetraploidy is poorly understood. The effects of this checkpoint on the long-term viability of cells appear to be cell line specific. In a comprehensive study, it was found that multiple non-transformed cell lines that were treated with cytochalasin-B to induce tetraploidy were able to undergo several rounds of mitosis upon washout, but eventually exited the cell cycle or died. It was found that the response of transformed cells containing p53 mutations to tetraploidy also varied, ranging from death, to repeated cytokinesis failure, to indefinitely viable tetraploid cells (Krzywicka-Racka and Sluder, 2011).
Using in vivo and in vitro models, we have studied the role of constitutive Cdk2 activity, due to Cyclin D1/Cdk2 complexes, in polyploidization. Mice with mammary-specific expression of a D1K2 fusion protein generate spontaneous mammary tumors. These tumors are aneuploid, exhibit centrosome amplification, aberrant mitosis, and have a sub-population of near tetraploid cells. This indicates that these cells harbor a mechanism for becoming tetraploid as well as a lack of a functional tetraploidy checkpoint. Indeed, treatment of cell lines derived from these tumors with the spindle poisons paclitaxel or nocodazole results in further genome amplification.
MCF10A, nontransformed human mammary epithelial cells, stably expressing the D1K2 fusion protein exhibit cell cycle deregulation caused by Rb hyperphosphorylation that results in an increase in population growth rates. Proliferation of this cell line in the presence of paclitaxel for 72 hours results in the formation of an 8N population that is not seen in similarly treated control cells. These results indicate that D1K2 is able to bypass a checkpoint that is present in MCF10A cells that serves to block paclitaxel induced polyploidization. We have previously shown that the D1K2 protein is capable of exerting effects through protein/protein interactions in addition to through its kinase activity, so we generated a cell line expressing a kinase-dead variant of D1K2. This cell line mirrors the control cell line when treated with paclitaxel, failing to generate an 8N population. Further, treatment of D1K2-expressing cells with the Cdk2 inhibitor CVT313 blocks tetraploidization, precluding the possibility that D1K2 weakens the SAC through activation of Cdk1 via sequestration of Cdk inhibitory proteins such as p21, and indicates that the effects of D1K2 are due to its kinase activity. However, since flow cytometry cannot discriminate between diploid 4N cells arrested in G2/M and 4N cells that have entered G1, these data cannot distinguish between effects on the SAC or the tetraploidy checkpoint.
Transcription of the SAC effector protein Mad2 is E2F-dependent and overexpression of Mad2 is capable of initiating tumorigenesis and chromosomal instability (Hernando et al., 2004; Sotillo et al., 2007). Inactivation of Rb leads to Mad2 upregulation (Hernando et al., 2004), therefore similar results would be expected through D1K2-induced Rb hyperphosphorylation, which would effectively ablate cells of functional Rb. We showed that D1K2 expression and kinase activity lead to a concomitant increase in Mad2 expression. While Mad2 is crucial in the functioning of the SAC, and even small decreases in its expression weaken the SAC (Janssen et al., 2009), it has been reported that increased Mad2 expression can also lead to an impaired SAC (Kabeche and Compton, 2012). The observed increase in expression is possibly the cause of the enhanced paclitaxel sensitivity in D1K2 cells as previous studies have shown that Mad2 is required for paclitaxel sensitivity (Sudo et al., 2004; Hao et al., 2010). It has previously been reported that Mad2 overexpression does not increase the sensitivity of MCF10A cells to paclitaxel; however, the experiments only examined treatments up to 36 hours (Sudo et al., 2004). We did not observe a differential effect at similar time points (data not shown) and the effects of Mad2 overexpression were not seen until after 72 hours. Partial knockdown of Mad2 in the D1K2 CL1 cell line failed to decrease sensitivity (data not shown), however we have observed this cell line to be much less adherent than the parental cell lines. Multiple studies have shown that cell adherence inversely correlates with paclitaxel sensitivity (Ahmed et al., 2007; Chen et al., 2010), so it is possible this effect masks that of decreasing Mad2 levels.
If D1K2 expression leads to paclitaxel-induced polyploidization by weakening the SAC through Mad2 upregulation, restoration of Mad2 levels in D1K2-expressing cells would be expected to restore cell cycle arrest. However, partial knockdown of Mad2 to near basal levels cooperated with D1K2 expression to increase the generation of 8N cells upon paclitaxel addition. Furthermore, immunoblot analysis of Mad2 expression upon long-term paclitaxel treatment showed that Mad2 levels increased before decreasing dramatically, regardless of D1K2 expression. This indicates that the cell lines in this study will eventually suffer mitotic slippage and enter G1 in the tetraploid state. The increased Mad2 level in D1K2-expressing cells serves to strengthen the SAC and prevent this phenomenon. This is evidenced by an increase in MPM2 phosphorylation in the D1K2 CL1 cell line compared to the Hygro cell line after treatment with paclitaxel. Recognizing phosphorylated mitotic proteins, the MPM2 antibody is a mitotic marker commonly used as a measure of SAC strength (Westendorf et al., 1994; Chin and Herbst, 2006). Perhaps a certain level of increased Mad2 expression strengthens the SAC, but any further overexpression causes a weakening. This could explain the difference between the results seen in this study and those in which direct overexpression of Mad2 causes a weakening of the SAC (Sotillo et al., 2007; Kabeche and Compton, 2012).
These data point to the conclusion that D1K2 is promoting a tetraploid population by exerting its effects on the tetraploidy checkpoint. Indeed, knockdown of p53 leads to an even greater population of 8N cells in the D1K2-expressing cell line, indicating that a p53-dependent mechanism is hampering the formation of this tetraploid population. In order to look more closely at the tetraploidy checkpoint, we generated tetraploid cells through failed cytokinesis induced by cytochalasin-B. This resulted in the formation of 8N cells in D1K2 expressing and shp53 cell lines, but not in the control cell line. Interestingly, whereas the shp53 cell line developed an 8N population in the presence of cytochalasin-B, this is in contrast to the result seen previously in the presence of paclitaxel (Fig. 4A). While the reason for this is unclear, it is possible that paclitaxel induced arrest in these cells independent of the SAC as has been seen previously (Trielli et al., 1996). This would also explain the apparent lack of a G2 arrest. p53 knockdown may induce the death of the 8N population through an unknown mechanism, or these cells may be capable of dividing normally in the presence of paclitaxel. Importantly, this is likely a drug-dependent effect and does not affect the conclusions drawn from the experiments with cytochalasin-B.
Visual examination of these cells showed that the majority of the control cells were binucleate, with the remainder being mononucleate. D1K2 and shp53 cells additionally had a high rate of multinucleation, with three or more nuclei. Presumably, control cells arrest after failed cytokinesis whereas if the tetraploidy checkpoint is abrogated or bypassed with p53 knockdown or D1K2 expression, respectively, the cells undergo another round of DNA replication and failed cytokinesis. Immunoblot analysis of lysates obtained from treated cells show that Rb phosphorylation is lost in the Hygro cell line after 48 hours of treatment, whereas the D1K2 CL1 cell line as well as the shp53 cell line maintain greater phosphorylation. After an additional 24 hours, Rb phosphorylation is lost in all cell lines other than shp53, indicating that D1K2 is not capable of bypassing the tetraploidy checkpoint indefinitely.
The ability of D1K2 to allow DNA replication of 4N cells is despite a greater increase in p53 induction, compared to the Hygro control. Levels of p53 appear to increase further between 48 and 72 hours, at which point D1K2 is no longer able to promote further DNA replication, demonstrating the possibility that the tetraploidy checkpoint functions in a step-wise manner. To our knowledge, this is the first evidence that p53 induction is proportional to DNA content, with an 8N DNA content eliciting a greater response than 4N. While the mechanism of sensing ploidy is currently unknown, these data suggest that it is sensitive to total quantities of DNA. Similarly, knockdown of p53 is not sufficient to allow replication of DNA past the 8N state, indicating that cells have an alternative mechanism for stopping the cell cycle. Concomitant expression of constitutively active Cdk2 along with ablation of the tetraploidy checkpoint through p53 knockdown is capable of bypassing both systems, allowing further DNA replication.
Ultimately, the significance of tetraploidy is determined by the fate of the affected cells. If a tetraploid state is fatal to a tumor cell, treatments that induce it should be beneficial. If the cells remain viable and maintain a tetraploid DNA content or revert to a diploid state, it will have little consequence. However, if tetraploidy leads to aneuploidy, it may possibly have negative consequences clinically. Our results show that tetraploid MCF10A cells are not viable; dying, failing to divide, or dividing once to produce unviable progeny. Thus, it is expected that treatment of tumors having constitutively active Cdk2 with certain therapeutics will induce tetraploidy that will ultimately lead to tumor death, consistent, and possibly complementary, with our observation of increased paclitaxel sensitivity due to Mad2 upregulation. While we did not visibly observe its occurrence, we cannot rule out the possibility that MCF10A cells are able to revert from a tetraploid state to a diploid state and remain viable. This phenomenon of halving the cellular chromosome content occurs in meiosis and has been observed in tetraploid hepatocytes (Duncan et al., 2009); however, the mechanism is unknown in the latter case. Even if a very small fraction of tumor cells are able to carry out this reduction and remain viable, it would likely be sufficient to cause tumor relapse, potentially with a dramatically different tumor phenotype. It will be interesting to explore this possibility in the future, as it holds substantial consequence to patient treatment.
It is important to note, however, that our model using the MCF10A cell line cannot account for effects on cells due to other genetic and epigenetic alterations that may exist in tumors. Our results indicate that tumors with p53 mutations along with Cdk2 activation may be particularly prone to polyploidy and its potential ramifications. While the concentration of paclitaxel used to induce polyploidy in this study is lethal to the vast majority of cells, a small number of viable cells remain 48 hours after drug washout (supplementary material Fig. S1). In treating a patient, a relapse of the tumor may occur due to this small surviving population that contains a potentially considerable difference in chromosome content.
These data have allowed us to put forth two models of how constitutive Cdk2 activity affects the mitotic and tetraploidy checkpoints. The hyperphosphorylation of Rb induced by D1K2 causes an upregulation of Mad2. This strengthens the mitotic checkpoint, delaying slippage into G1 with a tetraploid DNA content (Fig. 7A). In this case, D1K2 expression serves to prevent additional rounds of DNA replication.
Fig. 7.
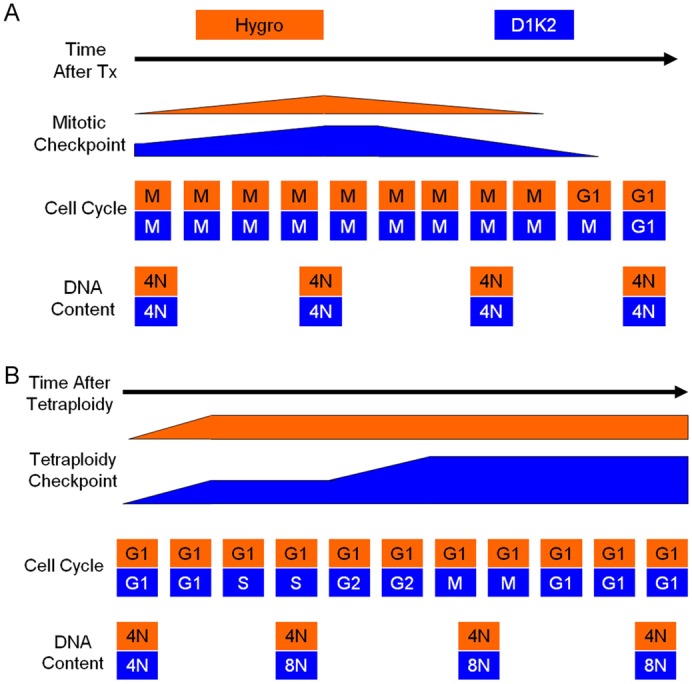
Current working models. (A) The hyperphosphorylation of Rb induced by D1K2 causes an upregulation of Mad2. This strengthens the mitotic checkpoint, delaying slippage into G1 with a tetraploid DNA content. (B) After tetraploidy occurs, an initial response of the tetraploidy checkpoint is sufficient to arrest cells without D1K2 expression. Cells with constitutive Cdk2 activity provided by D1K2 are capable of progressing to S phase, where they become octaploid. After a second failed cytokinesis, the tetraploidy checkpoint increases in strength, causing cell cycle arrest.
Once cells have reached G1 with a tetraploid DNA content, the p53-dependent tetraploidy checkpoint is induced. In our model (Fig. 7B), this initial response is sufficient to arrest cells without D1K2 expression. Cells with constitutive Cdk2 activity provided by D1K2 are capable of progressing to S phase, where they become octaploid. After a second failed cytokinesis, the tetraploidy checkpoint increases in strength, causing cell cycle arrest. Together, it appears that overactive Cdk2 produces competing effects in terms of the generation of polyploid cells when treated with spindle poisons. However, the preventive effects offered by a strengthened spindle assembly checkpoint are temporary, allowing a weakened tetraploidy checkpoint to be the dominant phenotype observed.
Materials and Methods
Cell culture and treatments
Tumor-derived cell lines were obtained as described previously (Corsino et al., 2007) and maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. MCF10A cell lines were maintained in the suggested (Soule et al., 1990) 50/50 mixture of Dulbecco’s modified Eagle’s medium and Ham’s F12 medium supplemented with 5% horse serum, 20 ng/ml EGF, 100 ng/ml cholera toxin, 10 µg/ml insulin, and 500 ng/ml hydrocortisone (Sigma-Aldrich, St. Louis, MO). The parental MCF10A cell line was obtained from ATCC (Manassas, VA). Nocodazole, paclitaxel, CVT313 (238803; Calbiochem, Billerica, MA), and cytochalasin-B (C6762; Sigma-Aldrich) were dissolved in DMSO.
Stable cell line generation
MCF10A Hygro, D1K2, and D1K2(KD) cell lines were generated using the pBabe vector system as described previously (Chytil et al., 2004). Stable knockdown cell lines were generated by co-transfecting shRNA constructs (Thermo Scientific, Waltham, MA) along with viral packaging plasmids PMD2G and PsPax2 obtained from Addgene (Cambridge, MA) into the 293T cell line using Lipofectamine Reagent (Invitrogen, Grand Island, NY). Medium from the transfected 293T cell line was then used to infect the target cell line, which was subsequently selected using 10 µg/ml Puromycin.
Propidium iodide staining and flow cytometry
Following treatment, cells were removed from the plate by trypsin digestion, washed three times with PBS, and resuspended in a solution containing 3.4 mM sodium citrate, 75 µM propidium iodide, 0.1% Triton X-100 and 5 µg/ml RNase A. Samples were analyzed on a Becton Dickinson FACSort flow cytometer. A total of 10,000 cells were counted for each sample. Data were analyzed using the ModFit program (Verity Software House Inc., Topsham, ME).
Cell sorting
Cells were trypsinized and resuspended in medium containing 5 µM DyeCycle Violet DNA dye (Invitrogen) and incubated at 37°C for 30 minutes. Cells were then incubated on ice until sorting was complete. Cells were sorted on a FACSAria instrument from BD Bioscience (San Jose, CA) according to DNA content based on absorbance of the dye.
Immunofluorescence microscopy
D1K2 tumor cells for immunofluorescence studies were plated onto glass coverslips in 6-well plates. After treatment, the cells were fixed with a solution containing 90% methanol and 10% MeS buffer (100 mM MeS, pH 6.9, 1 mM EGTA and 1 mM MgCl2). The coverslips were subsequently incubated with antibody buffer [5% goat serum in phosphate-buffered saline (PBS)] in a humidified chamber for 1 hour. Primary antibody staining was performed using an antibody for γ-Tubulin (sc-17787; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) at a 1∶200 dilution in antibody buffer for 2 hours. Following primary antibody incubation, the coverslips were washed three times with PBS, and incubated with a goat anti-mouse Cy3 secondary antibody (81-6515; Zymed, Carlsbad, CA) for 1 hour at a 1∶200 dilution in antibody buffer. Following three additional washes with PBS, coverslips were mounted onto slides with Vectashield + 4′,6-diamidino-2-phenylindole (DAPI; H-1200; Vector Laboratories, Burlingame, CA) to visualize nuclei. Images were captured using an upright microscope (Axioplan2; Zeiss, Thornwood, NY), and processed using Openlab 5.5 Improvision software.
MCF10A cell lines intended for immunofluorescence were plated in Petri dishes and treated with DMSO, paclitaxel, CVT313, or paclitaxel with CVT313 for 72 hours. Cells were then trypsinized, washed with PBS, and fixed with 2% paraformaldehyde in PBS. Cells were then quenched with 0.2% Tween 20 and 50 mM NH4Cl in PBS. After washing with PBS, cells were resuspended in antibody buffer for 1 hour, at which time an antibody directed towards pericentrin (ab4448, Abcam plc, Cambridge, MA) was added at a 1∶200 dilution for 2 hours. Cells were pelleted, washed with PBS, and resuspended in antibody buffer containing a 1∶200 dilution of a goat anti-rabbit Cy3 antibody (81-6115, Zymed) for 1 hour. After PBS washing, the cells were resuspended in PBS containing 0.1% Triton X-100 and spread evenly across coverslips. Upon drying, coverslips were mounted onto slides with Vectashield + DAPI to visualize nuclei and centrosomes.
Analysis of metaphase spreads
Preparation of metaphase spreads were performed according to basic protocol 2 from Current Protocols in Cell Biology (Bayani, 2004). The chromosomes were stained with DAPI and visualized and photographed using fluorescence microscopy as described above.
Immunoblot analysis
Immunoblotting was performed as described (Law et al., 2002), employing antibodies to NPM, p-NPM[T199], p-Rb 780, 795, or 807/811 (3542; 3541; 9307; 9301; 9308; Cell Signaling Technology, Inc., Danvers, MA), p-Rb 249/252 (44-584; Bio-Source International, Grand Island, NY), FLAG (F-3165; Sigma-Aldrich), Mad2 (Ab70385; Abcam plc), Actin, Cdk2, Rb, p21, or p53 (sc-1616-R; sc-163; sc-7905; sc-3997; sc-100; Santa Cruz).
Growth curves
For growth curves constructed using cell counts, cell lines were treated for 72 hours and then seeded at 10,000 cells/well in six-well plates. Cells were subsequently trypsinized and counted in triplicate (three separate wells) daily using a hemocytometer. For growth curves constructed using Crystal Violet staining, cell lines were seeded at 2500 cells/well in 24-well plates. Cells were quantitated daily by fixation using 4% paraformaldehyde in PBS for 20 minutes, staining with 0.1% Crystal Violet (C0775; Sigma-Aldrich) in 30/70 methanol/water for 30 minutes, followed by five washes using water. After drying overnight, retained Crystal Violet was eluted using methanol and quantified by measuring absorbance at 590 nm on a DU800 spectrophotometer (Beckman Coulter Inc., Brea, CA). Values were normalized to the absorbance observed on day 1. Samples producing absorbance higher than 2 were diluted with additional methanol to lower absorbance readings below 2. Plotted values for these samples are back-calculated.
Paclitaxel washout experiment
Cells were plated at 200,000 cells/p100 dish. The number of cells in each dish was counted in replicate plates before treatment with paclitaxel, after 72 hours of treatment, or after 72 hours of treatment followed by a 48-hour washout period, using a hemocytometer.
Tetraploid cell fate analysis
Cells were plated at a single-cell density in 96-well plates. After incubating overnight, they were treated with 4.5 µg/ml cytochalasin-B for 72 hours, followed by drug washout. Cells were visually inspected to identify multinucleate cells (>2 nuclei) and their fate was determined by re-examination 1 week after drug washout.
[3H]Thymidine incorporation
[3H]Thymidine incorporation assays were carried out as previously described (Corsino et al., 2007; Corsino et al., 2008), using a 2-hour [3H]thymidine pulse.
Supplementary Material
Acknowledgments
We thank Mr Brian Gray of the Rocky Point Lab, Gainesville, FL, for performing karyotype analyses and Craig Moneypenny of the University of Florida Interdiscplinary Center for Biotechnology Research for performing the cell sorting. We also thank Dr Thomas Rowe, Heather Brown and Serena Giovinazzi for helpful discussions, and Dr Scott Grieshaber for the gift of the Mad2 antibody.
Footnotes
Author contributions
S.J. designed and performed research as well as wrote the manuscript. B.D. performed research. P.C., M.L., P.N. and B.L. designed and performed research.
Funding
This work was supported by National Institutes of Health [grant number R01-CA93651 to B.L.]; Florida Department of Health [grant numbers 07BB-8 and 09BB-10 to B.L.]; Susan G. Komen for the Cure [grant number KG080510 to B.L.]; the University of Florida College of Medicine Alumni Fellowship (to S.J.); and the National Institutes of Health/National Cancer Institute T32 Training Grant in Cancer Biology [grant number CA09126 to P.C.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.117382/-/DC1
References
- Adon A. M., Zeng X., Harrison M. K., Sannem S., Kiyokawa H., Kaldis P., Saavedra H. I. (2010). Cdk2 and Cdk4 regulate the centrosome cycle and are critical mediators of centrosome amplification in p53-null cells. Mol. Cell. Biol. 30, 694–710 10.1128/MCB.00253-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed A. A., Mills A. D., Ibrahim A. E., Temple J., Blenkiron C., Vias M., Massie C. E., Iyer N. G., McGeoch A., Crawford R.et al. (2007). The extracellular matrix protein TGFBI induces microtubule stabilization and sensitizes ovarian cancers to paclitaxel. Cancer Cell 12, 514–527 10.1016/j.ccr.2007.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen P. R., Lohez O. D., Lacroix F. B., Margolis R. L. (2001). Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol. Biol. Cell 12, 1315–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balczon R., Bao L., Zimmer W. E., Brown K., Zinkowski R. P., Brinkley B. R. (1995). Dissociation of centrosome replication events from cycles of DNA synthesis and mitotic division in hydroxyurea-arrested Chinese hamster ovary cells. J. Cell Biol. 130, 105–115 10.1083/jcb.130.1.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayani J., Squire J. A.(2004). Preparation of Cytogenetic Specimens from Tissue Samples. Hoboken, NJ: John Wiley and Sons; [DOI] [PubMed] [Google Scholar]
- Chen Z., Indjeian V. B., McManus M., Wang L., Dynlacht B. D. (2002). CP110, a cell cycle-dependent CDK substrate, regulates centrosome duplication in human cells. Dev. Cell 3, 339–350 10.1016/S1534-5807(02)00258-7 [DOI] [PubMed] [Google Scholar]
- Chen Y. X., Wang Y., Fu C. C., Diao F., Song L. N., Li Z. B., Yang R., Lu J. (2010). Dexamethasone enhances cell resistance to chemotherapy by increasing adhesion to extracellular matrix in human ovarian cancer cells. Endocr. Relat. Cancer 17, 39–50 10.1677/ERC-08-0296 [DOI] [PubMed] [Google Scholar]
- Chin G. M., Herbst R. (2006). Induction of apoptosis by monastrol, an inhibitor of the mitotic kinesin Eg5, is independent of the spindle checkpoint. Mol. Cancer Ther. 5, 2580–2591 10.1158/1535-7163.MCT-06-0201 [DOI] [PubMed] [Google Scholar]
- Chytil A., Waltner–Law M., West R., Friedman D., Aakre M., Barker D., Law B. (2004). Construction of a cyclin D1-Cdk2 fusion protein to model the biological functions of cyclin D1-Cdk2 complexes. J. Biol. Chem. 279, 47688–47698 10.1074/jbc.M405938200 [DOI] [PubMed] [Google Scholar]
- Collecchi P., Santoni T., Gnesi E., Giuseppe Naccarato A., Passoni A., Rocchetta M., Danesi R., Bevilacqua G. (2000). Cyclins of phases G1, S and G2/M are overexpressed in aneuploid mammary carcinomas. Cytometry 42, 254–260 [DOI] [PubMed] [Google Scholar]
- Corsino P., Davis B., Law M., Chytil A., Forrester E., Nørgaard P., Teoh N., Law B. (2007). Tumors initiated by constitutive Cdk2 activation exhibit transforming growth factor beta resistance and acquire paracrine mitogenic stimulation during progression. Cancer Res. 67, 3135–3144 10.1158/0008-5472.CAN-06-3815 [DOI] [PubMed] [Google Scholar]
- Corsino P. E., Davis B. J., Nørgaard P. H., Parker N. N., Law M., Dunn W., Law B. K. (2008). Mammary tumors initiated by constitutive Cdk2 activation contain an invasive basal-like component. Neoplasia 10, 1240–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deangelis P., Stokke T., Smedshammer L., Lothe R., Meling G., Rofstad M., Chen Y., Clausen O. (1993). P53 expression is associated with a high-degree of tumor DNA aneuploidy and incidence of p53 gene mutation, and is localized to the aneuploid component in colorectal carcinomas. Int. J. Oncol. 3, 305–312 [DOI] [PubMed] [Google Scholar]
- Duensing A., Liu Y., Tseng M., Malumbres M., Barbacid M., Duensing S. (2006). Cyclin-dependent kinase 2 is dispensable for normal centrosome duplication but required for oncogene-induced centrosome overduplication. Oncogene 25, 2943–2949 10.1038/sj.onc.1209310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan A. W., Hickey R. D., Paulk N. K., Culberson A. J., Olson S. B., Finegold M. J., Grompe M. (2009). Ploidy reductions in murine fusion-derived hepatocytes. PLoS Genet. 5, e1000385 10.1371/journal.pgen.1000385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T., Bandi M., Nitta M., Ivanova E. V., Bronson R. T., Pellman D. (2005). Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 437, 1043–1047 10.1038/nature04217 [DOI] [PubMed] [Google Scholar]
- Ganem N. J., Godinho S. A., Pellman D. (2009). A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278–282 10.1038/nature08136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Hanse E. A., Nelsen C. J., Goggin M. M., Anttila C. K., Mullany L. K., Berthet C., Kaldis P., Crary G. S., Kuriyama R., Albrecht J. H. (2009). Cdk2 plays a critical role in hepatocyte cell cycle progression and survival in the setting of cyclin D1 expression in vivo. Cell Cycle 8, 2802–2809 10.4161/cc.8.17.9465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao X., Zhou Z., Ye S., Zhou T., Lu Y., Ma D., Wang S. (2010). Effect of Mad2 on paclitaxel-induced cell death in ovarian cancer cells. J. Huazhong Univ. Sci. Technolog. Med. Sci. 30, 620–625 10.1007/s11596-010-0553-y [DOI] [PubMed] [Google Scholar]
- Hernando E., Nahlé Z., Juan G., Diaz–Rodriguez E., Alaminos M., Hemann M., Michel L., Mittal V., Gerald W., Benezra R.et al. (2004). Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature 430, 797–802 10.1038/nature02820 [DOI] [PubMed] [Google Scholar]
- Hinchcliffe E. H., Li C., Thompson E. A., Maller J. L., Sluder G. (1999). Requirement of Cdk2-cyclin E activity for repeated centrosome reproduction in Xenopus egg extracts. Science 283, 851–854 10.1126/science.283.5403.851 [DOI] [PubMed] [Google Scholar]
- Janssen A., Kops G. J., Medema R. H. (2009). Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc. Natl. Acad. Sci. USA 106, 19108–19113 10.1073/pnas.0904343106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeche L., Compton D. A. (2012). Checkpoint-independent stabilization of kinetochore-microtubule attachments by Mad2 in human cells. Curr. Biol. 22, 638–644 10.1016/j.cub.2012.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywicka–Racka A., Sluder G. (2011). Repeated cleavage failure does not establish centrosome amplification in untransformed human cells. J. Cell Biol. 194, 199–207 10.1083/jcb.201101073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanni J. S., Jacks T. (1998). Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol. Cell. Biol. 18, 1055–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law B. K., Chytil A., Dumont N., Hamilton E. G., Waltner–Law M. E., Aakre M. E., Covington C., Moses H. L. (2002). Rapamycin potentiates transforming growth factor beta-induced growth arrest in nontransformed, oncogene-transformed, and human cancer cells. Mol. Cell. Biol. 22, 8184–8198 10.1128/MCB.22.23.8184-8198.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M., Sotillo R., Santamaría D., Galán J., Cerezo A., Ortega S., Dubus P., Barbacid M. (2004). Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118, 493–504 10.1016/j.cell.2004.08.002 [DOI] [PubMed] [Google Scholar]
- Matsumoto Y., Hayashi K., Nishida E. (1999). Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Curr. Biol. 9, 429–432 10.1016/S0960-9822(99)80191-2 [DOI] [PubMed] [Google Scholar]
- Minn A. J., Boise L. H., Thompson C. B. (1996). Expression of Bcl-xL and loss of p53 can cooperate to overcome a cell cycle checkpoint induced by mitotic spindle damage. Genes Dev. 10, 2621–2631 10.1101/gad.10.20.2621 [DOI] [PubMed] [Google Scholar]
- Nelsen C. J., Kuriyama R., Hirsch B., Negron V. C., Lingle W. L., Goggin M. M., Stanley M. W., Albrecht J. H. (2005). Short term cyclin D1 overexpression induces centrosome amplification, mitotic spindle abnormalities, and aneuploidy. J. Biol. Chem. 280, 768–776 [DOI] [PubMed] [Google Scholar]
- Nishimura T., Takahashi M., Kim H. S., Mukai H., Ono Y. (2005). Centrosome-targeting region of CG-NAP causes centrosome amplification by recruiting cyclin E-cdk2 complex. Genes Cells 10, 75–86 10.1111/j.1365-2443.2005.00816.x [DOI] [PubMed] [Google Scholar]
- Nowell P. C. (1976). The clonal evolution of tumor cell populations. Science 194, 23–28 10.1126/science.959840 [DOI] [PubMed] [Google Scholar]
- Okuda M., Horn H. F., Tarapore P., Tokuyama Y., Smulian A. G., Chan P. K., Knudsen E. S., Hofmann I. A., Snyder J. D., Bove K. E.et al. (2000). Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell 103, 127–140 10.1016/S0092-8674(00)00093-3 [DOI] [PubMed] [Google Scholar]
- Sherr C. J. (1996). Cancer cell cycles. Science 274, 1672–1677 10.1126/science.274.5293.1672 [DOI] [PubMed] [Google Scholar]
- Sluder G., Nordberg J. J. (2004). The good, the bad and the ugly: the practical consequences of centrosome amplification. Curr. Opin. Cell Biol. 16, 49–54 10.1016/j.ceb.2003.11.006 [DOI] [PubMed] [Google Scholar]
- Sotillo R., Hernando E., Díaz–Rodríguez E., Teruya–Feldstein J., Cordón–Cardo C., Lowe S. W., Benezra R. (2007). Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 11, 9–23 10.1016/j.ccr.2006.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soule H. D., Maloney T. M., Wolman S. R., Peterson W. D., Jr, Brenz R., McGrath C. M., Russo J., Pauley R. J., Jones R. F., Brooks S. C. (1990). Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 50, 6075–6086 [PubMed] [Google Scholar]
- Stewart Z. A., Leach S. D., Pietenpol J. A. (1999). p21(Waf1/Cip1) inhibition of cyclin E/Cdk2 activity prevents endoreduplication after mitotic spindle disruption. Mol. Cell. Biol. 19, 205–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudo T., Nitta M., Saya H., Ueno N. T. (2004). Dependence of paclitaxel sensitivity on a functional spindle assembly checkpoint. Cancer Res. 64, 2502–2508 10.1158/0008-5472.CAN-03-2013 [DOI] [PubMed] [Google Scholar]
- Sweeney K. J., Swarbrick A., Sutherland R. L., Musgrove E. A. (1998). Lack of relationship between CDK activity and G1 cyclin expression in breast cancer cells. Oncogene 16, 2865–2878 10.1038/sj.onc.1201814 [DOI] [PubMed] [Google Scholar]
- Tokuyama Y., Horn H. F., Kawamura K., Tarapore P., Fukasawa K. (2001). Specific phosphorylation of nucleophosmin on Thr(199) by cyclin-dependent kinase 2-cyclin E and its role in centrosome duplication. J. Biol. Chem. 276, 21529–21537 10.1074/jbc.M100014200 [DOI] [PubMed] [Google Scholar]
- Trielli M. O., Andreassen P. R., Lacroix F. B., Margolis R. L. (1996). Differential Taxol-dependent arrest of transformed and nontransformed cells in the G1 phase of the cell cycle, and specific-related mortality of transformed cells. J. Cell Biol. 135, 689–700 10.1083/jcb.135.3.689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ussar S., Voss T. (2004). MEK1 and MEK2, different regulators of the G1/S transition. J. Biol. Chem. 279, 43861–43869 10.1074/jbc.M406240200 [DOI] [PubMed] [Google Scholar]
- Weaver B. A., Silk A. D., Montagna C., Verdier–Pinard P., Cleveland D. W. (2007). Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 11, 25–36 10.1016/j.ccr.2006.12.003 [DOI] [PubMed] [Google Scholar]
- Westendorf J. M., Rao P. N., Gerace L. (1994). Cloning of cDNAs for M-phase phosphoproteins recognized by the MPM2 monoclonal antibody and determination of the phosphorylated epitope. Proc. Natl. Acad. Sci. USA 91, 714–718 10.1073/pnas.91.2.714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X., Shaikh F. Y., Harrison M. K., Adon A. M., Trimboli A. J., Carroll K. A., Sharma N., Timmers C., Chodosh L. A., Leone G.et al. (2010). The Ras oncogene signals centrosome amplification in mammary epithelial cells through cyclin D1/Cdk4 and Nek2. Oncogene 29, 5103–5112 10.1038/onc.2010.253 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.