

Summary
The prototypical transient receptor potential (TRP) channel is the major light-sensitive, and Ca2+-permeable channel in the microvillar photoreceptors of Drosophila. TRP channels are activated following hydrolysis of phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2] by the key effector enzyme phospholipase C (PLC). Mutants lacking TRP channels undergo light-dependent retinal degeneration, as a consequence of the reduced Ca2+ influx. It has been proposed that degeneration is caused by defects in the Ca2+-dependent visual pigment cycle, which result in accumulation of toxic phosphorylated metarhodopsin–arrestin complexes (MPP–Arr2). Here we show that two interventions, which prevent accumulation of MPP–Arr2, namely rearing under red light or eliminating the C-terminal rhodopsin phosphorylation sites, failed to rescue degeneration in trp mutants. Instead, degeneration in trp mutants reared under red light was rescued by mutation of PLC. Degeneration correlated closely with the light-induced depletion of PtdIns(4,5)P2 that occurs in trp mutants due to failure of Ca2+-dependent inhibition of PLC. Severe retinal degeneration was also induced in the dark in otherwise wild-type flies by overexpression of a bacterial PtdInsPn phosphatase (SigD) to deplete PtdIns(4,5)P2. In degenerating trp photoreceptors, phosphorylated Moesin, a PtdIns(4,5)P2-regulated membrane–cytoskeleton linker essential for normal microvillar morphology, was found to delocalize from the rhabdomere and there was extensive microvillar actin depolymerisation. The results suggest that compromised light-induced Ca2+ influx, due to loss of TRP channels, leads to PtdIns(4,5)P2 depletion, resulting in dephosphorylation of Moesin, actin depolymerisation and disintegration of photoreceptor structure.
Key words: Moesin; Actin; PtdIns(4,5)P2; TRP channels; Rhodopsin
Introduction
With 28 mammalian isoforms, the transient receptor potential (TRP) family is one of the largest and most diverse ion channel families (Montell, 2005; Ramsey et al., 2006). TRP channels were discovered in Drosophila photoreceptors (Hardie and Minke, 1992; Montell and Rubin, 1989), where they are the dominant light-sensitive channel and defining representative of the TRPC subfamily. The channels are present within ∼30,000 tightly packed microvilli, forming a light-guiding ‘rhabdomere’, and are activated by a canonical G-protein-coupled signalling cascade involving rhodopsin (Rh1, encoded by ninaE), Gq protein (Gαq) and phospholipase C (PLC, encoded by norpA). PLC hydrolyses the membrane phospholipid, phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2], leading, by a still unresolved mechanism, to activation of the light-sensitive channels (Hardie, 2012; Katz and Minke, 2009; Montell, 2012). Drosophila TRP is highly Ca2 selective and responsible for massive Ca2+ influx during the response to light (Hardie, 1996; Hardie and Minke, 1992; Peretz et al., 1994b). Fly photoreceptors also express a second light-sensitive channel, known as TRP-like (TRPL), with ∼10 times lower selectivity for Ca2+. TRPL channels mediate the residual response in trp mutants, but only ∼5% of the wild-type light-sensitive current (Niemeyer et al., 1996; Phillips et al., 1992; Reuss et al., 1997).
Numerous human diseases have been associated with TRP dysfunction, including neurodegenerative diseases, renal and cardiovascular disorders (for reviews, see Everett, 2011; Inoue et al., 2006; Nilius et al., 2007; Selvaraj et al., 2009; Zhu et al., 2011). Light-dependent retinal degeneration in Drosophila trp mutants was first reported 40 years ago (Cosens and Perry, 1972), and later hypothesised to be due to impaired Ca2+ influx (Liu et al., 2007; Wang et al., 2005). In Drosophila, retinal degeneration due to reduced Ca2+ levels is usually attributed to defects in the Ca2+-dependent visual pigment cycle. For example, defective rhodopsin recycling is believed to underlie retinal degeneration in mutants of genes such as rdgC, which encodes a Ca2+-dependent rhodopsin phosphatase (Byk et al., 1993; Steele et al., 1992), and blind mutants like norpA (lacking PLC), which have minimal light-induced Ca2+ influx (Alloway et al., 2000; Byk et al., 1993). These mutants accumulate toxic levels of hyper-phosphorylated metarhodopsin–arrestin (MPP–Arr2) complexes, which undergo clathrin-mediated endocytosis and are proposed to trigger cell death (Alloway et al., 2000; Kiselev et al., 2000) (see Discussion and Fig. 9 for further details).
Fig. 9.
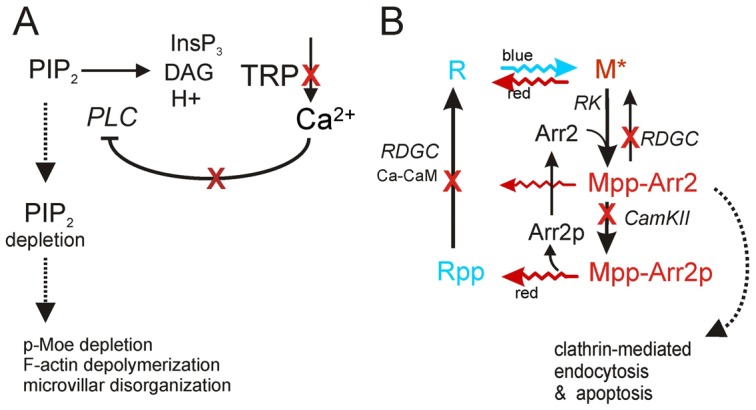
Mechanisms underlying retinal degeneration. (A) TRP channels are activated downstream of PtdIns(4,5)P2 hydrolysis by PLC. Loss of TRP channels in trp mutants eliminates the Ca2+ influx, which normally inhibits PLC (norpA gene) via Ca2+-dependent PKC (Gu et al., 2005) (red crosses indicate Ca2+ influx and sites of Ca2+-dependent feedback disrupted by the trp mutation). As a result, PtdIns(4,5)P2 in the microvilli is depleted even by red light, leading to degeneration. Downstream mechanisms include p-Moe dephosphorylation, possible defects in other actin organizing proteins, F-actin depolymerisation and microvillar disorganization. (B) Visual pigment cycle: rhodopsin (R) is photoisomerised by blue light to metarhodopsin (M). M is phosphorylated (MPP) by rhodopsin kinase (RK) and inactivated by binding to arrestin (Arr2), which is phosphorylated by CaMKII. Mpp is photoreisomerised to Rpp by red light, which prevents any possible accumulation of MPP–Arr2. Rpp and MPP are dephosphorylated by a Ca2+/CaM-dependent phosphatase encoded by rdgC. In low Ca2+, Arr2 is not phosphorylated, whilst RPP (or MPP) cannot be dephosphorylated (red crosses). Under white or blue light this results in accumulation of MPP–Arr2, which is a target for clathrin-mediated endocytosis. This is believed to underlie degeneration in norpA, rdgC and some other blind mutants (Alloway et al., 2000; Kiselev et al., 2000). This may also contribute to degeneration in trp mutants reared under white light (Wang et al., 2005). However because clathrin-mediated endocytosis is also PtdIns(4,5)P2 dependent this requires further investigation.
Because of the reduced Ca2+ influx in trp mutants, accumulation of MPP–Arr2 complexes has accordingly also been proposed as a mechanism of trp retinal degeneration (Wang et al., 2005). However, Ca2+ influx via TRP channels has many other roles in phototransduction; amongst these, it feeds back negatively to inhibit PLC. Consequently, in mutants lacking TRP channels, or following experimental manipulations that prevent Ca2+ influx, the unattenuated light-induced PLC activity rapidly depletes virtually the entire rhabdomeric pool of PtdIns(4,5)P2 (Hardie et al., 2001). This loss of PtdIns(4,5)P2 gives rise to the characteristic ‘transient receptor potential’ phenotype, whereby the response to light decays to baseline during continuous light in trp mutants (Cosens and Manning, 1969; Minke, 1982). This behaviour provides the experimenter with a unique ability to accurately control the level of PtdIns(4,5)P2 in a defined subcellular compartment in vivo in completely intact animals (Hardie et al., 2004; Hardie et al., 2001).
Because PtdIns(4,5)P2 is a key regulator of many vital cellular processes (Di Paolo and De Camilli, 2006; Hilgemann et al., 2001; Payrastre et al., 2001; Rohacs, 2009; Yin and Janmey, 2003), in the present study, we asked whether depletion of PtdIns(4,5)P2 might underlie retinal degeneration in trp mutants. We first showed that rearing flies under red light, which prevents accumulation of MPP–Arr2 complexes, rescued degeneration in norpA but not in trp. However, when PtdIns(4,5)P2 hydrolysis was genetically blocked in trp mutants by the norpA mutation, degeneration under red light was completely rescued. Furthermore, we demonstrated close quantitative correlation between retinal degeneration and the extent of PtdIns(4,5)P2 depletion in trp retina, whilst degeneration could be induced in the dark by overexpression of a bacterial PtdInsPn phosphatase. Lastly, we showed that Moesin, a PtdIns(4,5)P2-regulated protein required for microvillar formation and maintenance (Chorna-Ornan et al., 2005; Karagiosis and Ready, 2004), was severely affected in trp retinae exposed to light, whilst microvillar actin was extensively depolymerized, suggesting a possible molecular basis for the degeneration.
Results
The Ca2+-selective TRP channel is the major light-sensitive channel in Drosophila, and trp mutants have severely reduced light-induced Ca2+ influx (Hardie, 1996; Hardie and Minke, 1992; Peretz et al., 1994a). Another mutant with compromised Ca2+ influx is norpA, which lacks PLC and has no response to light. Both norpA and trp flies undergo severe light-dependent retinal degeneration, and toxic accumulation of stable MPP–Arr2 complexes has been proposed to underlie degeneration in both (Alloway et al., 2000; Wang et al., 2005). To determine the extent of similarity between degeneration in norpA and trp flies, we characterized and compared degeneration patterns using null alleles (norpAP24 and trp343) of both mutants.
Light-dependent retinal degeneration in trp and norpA mutants
Optical neutralization, allowing visualization of rhabdomeres in intact fly heads, was used to monitor degeneration in red-eyed flies reared under continuous white LED illumination. Wild-type flies showed no degeneration over 30 days under these conditions, validating our continuous illumination protocol for monitoring degeneration. In agreement with Cosens and Perry's original findings, trp mutants degenerated under the same conditions (Cosens and Perry, 1972). The degeneration was characterized by the outer rhabdomeres (R1–6) becoming gradually dimmer and less defined in form, reaching an apparent end point within 2 weeks, when they could no longer be discerned (Fig. 1). The central rhabdomere, R7, which responds only to ultraviolet light (Feiler et al., 1992), was always spared, and served as an internal control. norpA mutant flies degenerated with a similar time course to trp mutant flies; however, examination of the degenerating photoreceptors with transmission electron microscopy (TEM) revealed differences. In trp mutants, the degenerating photoreceptors had a severely disorganized and vesiculated microvillar structure giving the rhabdomeres a characteristic ‘frothy’ appearance (Fig. 1C; Fig. 3). By contrast in norpA mutant flies, although rhabdomeres became short and fragmented, any remaining microvilli still retained their distinctive morphology (Fig. 1C).
Fig. 1.

Retinal degeneration under white light. (A) Time course of retinal degeneration in wild type, norpAP24, and two independent trp mutants (trp343 and trpCM) under continuous white light (n = 5–7, means ± s.e.m.). (B) Representative optical neutralisation images of wild-type and mutant retinae after the indicated number of days (d) under continuous white light (WL) or darkness (Dark). (i,ii) Wild-type strain, Oregon R; (iii,iv) norpAP24; (v,vi) trp343 mutant. Scale bar: ∼16 µm. (C) Transmission electron microscopy (i,ii) Wild-type retina shows normal morphology after 5 days continuous white illumination. (iii,iv) norpAP24 retina exposed to white light for 5 days has short microvilli, but microvillar structure is still largely intact; (v,vi) trpCM retina exposed to white light for 5 days shows highly disintegrated microvilli giving a frothy appearance (black arrow). The rhabdomere of the UV-sensitive central photoreceptor (R7) in both mutants is spared from degeneration. Scale bars: ∼2 µm (Ci,iii,v); ∼500 nm (Cii,iv,vi).
Fig. 3.

Ultrastructural pathology of the degenerating photoreceptors of trp343 mutants. (A) Wild-type dark control (∼28 days). (B) Intact photoreceptors of trp343 mutants maintained in the dark for ∼30 days. (C–H) trp343 mutants exposed to red light for different time periods. (C) 7 days exposure. (D) 30 days exposure. trp retina shows disrupted rhabdomere structure of peripheral photoreceptors. R7, the UV-sensitive photoreceptor, is intact. Blue arrowheads indicate the cell junctions. (E,F) 7 days exposure. Degenerating trp photoreceptors with multivesicular bodies (MVBs), endosomes and a lysosome (L). (G) 14 days exposure. Photoreceptor cell with completely fragmented microvilli being engulfed by a pigment cell (P), identified by their homogenous, granular appearance. Some phagocytosed material (*) can also be observed, which is possibly the remnant of a neighbouring degenerated photoreceptor cell. (H) 30 days exposure. Large vacuoles have appeared in the photoreceptors, but the cell junctions (blue arrow) are intact. N, nucleus; R, rhabdomeres; G, Golgi complex. Scale bars: ∼2 µm (A–E,G,H); ∼500 nm (F).
Mutant flies kept in darkness for 4 weeks showed no degeneration, confirming that the degeneration seen in trp and norpA mutants is light dependent. As both norpA and trp mutants have compromised Ca2+ influx when exposed to light, the results are consistent with reduced Ca2+ playing an important role in the degeneration pathway; however, the ultrastructural differences hint at different downstream mechanisms.
Blocking MPP–Arr2 accumulation fails to rescue trp retinal degeneration
Both norpA and trp mutant retinae were previously suggested to degenerate due to defects in the Ca2+-dependent visual pigment cycle, resulting in toxic accumulation of MPP–Arr2 complexes (Alloway et al., 2000; Wang et al., 2005). A hallmark of this degeneration is that it can be rescued by exposing the flies to long wavelength light, which photoreconverts metarhodopsin back to rhodopsin thus preventing accumulation of MPP–Arr2 (Kiselev et al., 2000). We tested whether we could prevent degeneration in norpA and trp mutants in the same way by rearing flies under long wavelength (red) light. Whereas norpA retinae no longer showed any sign of degeneration under red light, those of trp flies continued to degenerate, albeit with a slightly slower time course (Fig. 2). Degeneration followed a similar time course in flies carrying the two independent alleles of trp (trpCM and trp343) and in both red-eyed and white-eyed backgrounds (Fig. 2A). This observation does not rule out the involvement of the MPP–Arr2 pathway in trp retinal degeneration under white light; however, it indicates that an alternate mechanism exists in trp mutant retinae, which induces degeneration even when MPP–Arr2 accumulation is prevented.
Fig. 2.

Effect of expressing phosphorylation-deficient ninaE (ninaEΔ356) and red light on retinal degeneration in trp and norpA mutants. (A) Time course of retinal degeneration in wild-type, norpAP24 and two independent trp (trpCM and trp343) mutants under continuous red light. Also shown is the time course of degeneration in white-eyed trp343 mutants (evaluated by counting the fraction of flies with intact deep pseudopupils, and normalised to the same scale). Exposure to red light rescues retinal degeneration in norpA but not in trp flies. (B) Representative optical neutralization images of red-eyed wild-type (i), norpAP24 (ii), and trp343 (iii) retinae. (C,D) Expression of ninaEΔ356 in an otherwise ninaE null (ninaEEI17) background suppressed norpA retinal degeneration, but failed to rescue trp degeneration. (C) Time course of retinal degeneration in all mutants under continuous white light (means ± s.e.m., n>15). (D) Representative optical neutralisation images of trp (i), norpAP24 (ii), trpCM, ninaEEI17, P-ninaEΔ356 (iii), norpAP24;;ninaEEI17, P-ninaEΔ356 (iv), ninaEEI17, P-ninaEΔ356 (v) retinae. Scale bars: ∼16 µm.
Another way to prevent accumulation of stable MPP–Arr2 complexes is by eliminating the C-terminal phosphorylation sites on rhodopsin. In norpA or rdgC mutants, expression of such a truncated, phosphorylation-deficient form of rhodopsin (ninaEΔ356) in an otherwise rhodopsin null background suppresses retinal degeneration (Alloway et al., 2000; Kiselev et al., 2000; Vinós et al., 1997). Because red light failed to rescue degeneration in trp mutants, we predicted that the ninaEΔ356 mutation would also fail to suppress degeneration. Indeed, trp, ninaEΔ356 double mutants still showed extensive degeneration under white light, even appearing to degenerate slightly faster than the trp mutant alone (Fig. 2C). As previously reported (Alloway et al., 2000), we confirmed that the ninaEΔ356 mutation substantially rescued degeneration in norpAP24 mutants, although in contrast to the previous report we found that degeneration was not entirely prevented under our conditions. Thus we observed a partial loss of rhabdomere integrity in norpAP24;;ninaEΔ356 double mutants under optical neutralization over a period of 2 weeks, whilst norpAP24 controls degenerated to an end point within 7 days (Fig. 2C). Overall, these findings confirm that there is an alternative mechanism for light-induced retinal degeneration in trp mutant flies that is independent of MPP–Arr2 internalization.
Morphological features of trp retinal degeneration
To understand this previously uncharacterized pathway better, we looked at TEM sections of trp photoreceptors exposed to red light for different durations (7, 14 and 30 days). The photoreceptor cells of control trp mutants, maintained in dark for 30 days, had normal organized microvillar structure similar to that of wild-type dark control (Fig. 3A,B). The large vacuolar structures observed in the dark control trp retinae are the space occupied by collapsed pigment cells, which are very sensitive to fixation conditions. After exposure to red light for 7 days or longer, the photoreceptors showed extensive vesiculation and distortion of the rhabdomeres (Fig. 3C,D), similar to that observed in trp mutants degenerating under white illumination. Multiple membrane bound vesicles could be observed in degenerating photoreceptors, some with heterogeneous electron translucent content (identified as endosomal vesicles), some with electron dense core (lysosomes), and some with multiple vesicles inside (multivesicular bodies or MVBs; Fig. 3E,F). Although there were occurrences of engulfment of degenerated photoreceptor cells by neighbouring pigmented glial cells (Fig. 3G), these were very rare. In most cases, the photoreceptor cell bodies were present with intact cell junctions (zonula adherens: arrows) between photoreceptors and well-defined Golgi (e.g. Fig. 3E). The cell junctions were evident even after ∼30 days of continuous light exposure; however, the cell bodies were full of vacuolar structures with rhabdomeres either highly distorted or completely lost (Fig. 3D,H). The nuclei in the degenerating photoreceptors appeared normal suggesting the cells were not progressing along a classical apoptotic pathway. As previously reported (Cosens and Perry, 1972) photoreceptors at different stages of degeneration could be observed even within a single ommatidium. These results showed that in trp retina, the microvillar rhabdomeres were the most severely affected part. Cell bodies, except for a few instances, were usually still present even after ∼30 days of light exposure.
Phospholipase C activity is required for trp retinal degeneration under red light
Another consequence of reduced Ca2+ influx in trp mutants is severe light-induced depletion of PtdIns(4,5)P2 in the rhabdomeres, due to the lack of Ca2+-dependent inhibition of PLC (Hardie et al., 2001). Because PtdIns(4,5)P2 is an important regulator of many cellular processes, including maintenance of the actin cytoskeleton (Janmey and Lindberg, 2004; Mao and Yin, 2007; Yin and Janmey, 2003) and membrane trafficking (Di Paolo and De Camilli, 2006), we hypothesized that PtdIns(4,5)P2 depletion might underlie degeneration in trp mutants. To test this, we generated norpA;;trp double mutants to prevent PtdIns(4,5)P2 depletion in trp mutants and assayed for retinal degeneration using optical neutralization.
In flies reared under white light, the norpA mutation failed to rescue trp retinal degeneration (i.e. in norpA;;trp double mutants), and even slightly accelerated it (Fig. 4). However, under these conditions the retina would be expected to degenerate due to accumulation of MPP–Arr2 complexes in the absence of Ca2+ influx, just as in single norpA mutants. The slight acceleration of degeneration in norpA;;trp mutants may be explained because there is a small residual Ca2+ influx via TRP channels in norpA null mutants (Hardie et al., 2003), which is blocked in the norpA;;trp double mutant. Strikingly however, the norpA mutation completely rescued trp degeneration under red light, both under optical neutralization as well as in terms of ultrastructural appearance, which appeared essentially wild type (Fig. 4C). This indicates that PLC activity is essential for this alternate mechanism of degeneration in trp mutants, and is consistent with the hypothesis that depletion of microvillar PtdIns(4,5)P2 underlies the degeneration.
Fig. 4.
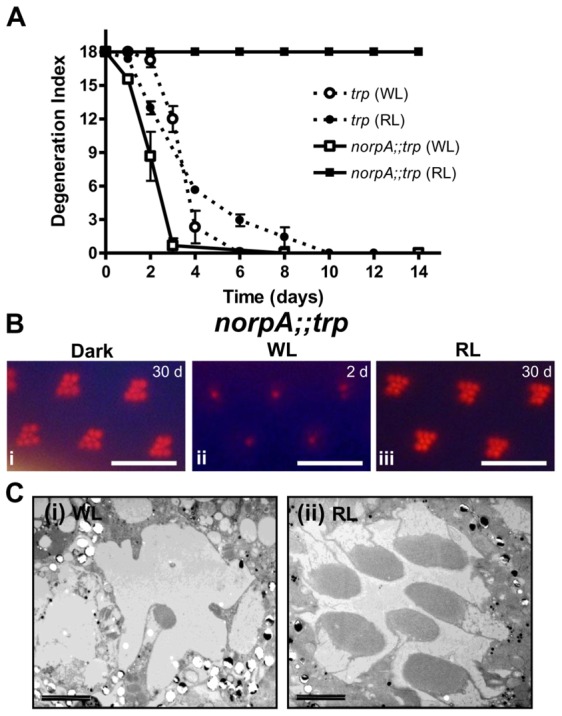
Eliminating PLC activity rescues retinal degeneration induced by red light in trp mutants. (A) Time course of retinal degeneration in norpAP24;;trp343 and trp343 flies under continuous white and red light (n>15, means ± s.e.m.). Retinal degeneration in trp mutants under red light was rescued by blocking PtdIns(4,5)P2 hydrolysis. (B) Representative optical neutralisation images of norpAP24;;trp343 retinae kept in the dark (i), white light (WL; ii) and red light (RL; iii). Scale bars: ∼16 µm. (C) Representative TEM of norpAP24;trpCM mutants reared for 6 days in white light (i); apart from R7, the rhabdomeres have largely degenerated; but under red light (ii) essentially wild-type morphology is preserved. Scale bars: 2 µm.
Intensity-dependent PtdIns(4,5)P2 depletion and retinal degeneration in trp mutants
PtdIns(4,5)P2 depletion in trp mutants manifests itself in a complete decay of the response to light during continuous illumination (hence the name ‘transient receptor potential’ channel). Following the decay, the photoreceptors are profoundly unresponsive (refractory), before recovering sensitivity as PtdIns(4,5)P2 is resynthesized in the dark over a period of 1–2 minutes (Fig. 5A; Hardie et al., 2001; Hardie et al., 2004). This behaviour is steeply intensity dependent, and under dim light PtdIns(4,5)P2 is not depleted in trp mutants, while the light response is maintained (Hardie et al., 2001). It was therefore important to determine whether the red light that induced retinal degeneration in trp mutants was sufficient to deplete PtdIns(4,5)P2.
Fig. 5.

PtdIns(4,5)P2 depletion and retinal degeneration in trp mutants. (A) ERG recording from a trp mutant using a standard ERG set up. trp mutants show response inactivation to 200 ms dim test flashes (arrowheads) after prolonged illumination of 30 s (bar), reflecting PtdIns(4,5)P2 depletion (Hardie et al., 2001). The response gradually recovered, representing regeneration of PtdIns(4,5)P2. (B,C) PtdIns(4,5)P2 depletion in trp mutants under degenerative condition. (B) Representative traces from a wild type, and a trp mutant before (dark-adapted) and ∼5 s after exposure to the red LED in a light box for 30 minutes. (Ci) ERG amplitude response to a test flash as a function of duration of exposure to red LED, normalized to dark-adapted response prior to exposure (V/Vmax). 30 mins exposure caused near complete response inactivation (PtdIns(4,5)P2 depletion) in trp mutants but not in wild-type flies. (Cii) Response recovery after 30 mins of red light exposure (n = 5, means ± s.e.m.). (D) Intensity dependence of response inactivation (V/Vmax), reflecting PtdIns(4,5)P2 depletion (i), and retinal degeneration (ii) in trp mutants. Severity of PtdIns(4,5)P2 depletion (means ± s.e.m., n indicated in brackets) and retinal degeneration in trp (n≥7, means ± s.e.m.) had similar intensity dependence.
To monitor the state of PtdIns(4,5)P2 depletion, electroretinogram (ERG) recordings were used to measure the residual sensitivity of trp mutant retinae after exposure to the same red light used for degeneration assays. Intact flies with chronically implanted wire electrodes were mounted inside the same box used for the degeneration assay and control ERG responses recorded to 1 second test flashes in the dark. The flies were then exposed to the red LEDs of the light box for different time periods and the ERG again recorded ∼5 seconds after turning off the red LEDs. Exposure to 1–5 minutes of red light was already sufficient to severely attenuate the light response in trp flies and after 30 minutes red light the response was essentially abolished, indicating near complete PtdIns(4,5)P2 depletion (Fig. 5B,Ci). By contrast wild-type flies showed virtually no loss of sensitivity under the same conditions. The recovery of the ERG response in the dark, reflecting PtdIns(4,5)P2 re-synthesis, was also monitored, and returned to pre-exposure levels with a similar time course (t1/2 = 59±15 seconds, mean ± s.e.m. n = 6; Fig. 5Cii) to that previously reported for PtdIns(4,5)P2 resynthesis (Hardie et al., 2004; Hardie et al., 2001). These results indicate that the red light used for degeneration assays was sufficient to induce depletion of PtdIns(4,5)P2 in trp retinae.
Next we varied the intensity of red light in the light box using neutral density filters and both retinal degeneration and PtdIns(4,5)P2 depletion were assayed under similar conditions. Fig. 5Di shows the residual sensitivity in trp retinae after 30 minutes exposure to different intensities of red light. The full intensity light (log 0.0 attenuation) resulted in total loss, 0.2 attenuation of intensity caused ∼75% loss, 0.4 and 0.8 attenuation of intensity led to only 30–40% loss of sensitivity. Similarly, trp retinae showed most severe degeneration under full intensity light; the severity was already substantially reduced with 0.2 attenuation, and with 0.4 attenuation, there was no sign of retinal degeneration over a 2-week period (Fig. 5Dii). These results establish that the threshold intensity for inducing degeneration closely matches the intensity required to deplete PtdIns(4,5)P2, providing further strong evidence for the role of PtdIns(4,5)P2 depletion in trp retinal degeneration.
Depletion of PtdIns(4,5)P2 by PIP2 phosphatase also induces retinal degeneration
To test whether depletion of PtdIns(4,5)P2 was sufficient to induce degeneration, the Salmonella typhimurium PtdInsPn phosphatase, SigD, was expressed in fly photoreceptors R1–6 under control of the Rh1 promoter. SigD dephosphorylates membrane PtdIns(4,5)P2 to PtdIns(5)P or PtdIns(4)P, and, unlike depletion by PLC, generates neither diacylglycerol (DAG) nor InsP3 (Mason et al., 2007; Terebiznik et al., 2002; Wei et al., 2008).
Under optical neutralisation, flies expressing only one copy of SigD had normal retinae on eclosion (Fig. 6Ai,iii). However, flies with two copies of the gene had degenerated retinae on eclosion, even if reared in dark. Because the Rh1 promoter is active only in the peripheral photoreceptors, only R1–6 cells showed degeneration whereas R7 remained intact. To rule out the possibility that the degeneration was due to disruption (as a result of P-element insertion) in a gene required for eye function, a heteroallelic combination of two SigD-expressing lines with independent P-inserts was tested and a similar phenotype was observed (data not shown). By contrast, control flies expressing two copies of a catalytically inactive form of the enzyme, SigDdead, had intact photoreceptors and rhabdomeres.
Fig. 6.
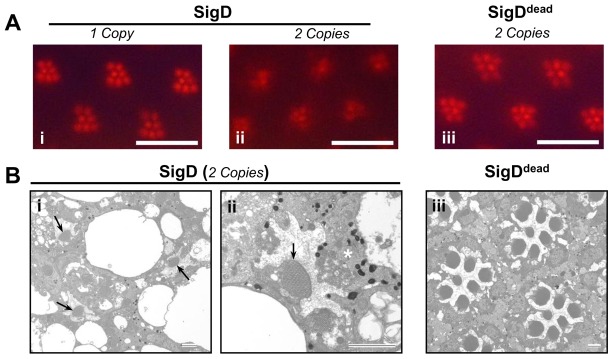
Retinal degeneration in flies expressing the Salmonella PtdInsPn-phosphatase gene, SigD. Optical neutralisation (A) and TEM (B) images of fly retinae expressing Salmonella SigD and its catalytically inactive form SigDdead, under the Rh1 promoter. (Aii,Bi,ii) Transgenic flies expressing two copies SigD show severe retinal degeneration, with fragmented rhabdomeres and degenerating photoreceptors being engulfed by a neighbouring pigment cell (*). (B,i,ii) R7 photoreceptors (arrows), where SigD is not expressed, are normal. (Aiii,Biii) The retina is normal in flies expressing the catalytically inactive SigDdead Scale bars: ∼16 mm (A); ∼2 µm (B).
TEM of newly eclosed Rh1-SigD-expressing flies showed that the degeneration was associated with severe ultrastructural defects. The rhabdomere structures were lost in most cases and if present, were completely disorganised. Degenerated photoreceptors were also observed becoming engulfed by neighbouring pigmented glial cells (Fig. 6Bii). Control flies, expressing a catalytically inactive form of SigD (SigDdead), had normal photoreceptor structure (Fig. 6Aiii,Biii). These results indicate that PtdIns(4,5)P2 depletion in fly retinae is sufficient to induce severe retinal degeneration even in the dark and irrespective of the nature of the by-product (DAG and InsP3 on the one hand or PtdIns(4)P or PtdIns(5)P on the other).
Depletion of Moesin from rhabdomeres in trp retina
The correlation of trp retinal degeneration with PtdIns(4,5)P2 depletion, and the fact that degeneration is associated with a breakdown of the microvillar structure drew our attention to the Ezrin/Radixin/Moesin (ERM) family of PtdIns(4,5)P2-regulated proteins. ERM proteins are crosslinkers between cortical actin and plasma membrane, and play a key role in formation of membrane projections, such as microvilli and filopodia (Berryman et al., 1993; Bretscher, 1999; Bretscher et al., 1997; Fehon et al., 2010; Fiévet et al., 2007; Neisch and Fehon, 2011). In the presence of PtdIns(4,5)P2 the active, phosphorylated form of the protein is attached to the membrane (Roch et al., 2010); upon PtdIns(4,5)P2 hydrolysis the proteins are dephosphorylated and the inactive form is released to cytosol (Barret et al., 2000; Yonemura et al., 2002). Drosophila has only a single ERM protein, dMoesin (McCartney and Fehon, 1996; Polesello et al., 2002), which is required, inter alia, for morphogenesis and maintenance of microvillar structure in the photoreceptors (Chorna-Ornan et al., 2005; Karagiosis and Ready, 2004). Moesin localises to the base of the rhabdomere in wild-type flies in the dark (Karagiosis and Ready, 2004), but becomes dephosphorylated and moves to cytosol under bright illumination (Chorna-Ornan et al., 2005). This suggested Moesin as a candidate link between PtdIns(4,5)P2 depletion and microvillar disintegration.
To explore this possibility we examined Moesin localization using live imaging of dissociated ommatidia expressing GFP-tagged dMoesin (Polesello et al., 2002). Under control conditions Moe–GFP was initially most strongly localized in a narrow band at the base of the microvillar rhabdomere as previously reported using anti-Moe antibodies (Fig. 7A). This then rapidly (<1 minute) disappeared indicating translocation to the cell body (see supplementary material Movie 1). Because the intense blue excitation light required to visualise the Moe–GFP, is a super-saturating stimulus for the photoreceptors, we suspected that it may have been sufficiently bright to deplete microvillar PtdIns(4,5)P2 even in a wild-type background. This was confirmed using a GFP-tagged PLCδ1 pleckstrin homology (PH–GFP) domain (Várnai and Balla, 1998) to monitor PtdIns(4,5)P2 hydrolysis. Under the same conditions this probe initially localized to the rhabdomeres, but then translocated to the cytosol within only a few seconds (Fig. 7Aii; supplementary material Movie 2).
Fig. 7.

Depletion of dMoesin from rhabdomere bases following red light exposure in the absence of TRP channels. (A) (i) Dissociated live ommatidia (∼2/3 of ommatidial length imaged, distal ends downwards) expressing GFP-tagged Moesin after continuous red illumination for 3–20 min in control bath (top row) and in the presence of 100 µM La3+, which phenocopies the trp mutation by blocking all TRP channels (bottom row). Note the prominent stripes of fluorescence at the base of the rhabdomeres (small arrows), which are no longer seen after red illumination in the presence of La3+ (two examples shown; representative of 18 control and 24 La3+-treated ommatidia). (ii) Ommatidia expressing the PtdIns(4,5)P2-binding PH domain from PLCδ1 (PH–GFP). In control solutions PH–GFP remained localized to the rhabdomeres (r) after red illumination; but in La3+-treated ommatidia (below) it had translocated to the cell body (c), indicating severe depletion of PtdIns(4,5)P2 in the rhabdomeres (representative of 4–7 ommatidia in each condition). See also supplementary material Movies 1 and 2. Scale bars: 5 µm. (B) Representative intensity profiles (averaged line scans from rhabdomere to periphery at 90° to rhabdomere long axis, as indicated by large arrows in A from five control ommatidia (i) and five ommatidia exposed to La3+ (ii); a sharp peak representing the stripe of Moesin (arrow) adjacent to the rhabdomere (indicated by double arrow) is only apparent in control. (C) Transverse confocal slices of ommatidia from fixed whole-mount preparations of eyes exposed to red light for 2 hours. The retinae were probed with anti p-Moe (green), and Rhodamine-conjugated phalloidin (red), which labels actin in the rhabdomeres. Left: p-Moe alone; right: merged images. (i) In the wild-type retina, p-Moe is concentrated in a band at the base of the rhabdomeres (white arrowhead). (ii) In the trp343 retina, the white arrow indicates the absence of p-Moe from the bases of rhabdomeres R1–R6 . R7, which does not respond to red light retains the band at the base (white arrowhead). (iii) Localization of p-Moe to the base of the rhabdomeres was rescued in norpAP24;;trp343 double mutants. Scale bars: ∼2 µm. (D) Quantification of the fluorescence intensity in the basal band of p-Moe immunofluorescence in R1–R6 normalised to that in R7, which served as an internal standard (means ± s.e.m. of nine ommatidia from three flies).
Next we pre-adapted the ommatidia for between 3 and 20 minutes with red illumination (LED: 630 nm peak wavelength) with an intensity (equivalent to ∼20,000 effectively absorbed photons per second) sufficient to induce response decay [PtdIns(4,5)P2 depletion] in trp mutants. In control solutions Moe–GFP remained concentrated in a narrow stripe at the base of the rhabdomeres immediately following such pre-illumination (Fig. 7Ai,Bi). However, if the ommatidia were first perfused with La3+, which accurately phenocopies the trp mutation by blocking TRP channels without affecting TRPL channels (Reuss et al., 1997), these Moe–GFP stripes were either no longer visible or much reduced in contrast indicating delocalization from the rhabdomere base to the cytosol (Fig. 7Ai,Bii). Similar experiments using PLCδ1 PH–GFP, confirmed that PtdIns(4,5)P2 was depleted from the rhabdomeres by similar exposure to red light in La3+-treated ommatidia, but not in control solutions (Fig. 7Aii).
Activation of Moesin and other ERM proteins is believed to require PtdIns(4,5)P2 binding followed by phosphorylation. To explore the role of phosphorylation we looked at the localization of phosphorylated Moesin (p-Moe) in trp retina upon red light exposure, probing wild-type and mutant retinae with anti p-Moe antibody. Fig. 6B shows immunolocalization of p-Moe in wild-type, trp and norpA;;trp flies exposed to red light for 2 hours. In wild-type flies, or in trp flies reared in the dark, p-Moe localized to the rhabdomere base; but was lost from the rhabdomere bases of trp flies illuminated with red light. Localization to the rhabdomere bases in red light was restored in norpA;;trp double mutants, where PtdIns(4,5)P2 hydrolysis was blocked by mutation in norpA gene. These results show that active p-Moe disappears from the rhabdomere bases of trp mutant retina upon exposure to red light in a PLC-dependent manner. Because PtdIns(4,5)P2 is known to play a key role in activation (or phosphorylation) and association of Moesin with membrane in other systems (Barret et al., 2000; Yonemura et al., 2002), we propose that the light-induced depletion of microvillar PtdIns(4,5)P2 in trp results in dephosphorylation of p-Moe and translocation of Moesin from the rhabdomere base to the cytosol.
Actin cytoskeleton in trp mutants
Dynamic regulation of ERM proteins is important for cortical actin reorganization in response to various signals (Tamma et al., 2005; Treanor et al., 2011). Therefore we also examined actin cytoskeleton organization in degenerating trp retina at different times after exposure to red light. The retinae were dissected and stained with phalloidin, which specifically binds to F-actin, and counterstained with anti-Rh1 antibody. Phalloidin staining in the R1–6 (but not R7, which serves as an internal standard) rhabdomeres of trp retinae was progressively lost with a clear reduction within 2–4 hours of red light exposure and almost complete loss of signal by 8 hours (Fig. 8), although Rh1 immunostaining remained intact. By contrast, wild-type flies exposed to red light for 24 hours, as well as trp flies maintained in dark for the same amount of time, showed normal phalloidin staining. These results indicate severe microvillar actin depolymerisation in trp retina when exposed to light over a few hours.
Fig. 8.

Loss of F-actin in trp mutant retina exposed to light. (A,A′) Transverse confocal slices of wild-type ommatidium after 24 hours of red light (LED-light box) exposure. The retinae were doubly probed with Rhodamine-conjugated phalloidin (red) and a monoclonal antibody against rhodopsin (4C5, green). (A′) Overlay of both the markers in a wild-type retina. (B–F) Phalloidin labelling of trp343 retinae after exposure to red light for 2 hours (B), 4 hours (C), 8 hours (D), 24 hours (E), and kept in the dark for 24 hours (F). (B′–F′) Merged images (overlay of phalloidin and rhodopsin staining) of the ommatidia in B–F. Scale bars ∼2 µm, n = 3 to 4. (G) Quantification of phalloidin fluorescence intensity in rhabdomeres R1–R6, expressed as a fraction of fluorescence intensity in R7, which served as an internal standard (means ± s.e.m. of nine ommatidia from three flies).
Discussion
The role of TRP channels in Drosophila phototransduction as the major light-sensitive Ca2+ permeable channel is well established (Hardie and Minke, 1992; Niemeyer et al., 1996; Reuss et al., 1997), but the mechanisms underlying retinal degeneration in trp mutants are poorly understood. Previous studies had implicated reduced Ca2+ influx (Liu et al., 2007; Wang et al., 2005) and subsequent defects in the visual pigment cycle leading to degeneration via accumulation of MPP–Arr2 complexes (Wang et al., 2005). However, the present study revealed an alternative Ca2+-dependent mechanism, independent of the pigment cycle, and which can be studied in isolation by rearing flies under red light. This mechanism is strictly PLC dependent and our evidence suggests that it is mediated by the unattenuated hydrolysis of PtdIns(4,5)P2 in the absence of Ca2+ influx. Depletion of this important phospholipid has downstream effects, including disruption of the microvillar actin cytoskeleton, possibly mediated, at least in part via the PtdIns(4,5)P2-regulated ERM protein, Moesin. Internalization of MPP–Arr2 complexes may also contribute to degeneration in trp mutants reared under white light; however, this needs further investigation; not least because PtdIns(4,5)P2 is also required for recruitment of components involved in clathrin-mediated endocytosis (Itoh and Takenawa, 2004; Rohde et al., 2002; Simonsen et al., 2001).
Reduced Ca2+ influx is a common factor underlying retinal degeneration
Ca2+ influx regulates multiple targets in the photoreceptors, including components of the visual pigment cycle. In this pathway, M binds to Arr2 and is phosphorylated by rhodopsin kinase to form MPP–Arr2 complexes (Fig. 9B). These complexes can undergo clathrin-mediated endocytosis unless Arr2 is phosphorylated by Ca2+-dependent CaMKII (Kiselev et al., 2000; Matsumoto et al., 1994). Ca2+/CaMKII-dependent Arr2 phosphorylation is also reported to be required for the dissociation of Arr2 from phosphorylated R (RPP), after it has been photo-reisomerised from MPP (Alloway and Dolph, 1999). A second Ca2+/CaM-dependent enzyme, rhodopsin phosphatase, (RDGC), then dephosphorylates RPP to recreate the ground state R. rdgC mutants accumulate stable hyperphosphorylated MPP–Arr2 complexes, which are proposed to induce degeneration when generated in excess (Kiselev et al., 2000). MPP–Arr2 complexes also accumulate in norpA mutants (Alloway et al., 2000; Orem and Dolph, 2002), suggesting the existence of a signalling pathway that can trigger cell death in any fly mutant with severely compromised Ca2+ influx (Alloway et al., 2000; Byk et al., 1993; Orem and Dolph, 2002).
trp mutants have greatly reduced light-induced Ca2+ influx, not only because of the lower Ca2+ permeability of the remaining TRPL channels, but also because the response to light in trp mutants decays to absolute baseline during maintained illumination, effectively preventing any further Ca2+ influx. The reduced Ca2+ influx has been implicated in trp retinal degeneration because the degeneration can be largely rescued by mutations of the Na+/Ca2+ exchanger (Wang et al., 2005) and because a point mutation in the TRP channel pore (trpD621G) that eliminated Ca2+ permeation (while still allowing Na+ influx) resulted in a severe retinal degeneration phenotype (Liu et al., 2007). The MPP–Arr2 pathway was suggested to induce degeneration in trp mutants, because arr2 mutations partially suppressed the degeneration (Wang et al., 2005). However, lack of full suppression suggested the existence of yet another pathway in trp retina.
Toxic accumulation of MPP–Arr2 complexes can be prevented by exposing flies to long wavelength light (Kiselev et al., 2000), which photoisomerises M to R. As expected, degeneration in norpA mutants was completely rescued by rearing flies in red light. However, degeneration in trp flies was rescued neither by red light, nor by the phosphorylation-deficient rhodopsin mutation, ninaEΔ356, indicating the existence of an alternate pathway for degeneration independent of the visual cycle. Importantly, the degeneration in trp flies under red light was completely rescued when PLC activity was eliminated in norpA;;trp double mutants, indicating that this alternate pathway is activated downstream of PtdIns(4,5)P2 hydrolysis.
Depletion of PtdIns(4,5)P2 in trp retinae correlates with retinal degeneration
Electrophysiological recordings from trp mutant retina show complete response decay during prolonged illumination, followed by a prolonged refractory period (Cosens and Manning, 1969; Hardie et al., 2001). Studies using a genetically targeted PtdIns(4,5)P2 biosensor (Kir2.1 channels), revealed that response decay and inactivation in trp mutants reflects severe depletion of PtdIns(4,5)P2 in the photoreceptors due to failure of Ca2+ and PKC-dependent inhibition of PLC (Gu et al., 2005; Hardie et al., 2001). ERG recordings performed on trp flies exposed to the same red light used for degeneration assays confirmed that PtdIns(4,5)P2 was depleted with the same absolute intensity dependence, suggesting that PtdIns(4,5)P2 depletion plays a critical role in trp degeneration. That PtdIns(4,5)P2 depletion (without, e.g. generation of DAG and InsP3) is sufficient to induce retinal degeneration was strongly supported by expressing the bacterial PtdInsPn</emph> phosphatase, SigD, in otherwise wild-type photoreceptors. Even in dark-reared flies, this resulted in retinal degeneration that was more severe than in light-reared trp mutants. The greater severity of the degeneration might be explained by a difference in the PtdIns(4,5)P2 pool depleted in the SigD-expressing flies. In trp mutants only the light-sensitive plasma membrane PtdIns(4,5)P2 pool in the microvillar rhabdomeres would be depleted, whereas expression of SigD would potentially deplete PtdIns(4,5)P2 in both plasma and endo-membranes of the entire cell.
While PtdIns(4,5)P2 depletion had not previously been considered as a mechanism underlying degeneration in trp mutants, with hindsight this proposal should not be surprising and such a mechanism may be more widespread. Thus besides its role in signal transduction, PtdIns(4,5)P2 is increasingly recognized as a key regulator of numerous vital cellular processes such as membrane trafficking, cytoskeleton organization, ion channel activation, cell proliferation and growth, and cell death (Di Paolo and De Camilli, 2006; Hilgemann et al., 2001; Payrastre et al., 2001; Rohacs, 2009; Yin and Janmey, 2003). Furthermore, mutants in virtually any component of the PtdIns(4,5)P2 recycling machinery in Drosophila, including DAG kinase [rdgA mutant (Masai et al., 1993)], CDP-DAG synthase [cds (Wu et al., 1995)], PtdIns transport protein [rdgB (Vihtelic et al., 1991)] and PtdIns synthase [dpis (Wang and Montell, 2006)] all undergo retinal degeneration, although the aetiology can be complex and may differ in different mutants (review by Raghu et al., 2012).
Moesin and actin
The severe breakdown of microvillar structure in degenerating trp retina drew our attention to dMoesin, the sole Drosophila representative of the ERM protein family, which regulates microvillar organisation in several tissues (Bretscher et al., 2002; Oshiro et al., 1998; Takeuchi et al., 1994). These proteins are activated by binding to PtdIns(4,5)P2 and phosphorylation of a conserved C-terminal threonine, whereupon they form associations between actin cytoskeleton and plasma membrane. When PtdIns(4,5)P2 levels drop, they are dephosphorylated and released from the membrane. Cytoplasmic translocation of ERM proteins has been shown to result in microvillar breakdown due to gross dissociation of actin-based cytoskeleton from plasma membrane in mouse L-cell fibroblasts (Kondo et al., 1997). Drosophila Moesin is required for morphogenesis and maintenance of the microvillar rhabdomeres and mutants have severely disrupted rhabdomeres (Chorna-Ornan et al., 2005; Karagiosis and Ready, 2004). In the present study, the active phosphorylated form of Moesin (p-Moe) was rapidly lost from the rhabdomeres of trp mutant retina exposed to red light, and because this was PLC dependent we attribute it to the depletion of PtdIns(4,5)P2 in trp photoreceptors.
In Drosophila photoreceptors, intense illumination has been reported to induce actin re-organization (Kosloff et al., 2003) and Moesin has been suggested to play a role in this process (Chorna-Ornan et al., 2005). In this study, trp R1–6 rhabdomeres showed near complete loss of phalloidin staining within 4–8 hours of red light exposure, indicating depolymerisation of the microvillar cytoskeleton, which includes a central actin filament in each microvillus. However, whether the loss of p-Moe directly induces actin depolymerisation in trp mutants needs to be further investigated. Membrane PtdIns(4,5)P2 levels regulate actin organisation by modulating a plethora of proteins, including profilin, gelsolin, villin, cofilin, CapZ and N-WASP. In general, low PtdIns(4,5)P2 levels promote actin depolymerisation while high concentrations of PtdIns(4,5)P2 favour actin polymerization (Janmey and Lindberg, 2004). It is possible that in the wild-type fly retina, the overall morphology of microvilli is maintained by a balance between actin polymerization and depolymerisation. However, in trp mutants depletion of PtdIns(4,5)P2 disturbs this balance and promotes depolymerisation resulting in breaking down of microvillar structure.
Conclusions
Low intracellular Ca2+ in trp mutants during illumination with white light may result in accumulation of stable MPP–Arr2 complexes as suggested in a previous study (Wang et al., 2005). However, it also causes depletion of PtdIns(4,5)P2 due to failure of Ca2+ and PKC inhibition of PLC (Fig. 9) (Gu et al., 2005; Hardie et al., 2001). The ability to chronically deplete PtdIns(4,5)P2 by controlled illumination in trp mutants provides a powerful tool for investigating the consequences of PtdIns(4,5)P2 dyshomeostasis in vivo. Here we found that PtdIns(4,5)P2 depletion results in a previously unrecognized form of retinal degeneration that can be simply isolated by rearing trp mutant flies under red light. Depletion of PtdIns(4,5)P2 results in loss of p-Moe from the rhabdomere bases and microvillar destabilization due to actin depolymerisation (Fig. 9). The latter may be as a consequence of loss of p-Moe, but may also involve other PtdIns(4,5)P2-regulated proteins involved in actin organization. Moesin depletion has been reported to trigger apoptosis in Drosophila epithelial cells via upregulation of RhoA and activation of the JNK pathway (Neisch et al., 2010). Both actin depolymerisation and microvillar breakdown due to loss of membrane associated ERM have also been shown to trigger cell death processes in several independent mammalian studies (Genescà et al., 2006; Kondo et al., 1997; Martin and Leder, 2001; Suria et al., 1999). Whether these events are sufficient to trigger trp degeneration or whether the degeneration pathway in these mutants involves other parallel pathways needs further investigation. Similar pathways could also play important roles in retinal degeneration associated with mutations in PI recycling genes, such as rdgB, cds, rdgA and dpis. Interestingly, PtdIns(4,5)P2 depletion mediated via effects of RhoA on PtdIns(4)P5-kinase has been implicated in cardiac hypertrophy (Howes et al., 2003; Miyamoto et al., 2010). Cardiomyocytes are also known to express high levels of TRP channels which play important roles in their functioning and maintenance (Vennekens, 2011; Watanabe et al., 2008). Red light-mediated retinal degeneration in trp mutants provides an experimentally amenable and genetically tractable model of degeneration with potential to provide insights into the mechanisms underlying diseases associated with TRP channel dysfunction and phosphoinositide dyshomeostasis.
Materials and Methods
Fly stocks
Fly stocks Drosophila melanogaster were raised on standard corn meal agar at 25°C in the dark. Oregon-R-S was the wild-type strain. Mutant strains included flies carrying norpAP24 a null allele of PLC, trp343, a null allele of the TRP channel and trpCM, which is a functionally null allele in flies reared at 25°C (Cosens and Manning, 1969; Reuss et al., 1997). No difference was observed in the retinal degeneration behaviour of flies with the two trp alleles, or their rescue under red light by norpA. Rhodopsin (ninaE) mutants lacking phosphorylation sites were made by expressing a third chromosome truncated rhodopsin transgene lacking C-terminal phosphorylation sites (P-ninaEΔ356) in a null ninaEEI17 background (Vinós et al., 1997) – [P-ninaEΔ356],ninaEEI17, referred to in short as ninaEΔ356. Standard crosses were set up to generate the following double mutants and transgenic flies – norpAP24;;trp343, norpAP24;;trpCM, norpAP24;;ninaEΔ35617 and ninaEΔ356,trpCM.
Transgenic flies
Salmonella typhimurium PtdInsPn phosphatase SigDΔN29 (SigD) and its catalytically inactive form, SigDdead (Cys462→Ser462) in tv3 vector (Wei et al., 2008) were subcloned into the pCaSper4 (pC4) vector under an Rh1 promoter for P-element transformation. The 3 kb Rh1 promoter was first cloned in pC4 vector between KpnI–NotI restriction sites. The 1.7 kb SigD/SigDdead gene was then cloned between NotI–BamHI restriction sites and finally the 0.8 kb SV40 polylinker was cloned in BamHI site. The clones obtained were sequenced and confirmed at the sequencing facility of Department of Biochemistry, University of Cambridge. Concentrated plasmid DNA preps (∼800–1000 ng/µl) were made using QIAfilter Plasmid Midi Kit and sent to BestGene Inc., USA for generation of transgenic flies. Flies expressing GFP-tagged Moesin under UAS control (Polesello et al., 2002) were crossed to flies expressing Rh1-Gal4 to induce specific expression in R1–6 photoreceptors.
Retinal degeneration assay
For retinal degeneration assays, initially dark-adapted flies (∼1 day after eclosion) of each genotype were kept in a light box in an incubator maintained at 25°C. The light box consisted of a 12×12 cm arena with white walls fitted with six evenly spaced red (peak wavelength ∼630 nm, 600 mcd) and white (major peak at ∼460 nm, minor peak at ∼560 nm, 320 mcd) LEDs. Flies were exposed to either continuous red light (effective luminance ∼250 cd/m2), continuous white light (∼100 cd/m2) or maintained in the dark in the same box. Plastic foil neutral density filters (Stage Electrics, Birmingham UK) were used to attenuate the light for intensity-dependent degeneration experiments. The extent of degeneration in the flies was monitored using optical neutralization (Franceschini and Kirschfeld, 1971), a method to analyse the integrity of rhabdomeres in single ommatidia. Fly heads were cut off and mounted on glass slide with eyes facing upwards using clear nail varnish. Rhabdomeres were observed with 40× oil immersion objective under antidromic illumination. The ommatidia that are axially aligned with the incident light allow the light to pass through them and seven rhabdomeres can be seen in each ommatidium. A score from 0 to 3 was given to each R1–6 rhabdomere depending on the state of its degeneration: 3 = no degeneration; 0 = rhabdomeres totally lost; and 1 or 2 = partial degeneration.
Thus the total score of a healthy ommatidium is 18. The degeneration score for a fly was the average score of 7–8 axially aligned ommatidia. For the time course of retinal degeneration the means ± s.e.m. value of degeneration score for more than seven flies is plotted against time. The ommatidia were scored from images were taken using Olympus DP12-2 digital camera.
Electroretinogram
To assess PtdIns(4,5)P2 depletion in trp retina under conditions used to induce retinal degeneration, the ERG was recorded from dark-reared flies with chronically fixed wire electrodes, mounted in the light box at the same position as vials of flies used for degeneration experiments. Loss of response following trp decay reflects PtdIns(4,5)P2 depletion and the recovery of the response following decay reflects PtdIns(4,5)P2 re-synthesis (Hardie et al., 2001). Flies were first introduced into truncated 200 µl Gilson tips (Villiers-de-Bel, France) and gently puffed, so that the head protruded from the small opening. The head was then immobilised using low melting point wax. A 50 µm platinum wire (Advent Research Materials, Halesworth, UK) was fixed with wax to the cornea via conducting gel, while an Ag–AgCl reference wire contacted gel covering the abdomen. Flies were exposed to the red LED in the light box for different durations and then the ERG was recorded with 1 second test flashes from an orange LED. Calibrated neutral density filter foil was used to vary the intensity of the red light box LED. The ERG was recorded using a Neurolog amplifier (Digitimer, Welwyn Garden, UK) and sampled and analysed using pClamp software (Molecular Devices, CA).
Transmission electron microscopy
Retinae from flies exposed to light or kept in dark were dissected under red light in ice-cold fixative (2% formaldehyde and 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.3. After 4 hours fixation at 4°C followed by buffer wash, the eyes were then postfixed in 1% OsO4 for 1 hour, stained en bloc in uranyl acetate for 1 hour, dehydrated in an alcohol series and embedded in Spurr's resin. Ultrathin sections were cut and stained with uranyl acetate and lead citrate (Raghu et al., 2009). The sections were viewed on a transmission electron microscope (Phillips CM100).
Immunolocalisation
For immunofluorescence, retinae from white eyed flies were dissected under red light in 4% formaldehyde (Polysciences Inc.) diluted in 1× phosphate buffered saline (PBS). The cornea was removed from the retinae for better penetration of the antibodies. They were then incubated in 4% formaldehyde for another 20 minutes at room temperature and washed three times with 1× PBS+0.3% Triton X-100 for 10–15 minutes. The tissues were then incubated in blocking solution [5% fetal calf serum (FCS) in 1× PBS+0.3% Triton X-100] for 1 hour at room temperature, after which the tissues were incubated with primary antibodies diluted in blocking solution [anti-pMoe (Karagiosis and Ready, 2004) – 1∶250, anti-Rh1 – 1∶250 (4C5, Developmental Studies Hybridoma Bank)], overnight (∼16 hours) at 4°C. Appropriate secondary antibodies conjugated with a fluorophore were used at 1∶1000 dilutions [anti-rabbit IgG–Chromeo 488™ (AbCam), anti-mouse IgG–Alexa-Fluor-488 (Molecular Probes)] and incubated overnight at 4°C. During the incubation with secondary antibody Rhodamine–phalloidin (Invitrogen) was also added to the tissues to stain the F-actin (1 unit in 200 µl of blocking solution). After three washes in 1× PBS+0.3% Triton X-100 the tissues were quickly washed in 1× PBS, mounted in mounting medium (0.25% n-propyl gallate in 50% glycerol in 1× PBS). The whole-mounted preparations were viewed under a Leica TCS (SP2) confocal microscope using 488 nm and 561nm wavelength lasers.
GFP imaging
For GFP fluorescence in live ommatidia, retina were dissected from flies expressing GFP-tagged constructs under control of the Rh1 (ninaE) promoter. Dissociated ommatidia were obtained by mechanical trituration and imaged using a 40× oil immersion objective and a Scion Firewire camera on an inverted Nikon microscope as previously described (e.g. Hardie et al., 2012). The standard bath solution contained (in mM) 120 NaCl, 5 KCl, 1.5 CaCl2, 4 MgCl2, 5 alanine, 20 proline, 10 N-Tris-(hydroxymethyl)-methyl-2-amino-ethanesulphonic acid (TES), pH 7.15. Flies expressing UAS-Moesin-GFP were obtained from Dr F Payre and crossed to flies expressing Rh1Gal4 to drive expression in the photoreceptors. Flies expressing the GFP-tagged PLCδ1-pleckstrin homology domain (PH-GFP) under control the trp-promoter element were obtained from Dr P. Raghu.
Supplementary Material
Acknowledgments
Gifts of fly stocks (UAS-Moe-GFP from Dr F. Payre; PH-GFP from Dr P. Raghu) are gratefully acknowledged.
Footnotes
Author contributions
R.C.H. conceived the project. All authors contributed to further experimental design. S.S. and T.B. performed degeneration assays and electron microscopy. D.F.R., H.X. and S.S. performed immunocytochemistry. S.S. also generated transgenic flies and performed ERG analysis. R.C.H. performed live ommatidia imaging. S.S. and R.C.H. wrote the paper with feedback from other authors.
Funding
This research was supported by grants from the Biotechnology and Biological Sciences Research Council [grant numbers BB/G006865/1, BB/D007585/1 to R.C.H., T.B.]; the Cambridge–Nehru Trust [Scholarship, ID 300752357 to S.S.]; and National Institutes of Health [grant number NEI10306 to D.F.R., H.X.]. Deposited in PMC after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.120592/-/DC1
References
- Alloway P. G., Dolph P. J. (1999). A role for the light-dependent phosphorylation of visual arrestin. Proc. Natl. Acad. Sci. USA 96, 6072–6077 10.1073/pnas.96.11.6072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alloway P. G., Howard L., Dolph P. J. (2000). The formation of stable rhodopsin-arrestin complexes induces apoptosis and photoreceptor cell degeneration. Neuron 28, 129–138 10.1016/S0896-6273(00)00091-X [DOI] [PubMed] [Google Scholar]
- Barret C., Roy C., Montcourrier P., Mangeat P., Niggli V. (2000). Mutagenesis of the phosphatidylinositol 4,5-bisphosphate (PIP(2)) binding site in the NH(2)-terminal domain of ezrin correlates with its altered cellular distribution. J. Cell Biol. 151, 1067–1080 10.1083/jcb.151.5.1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berryman M., Franck Z., Bretscher A. (1993). Ezrin is concentrated in the apical microvilli of a wide variety of epithelial cells whereas moesin is found primarily in endothelial cells. J. Cell Sci. 105, 1025–1043 [DOI] [PubMed] [Google Scholar]
- Bretscher A. (1999). Regulation of cortical structure by the ezrin-radixin-moesin protein family. Curr. Opin. Cell Biol. 11, 109–116 10.1016/S0955-0674(99)80013-1 [DOI] [PubMed] [Google Scholar]
- Bretscher A., Reczek D., Berryman M. (1997). Ezrin: a protein requiring conformational activation to link microfilaments to the plasma membrane in the assembly of cell surface structures. J. Cell Sci. 110, 3011–3018 [DOI] [PubMed] [Google Scholar]
- Bretscher A., Edwards K., Fehon R. G. (2002). ERM proteins and merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 3, 586–599 10.1038/nrm882 [DOI] [PubMed] [Google Scholar]
- Byk T., Bar–Yaacov M., Doza Y. N., Minke B., Selinger Z. (1993). Regulatory arrestin cycle secures the fidelity and maintenance of the fly photoreceptor cell. Proc. Natl. Acad. Sci. USA 90, 1907–1911 10.1073/pnas.90.5.1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorna–Ornan I., Tzarfaty V., Ankri–Eliahoo G., Joel–Almagor T., Meyer N. E., Huber A., Payre F., Minke B. (2005). Light-regulated interaction of Dmoesin with TRP and TRPL channels is required for maintenance of photoreceptors. J. Cell Biol. 171, 143–152 10.1083/jcb.200503014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosens D. J., Manning A. (1969). Abnormal electroretinogram from a Drosophila mutant. Nature 224, 285–287 10.1038/224285a0 [DOI] [PubMed] [Google Scholar]
- Cosens D., Perry M. M. (1972). The fine structure of the eye of a visual mutant, A-type of Drosophila melanogaster. J. Insect Physiol. 18, 1773–1786 10.1016/0022-1910(72)90109-6 [DOI] [PubMed] [Google Scholar]
- Di Paolo G., De Camilli P. (2006). Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657 10.1038/nature05185 [DOI] [PubMed] [Google Scholar]
- Everett K. V. (2011). Transient receptor potential genes and human inherited disease. Adv. Exp. Med. Biol. 704, 1011–1032 10.1007/978-94-007-0265-3_52 [DOI] [PubMed] [Google Scholar]
- Fehon R. G., McClatchey A. I., Bretscher A. (2010). Organizing the cell cortex: the role of ERM proteins. Nat. Rev. Mol. Cell Biol. 11, 276–287 10.1038/nrm2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiler R., Bjornson R., Kirschfeld K., Mismer D., Rubin G. M., Smith D. P., Socolich M., Zuker C. S. (1992). Ectopic expression of ultraviolet-rhodopsins in the blue photoreceptor cells of Drosophila: visual physiology and photochemistry of transgenic animals. J. Neurosci. 12, 3862–3868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiévet B., Louvard D., Arpin M. (2007). ERM proteins in epithelial cell organization and functions. Biochim. Biophys. Acta 1773, 653–660 10.1016/j.bbamcr.2006.06.013 [DOI] [PubMed] [Google Scholar]
- Franceschini N., Kirschfeld K.1971). Etude optique in vivo des éléments photorécepteurs dans l’oeil composé de Drosophila. Kybernetik 81–13 10.1007/BF02215177 [DOI] [PubMed] [Google Scholar]
- Genescà M., Sola A., Hotter G. (2006). Actin cytoskeleton derangement induces apoptosis in renal ischemia/reperfusion. Apoptosis 11, 563–571 10.1007/s10495-006-4937-1 [DOI] [PubMed] [Google Scholar]
- Gu Y., Oberwinkler J., Postma M., Hardie R. C. (2005). Mechanisms of light adaptation in Drosophila photoreceptors. Curr. Biol. 15, 1228–1234 10.1016/j.cub.2005.05.058 [DOI] [PubMed] [Google Scholar]
- Hardie R. C. (1996). INDO-1 measurements of absolute resting and light-induced Ca2+ concentration in Drosophila photoreceptors. J. Neurosci. 16, 2924–2933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie R. C. (2012). Phototransduction mechanisms in Drosophila microvillar photoreceptors. Wiley Interdiscip. Rev. Membr. Transp. Signal 1, 162–187 10.1002/wmts.20 [DOI] [Google Scholar]
- Hardie R. C., Minke B. (1992). The trp gene is essential for a light-activated Ca2+ channel in Drosophila photoreceptors. Neuron 8, 643–651 10.1016/0896-6273(92)90086-S [DOI] [PubMed] [Google Scholar]
- Hardie R. C., Raghu P., Moore S., Juusola M., Baines R. A., Sweeney S. T. (2001). Calcium influx via TRP channels is required to maintain PIP2 levels in Drosophila photoreceptors. Neuron 30, 149–159 10.1016/S0896-6273(01)00269-0 [DOI] [PubMed] [Google Scholar]
- Hardie R. C., Gu Y., Martin F., Sweeney S. T., Raghu P. (2004). In vivo light-induced and basal phospholipase C activity in Drosophila photoreceptors measured with genetically targeted phosphatidylinositol 4,5-bisphosphate-sensitive ion channels (Kir2.1). J. Biol. Chem. 279, 47773–47782 10.1074/jbc.M407525200 [DOI] [PubMed] [Google Scholar]
- Hardie R. C., Satoh A. K., Liu C. H. (2012). Regulation of arrestin translocation by Ca2+ and myosin III in Drosophila photoreceptors. J. Neurosci. 32, 9205–9216 10.1523/JNEUROSCI.0924-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann D. W., Feng S., Nasuhoglu C. (2001). The complex and intriguing lives of PIP2 with ion channels and transporters. Sci. STKE 2001, re19 10.1126/stke.2001.111.re19 [DOI] [PubMed] [Google Scholar]
- Howes A. L., Arthur J. F., Zhang T., Miyamoto S., Adams J. W., Dorn G. W., 2nd, Woodcock E. A., Brown J. H. (2003). Akt-mediated cardiomyocyte survival pathways are compromised by G alpha q-induced phosphoinositide 4,5-bisphosphate depletion. J. Biol. Chem. 278, 40343–40351 10.1074/jbc.M305964200 [DOI] [PubMed] [Google Scholar]
- Inoue R., Jensen L. J., Shi J., Morita H., Nishida M., Honda A., Ito Y. (2006). Transient receptor potential channels in cardiovascular function and disease. Circ. Res. 99, 119–131 10.1161/01.RES.0000233356.10630.8a [DOI] [PubMed] [Google Scholar]
- Itoh T., Takenawa T. (2004). Regulation of endocytosis by phosphatidylinositol 4,5-bisphosphate and ENTH proteins. Curr. Top. Microbiol. Immunol. 282, 31–47 10.1007/978-3-642-18805-3_2 [DOI] [PubMed] [Google Scholar]
- Janmey P. A., Lindberg U. (2004). Cytoskeletal regulation: rich in lipids. Nat. Rev. Mol. Cell Biol. 5, 658–666 10.1038/nrm1434 [DOI] [PubMed] [Google Scholar]
- Karagiosis S. A., Ready D. F. (2004). Moesin contributes an essential structural role in Drosophila photoreceptor morphogenesis. Development 131, 725–732 10.1242/dev.00976 [DOI] [PubMed] [Google Scholar]
- Katz B., Minke B. (2009). Drosophila photoreceptors and signaling mechanisms. Front. Cell Neurosci. 3, 2 10.3389/neuro.03.002.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselev A., Socolich M., Vinós J., Hardy R. W., Zuker C. S., Ranganathan R. (2000). A molecular pathway for light-dependent photoreceptor apoptosis in Drosophila. Neuron 28, 139–152 10.1016/S0896-6273(00)00092-1 [DOI] [PubMed] [Google Scholar]
- Kondo T., Takeuchi K., Doi Y., Yonemura S., Nagata S., Tsukita S. (1997). ERM (ezrin/radixin/moesin)-based molecular mechanism of microvillar breakdown at an early stage of apoptosis. J. Cell Biol. 139, 749–758 10.1083/jcb.139.3.749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosloff M., Elia N., Joel–Almagor T., Timberg R., Zars T. D., Hyde D. R., Minke B., Selinger Z. (2003). Regulation of light-dependent Gqα translocation and morphological changes in fly photoreceptors. EMBO J. 22, 459–468 10.1093/emboj/cdg054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. H., Wang T., Postma M., Obukhov A. G., Montell C., Hardie R. C. (2007). In vivo identification and manipulation of the Ca2+ selectivity filter in the Drosophila transient receptor potential channel. J. Neurosci. 27, 604–615 10.1523/JNEUROSCI.4099-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y. S., Yin H. L. (2007). Regulation of the actin cytoskeleton by phosphatidylinositol 4-phosphate 5 kinases. Pflugers Arch. 455, 5–18 10.1007/s00424-007-0286-3 [DOI] [PubMed] [Google Scholar]
- Martin S. S., Leder P. (2001). Human MCF10A mammary epithelial cells undergo apoptosis following actin depolymerization that is independent of attachment and rescued by Bcl-2. Mol. Cell. Biol. 21, 6529–6536 10.1128/MCB.21.19.6529-6536.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masai I., Okazaki A., Hosoya T., Hotta Y. (1993). Drosophila retinal degeneration A gene encodes an eye-specific diacylglycerol kinase with cysteine-rich zinc-finger motifs and ankyrin repeats. Proc. Natl. Acad. Sci. USA 90, 11157–11161 10.1073/pnas.90.23.11157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason D., Mallo G. V., Terebiznik M. R., Payrastre B., Finlay B. B., Brumell J. H., Rameh L., Grinstein S. (2007). Alteration of epithelial structure and function associated with PtdIns(4,5)P2 degradation by a bacterial phosphatase. J. Gen. Physiol. 129, 267–283 10.1085/jgp.200609656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto H., Kurien B. T., Takagi Y., Kahn E. S., Kinumi T., Komori N., Yamada T., Hayashi F., Isono K., Pak W. L.et al. (1994). Phosrestin I undergoes the earliest light-induced phosphorylation by a calcium/calmodulin-dependent protein kinase in Drosophila photoreceptors. Neuron 12, 997–1010 10.1016/0896-6273(94)90309-3 [DOI] [PubMed] [Google Scholar]
- McCartney B. M., Fehon R. G. (1996). Distinct cellular and subcellular patterns of expression imply distinct functions for the Drosophila homologues of moesin and the neurofibromatosis 2 tumor suppressor, merlin. J. Cell Biol. 133, 843–852 10.1083/jcb.133.4.843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minke B. (1982). Light-induced reduction in excitation efficiency in the trp mutant of Drosophila. J. Gen. Physiol. 79, 361–385 10.1085/jgp.79.3.361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S., Del Re D. P., Xiang S. Y., Zhao X., Florholmen G., Brown J. H. (2010). Revisited and revised: is RhoA always a villain in cardiac pathophysiology? J. Cardiovasc. Transl. Res. 3, 330–343 10.1007/s12265-010-9192-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell C. (2005). The TRP superfamily of cation channels. Sci. STKE 2005, re3 10.1126/stke.2722005re3 [DOI] [PubMed] [Google Scholar]
- Montell C. (2012). Drosophila visual transduction. Trends Neurosci. 35, 356–363 10.1016/j.tins.2012.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell C., Rubin G. M. (1989). Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron 2, 1313–1323 10.1016/0896-6273(89)90069-X [DOI] [PubMed] [Google Scholar]
- Neisch A. L., Fehon R. G. (2011). Ezrin, Radixin and Moesin: key regulators of membrane-cortex interactions and signaling. Curr. Opin. Cell Biol. 23, 377–382 10.1016/j.ceb.2011.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neisch A. L., Speck O., Stronach B., Fehon R. G. (2010). Rho1 regulates apoptosis via activation of the JNK signaling pathway at the plasma membrane. J. Cell Biol. 189, 311–323 10.1083/jcb.200912010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemeyer B. A., Suzuki E., Scott K., Jalink K., Zuker C. S. (1996). The Drosophila light-activated conductance is composed of the two channels TRP and TRPL. Cell 85, 651–659 10.1016/S0092-8674(00)81232-5 [DOI] [PubMed] [Google Scholar]
- Nilius B., Owsianik G., Voets T., Peters J. A. (2007). Transient receptor potential cation channels in disease. Physiol. Rev. 87, 165–217 10.1152/physrev.00021.2006 [DOI] [PubMed] [Google Scholar]
- Orem N. R., Dolph P. J. (2002). Loss of the phospholipase C gene product induces massive endocytosis of rhodopsin and arrestin in Drosophila photoreceptors. Vision Res. 42, 497–505 10.1016/S0042-6989(01)00229-2 [DOI] [PubMed] [Google Scholar]
- Oshiro N., Fukata Y., Kaibuchi K. (1998). Phosphorylation of moesin by rho-associated kinase (Rho-kinase) plays a crucial role in the formation of microvilli-like structures. J. Biol. Chem. 273, 34663–34666 10.1074/jbc.273.52.34663 [DOI] [PubMed] [Google Scholar]
- Payrastre B., Missy K., Giuriato S., Bodin S., Plantavid M., Gratacap M. (2001). Phosphoinositides: key players in cell signalling, in time and space. Cell. Signal. 13, 377–387 10.1016/S0898-6568(01)00158-9 [DOI] [PubMed] [Google Scholar]
- Peretz A., Sandler C., Kirschfeld K., Hardie R. C., Minke B. (1994a). Genetic dissection of light-induced Ca2+ influx into Drosophila photoreceptors. J. Gen. Physiol. 104, 1057–1077 10.1085/jgp.104.6.1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz A., Suss–Toby E., Rom–Glas A., Arnon A., Payne R., Minke B. (1994b). The light response of Drosophila photoreceptors is accompanied by an increase in cellular calcium: effects of specific mutations. Neuron 12, 1257–1267 10.1016/0896-6273(94)90442-1 [DOI] [PubMed] [Google Scholar]
- Phillips A. M., Bull A., Kelly L. E. (1992). Identification of a Drosophila gene encoding a calmodulin-binding protein with homology to the trp phototransduction gene. Neuron 8, 631–642 10.1016/0896-6273(92)90085-R [DOI] [PubMed] [Google Scholar]
- Polesello C., Delon I., Valenti P., Ferrer P., Payre F. (2002). Dmoesin controls actin-based cell shape and polarity during Drosophila melanogaster oogenesis. Nat. Cell Biol. 4, 782–789 10.1038/ncb856 [DOI] [PubMed] [Google Scholar]
- Raghu P., Coessens E., Manifava M., Georgiev P., Pettitt T., Wood E., Garcia–Murillas I., Okkenhaug H., Trivedi D., Zhang Q.et al. (2009). Rhabdomere biogenesis in Drosophila photoreceptors is acutely sensitive to phosphatidic acid levels. J. Cell Biol. 185, 129–145 10.1083/jcb.200807027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghu P., Yadav S., Mallampati N. B. (2012). Lipid signaling in Drosophila photoreceptors. Biochim. Biophys. Acta 1821, 1154–1165 10.1016/j.bbalip.2012.03.008 [DOI] [PubMed] [Google Scholar]
- Ramsey I. S., Delling M., Clapham D. E. (2006). An introduction to TRP channels. Annu. Rev. Physiol. 68, 619–647 10.1146/annurev.physiol.68.040204.100431 [DOI] [PubMed] [Google Scholar]
- Reuss H., Mojet M. H., Chyb S., Hardie R. C. (1997). In vivo analysis of the drosophila light-sensitive channels, TRP and TRPL. Neuron 19, 1249–1259 10.1016/S0896-6273(00)80416-X [DOI] [PubMed] [Google Scholar]
- Roch F., Polesello C., Roubinet C., Martin M., Roy C., Valenti P., Carreno S., Mangeat P., Payre F. (2010). Differential roles of PtdIns(4,5)P2 and phosphorylation in moesin activation during Drosophila development. J. Cell Sci. 123, 2058–2067 10.1242/jcs.064550 [DOI] [PubMed] [Google Scholar]
- Rohacs T. (2009). Phosphoinositide regulation of non-canonical transient receptor potential channels. Cell Calcium 45, 554–565 10.1016/j.ceca.2009.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde G., Wenzel D., Haucke V. (2002). A phosphatidylinositol (4,5)-bisphosphate binding site within mu2-adaptin regulates clathrin-mediated endocytosis. J. Cell Biol. 158, 209–214 10.1083/jcb.200203103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj S., Sun Y., Singh B. B. (2009). TRPC Channels and their Implication in Neurological Diseases. CNS Neurol. Disord. Drug Targets. 9, 94–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen A., Wurmser A. E., Emr S. D., Stenmark H. (2001). The role of phosphoinositides in membrane transport. Curr. Opin. Cell Biol. 13, 485–492 10.1016/S0955-0674(00)00240-4 [DOI] [PubMed] [Google Scholar]
- Steele F. R., Washburn T., Rieger R., O'Tousa J. E. (1992). Drosophila retinal degeneration C (rdgC) encodes a novel serine/threonine protein phosphatase. Cell 69, 669–676 10.1016/0092-8674(92)90230-A [DOI] [PubMed] [Google Scholar]
- Suria H., Chau L. A., Negrou E., Kelvin D. J., Madrenas J. (1999). Cytoskeletal disruption induces T cell apoptosis by a caspase-3 mediated mechanism. Life Sci. 65, 2697–2707 10.1016/S0024-3205(99)00538-X [DOI] [PubMed] [Google Scholar]
- Takeuchi K., Sato N., Kasahara H., Funayama N., Nagafuchi A., Yonemura S., Tsukita S., Tsukita S. (1994). Perturbation of cell adhesion and microvilli formation by antisense oligonucleotides to ERM family members. J. Cell Biol. 125, 1371–1384 10.1083/jcb.125.6.1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamma G., Klussmann E., Oehlke J., Krause E., Rosenthal W., Svelto M., Valenti G. (2005). Actin remodeling requires ERM function to facilitate AQP2 apical targeting. J. Cell Sci. 118, 3623–3630 10.1242/jcs.02495 [DOI] [PubMed] [Google Scholar]
- Terebiznik M. R., Vieira O. V., Marcus S. L., Slade A., Yip C. M., Trimble W. S., Meyer T., Finlay B. B., Grinstein S. (2002). Elimination of host cell PtdIns(4,5)P(2) by bacterial SigD promotes membrane fission during invasion by Salmonella. Nat. Cell Biol. 4, 766–773 10.1038/ncb854 [DOI] [PubMed] [Google Scholar]
- Treanor B., Depoil D., Bruckbauer A., Batista F. D. (2011). Dynamic cortical actin remodeling by ERM proteins controls BCR microcluster organization and integrity. J. Exp. Med. 208, 1055–1068 10.1084/jem.20101125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Várnai P., Balla T. (1998). Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J. Cell Biol. 143, 501–510 10.1083/jcb.143.2.501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennekens R. (2011). Emerging concepts for the role of TRP channels in the cardiovascular system. J. Physiol. 589, 1527–1534 10.1113/jphysiol.2010.202077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihtelic T. S., Hyde D. R., O'Tousa J. E. (1991). Isolation and characterization of the Drosophila retinal degeneration B (rdgB) gene. Genetics 127, 761–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinós J., Jalink K., Hardy R. W., Britt S. G., Zuker C. S. (1997). A G protein-coupled receptor phosphatase required for rhodopsin function. Science 277, 687–690 10.1126/science.277.5326.687 [DOI] [PubMed] [Google Scholar]
- Wang T., Montell C. (2006). A phosphoinositide synthase required for a sustained light response. J. Neurosci. 26, 12816–12825 10.1523/JNEUROSCI.3673-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Jiao Y., Montell C. (2005). Dissecting independent channel and scaffolding roles of the Drosophila transient receptor potential channel. J. Cell Biol. 171, 685–694 10.1083/jcb.200508030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H., Murakami M., Ohba T., Takahashi Y., Ito H. (2008). TRP channel and cardiovascular disease. Pharmacol. Ther. 118, 337–351 10.1016/j.pharmthera.2008.03.008 [DOI] [PubMed] [Google Scholar]
- Wei H. C., Rollins J., Fabian L., Hayes M., Polevoy G., Bazinet C., Brill J. A. (2008). Depletion of plasma membrane PtdIns(4,5)P2 reveals essential roles for phosphoinositides in flagellar biogenesis. J. Cell Sci. 121, 1076–1084 10.1242/jcs.024927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Niemeyer B., Colley N., Socolich M., Zuker C. S. (1995). Regulation of PLC-mediated signalling in vivo by CDP-diacylglycerol synthase. Nature 373, 216–222 10.1038/373216a0 [DOI] [PubMed] [Google Scholar]
- Yin H. L., Janmey P. A. (2003). Phosphoinositide regulation of the actin cytoskeleton. Annu. Rev. Physiol. 65, 761–789 10.1146/annurev.physiol.65.092101.142517 [DOI] [PubMed] [Google Scholar]
- Yonemura S., Matsui T., Tsukita S., Tsukita S. (2002). Rho-dependent and -independent activation mechanisms of ezrin/radixin/moesin proteins: an essential role for polyphosphoinositides in vivo. J. Cell Sci. 115, 2569–2580 [DOI] [PubMed] [Google Scholar]
- Zhu Z., Luo Z., Ma S., Liu D. (2011). TRP channels and their implications in metabolic diseases. Pflugers Arch. 461, 211–223 10.1007/s00424-010-0902-5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.