

Abstract
Background
Glioblastoma multiforme (GBM), the most aggressive malignant primary brain tumor of the central nervous system, is characterized by a relentless disease recurrence despite continued advancement in surgery, radiotherapy, and chemotherapy. Resistance to temozolomide (TMZ), a standard chemotherapeutic agent for GBM, remains a major challenge. Understanding the mechanisms behind TMZ resistance can direct the development of novel strategies for the prevention, monitoring, and treatment of tumor relapse.
Methods and results
Our research platform, based on the establishment of 2 pairs of TMZ-sensitive/resistant GBM cells (D54-S and D54-R; U87-S and U87-R), has successfully identified prolyl 4-hydroxylase, beta polypeptide (P4HB) over-expression to be associated with an increased IC50 of TMZ. Elevated P4HB expression was verified using in vivo xenografts developed from U87-R cells. Clinically, we found that P4HB was relatively up-regulated in the recurrent GBM specimens that were initially responsive to TMZ but later developed acquired resistance, when compared with treatment-naive tumors. Functionally, P4HB inhibition by RNAi knockdown and bacitracin inhibition could sensitize D54-R and U87-R cells to TMZ in vitro and in vivo, whereas over-expression of P4HB in vitro conferred resistance to TMZ in both D54-S and U87-S cells. Moreover, targeting P4HB blocked its protective function and sensitized glioma cells to TMZ through the PERK arm of the endoplasmic reticulum stress response.
Conclusions
Our study identified a novel target together with its functional pathway in the development of TMZ resistance. P4HB inhibition may be used alone or in combination with TMZ for the treatment of TMZ-resistant GBM.
Keywords: chemoresistance, ER stress response, glioma, P4HB, temozolomide
Glioblastoma multiforme (GBM) is one of the most aggressive malignancies affecting the central nervous system (CNS), and it accounts for over 60% of all primary brain tumors in adults.1 GBM is characterized by rapid and infiltrative growth, early recurrence, and resistance to conventional therapies. Despite aggressive surgical resection using state-of-the-art techniques and intensive adjuvant radio- and chemotherapy, clinical outcome remains poor, with a median survival of <12 months.2
Temozolomide (TMZ), a standard treatment for newly diagnosed and recurrent GBMs, can improve patients' survival by inducing tumor cell death through the transfer of O6-methylguanine, which activates the mismatch repair mechanism and ultimately triggers cytotoxicity and apoptosis.3 TMZ-resistant phenotypes are common and may arise from a variety of mechanisms.4,5 The most well-recognized cause is through the action of O6-methylguanine–DNA methyltransferase (MGMT), a DNA repair enzyme, which when highly expressed, can remove the alkyl group from the O6-guanine, thereby antagonizing the therapeutic effect of TMZ.6 Other resistance-related mechanisms include the deficit of DNA repair systems (eg, base excision repair [BER] and mismatch repair [MMR] systems)7,8 and the activation of different pro-survival pathways (eg, Akt, NFĸB, PI3K, p53, and β-catenin).9–12 De novo or acquired TMZ resistance accounts for the majority of disease progression during treatment or after an initial period of favorable response.13 Understanding the molecular mechanisms behind TMZ resistance and developing effective therapeutic measures are currently 2 major challenges in neuro-oncology.14
Endoplasmic reticulum stress response (ERSR) may be triggered by various physiological and pathological conditions, such as hypoxia, nutrient deprivation, disulfide bond formation, infection, and neoplasia.15 These conditions may cause an excessive and uncontrolled accumulation of improperly folded or unfolded proteins that eventually lead to the activation of the unfolded protein response (UPR) signaling. The latter activates the transcription of UPR target genes, such as endoplasmic reticulum (ER) chaperone, folding enzymes, and antioxidants, in an attempt to restore ER homeostasis.16 Low-grade ER stress response is potentially cell protective17 but, when prolonged, may trigger programmed cell death pathways, usually apoptosis,18 and autophagic cell death.19
Tumor cells are commonly engaged in low stress conditions resulting from the surrounding hypoxic and low nutrient environments. The protective mechanisms induced by ERSR appear to be chronically activated and may shield tumor cells from other insults, including drug-induced toxicity.20 GBM is constantly under hypoxic stress and may therefore develop an adaptive response that confers chemoresistance.21 Recently, several reported studies have used ERSR as the therapeutic target to treat glioma.22,23 The majority of studies focused on ERSR-induced autophagy as GBM is preferentially more resistant to apoptosis but less so to autophagy.24 Although TMZ is proautophagic and autophagy is presently considered a promising target in glioma therapies, chemoresistance remains a concern.25 Because molecular pathway intersections occur between autophagy and apoptosis, targeting the latter may provide an alternative approach in overcoming TMZ resistance.23
Our previous proteomic profiling study has identified a potential target, prolyl 4-hydroxylase, beta polypeptide (P4HB), which is associated with TMZ resistance in GBM cells.26 P4HB, also known as protein disulphide isomerase (PDI; GenBank accession number: NM_000918.3), is a multifunctional protein that catalyses the formation and rearrangement of disulfide bonds. It acts as a molecular chaperone that helps ameliorate misfolded proteins in response to ER stress.27 On the basis of its role in UPR and our previous finding of upregulated P4HB expression in TMZ-resistant cells, we hypothesized that inhibiting P4HB activity will sensitize GBM cells to TMZ by abrogating the pro-survival ERSR pathway. We also investigated the functional and therapeutic roles of P4HB inhibition and the mechanisms involved in TMZ resistance in GBM. Our study provided correlative in vitro, in vivo, and clinical data that implicate that P4HB occupies an important role in TMZ resistance and may serve as a potential target in overcoming or preventing the onset of chemoresistance in GBM.
Materials and Methods
Chronic Exposure of GBM Cells to TMZ
Our methods of developing TMZ-resistant subclones of GBM have been previously described.26 In brief, human GBM cell lines, D54-MG (Duke University Medical Center), and U87-MG (American Type Culture Collection, Manassas, VA) were exposed to TMZ (Temodal, Schering-Plough, Whitehouse Station, NJ) until stable TMZ-resistant subclones (D54-R and U87-R) were derived from the parental TMZ-sensitive cells (D54-S and U87-S). A resistant phenotype was confirmed by inhibitory concentration (IC50) testing, and the resistant subclones were maintained in a low dose (100 μM) of TMZ.
Small Interfering RNA (siRNA), Short Hairpin RNA (shRNA) Transfection, and Gene Over-Expression
To study the effect of P4HB inhibition, siRNA targeting P4HB (siP4HB_1: 5′-CAGGACGGTCATTGATTACAA-3′ and siP4HB_2: 5′-AAGATGAACTGTAATACGCAA-3′) and negative-control siRNA (siCtrl; both from Qiagen, Hilden, Germany) were used. GBM cells were transfected with 100 pmol of siRNA on 6-well plate (8000 cells/well) by using the FuGene6 Transfection Reagent (Roche Diagnostics, Indianapolis, IN) according to the manufacturer's protocol. TMZ was added to the cells 24 h after transfection and incubated for an additional 72 h before subsequent analysis. In addition, D54-R and U87-R cells were manipulated with P4HB-specific shRNA (shP4HB) encoding pGFP-V-RS plasmid (OriGene Technologies, Inc., Rockville, MD). Green fluorescent protein (GFP) was used to monitor transfection efficiency. The transfected clones were selected using 1 μg/mL of puromycin (Invitrogen Corporation, Grand Island, NY), and harvested by trypsinization in the cloning rings. Scrambled-control (Scr-Ctrl) was established by transfecting a nonspecific shRNA in parallel with shP4HB.
To study the effect of P4HB over-expression, human P4HB cDNA clone (OriGene Technologies, Inc) was transiently transfected into D54-S and U87-S cells at a density of 10 000 cells/well. After 24 h, TMZ was added to the cells for an incubation period of 72 h before subsequent analysis.
Clonogenic and Survival Assays
The inhibitory effects of TMZ were assayed by colony formation and cell survival assays. For the former, 500 cells/well were seeded on 6-well plates. After the cells were fully attached, TMZ was added for 48 h, followed by purging with fresh medium. A cultivation period of 14 days was allowed before colonies were stained with crystal violet solution (5 g/L; Sigma-Aldrich, Saint Louis, MO). The number of colonies formed reflected the sensitivity of cells to TMZ, in comparison with the DMSO control.
A sulforhodamine B assay (Sigma-Aldrich) was performed to assess cell survival in the absence or presence of serial concentrations of TMZ and with or without bacitracin, which is an inhibitor of P4HB. Seeded cells (5000 cells/well) were cultured in 96-well plates for 72 h. Cellular protein content was used to reflect surviving cell density, as measured by absorbance of optical density at 490 nm. The percentages of surviving cells were calculated after normalization to the controls (cells without prior drug treatment). The half maximal IC50 value was calculated by derivation of the best-fit line, using 3 independent experiments performed in triplicates.
Cellular Fractionation and Immunoblotting
Fractionation of cells was performed according to protocol using the ProteoExtract Subcellular Proteome Extraction Calbiochem Kit (Merck Millipore, Merck KGaA, Darnstadt, Germany) to allow stepwise extraction of mitochondrial and cytosolic protein fractions. In brief, adherent cells were washed twice with phosphate-buffered saline, followed by gentle agitation in 3 mL Extraction Buffer I with 5 μL Protease Inhibitor Cocktail for 30 min at 4°C. The supernatant was collected as the cytosolic fraction. The remaining cells were resuspended in 3 mL Extraction Buffer II with 5 μL Protease Inhibitor Cocktail for 30 min at 4°C, and the supernatant was collected as the mitochondrial fraction. Proteins from fractionated cell lysates or unfractionated whole-cell lysates (40 µg) were resolved on 12% SDS gels and electroblotted on nitrocellulose membranes as previously described.28 The antibodies used were against cytochrome c, GRP 78, and phospho-PERK (Santa Cruz Biotechnology Inc., CA); rabbit monoclonal Bax, Bcl-2, total/cleaved caspase-3, total/cleaved caspase-7, COX IV, GAPDH, IRE-α, PERK, and P4HB (all from Cell Signaling Technology, Inc., Danvers, MA).
Flow Cytometric Analysis and TUNEL Assay
Flow cytometric analysis was performed to assess apoptosis in 5 × 105 treated GBM cells by double staining with 5 µL Annexin-V-FITC and 0.1 µg propidium iodide (Invitrogen) in 100 µL binding buffer (10 mmol/L HEPES, 140 mmol/L NaCl, and 2.5 mmol/L CaCl2 [pH, 7.4]). Samples were incubated at room temperature in dark for 15 min. Immediately before analysis by Beckman Coulter Cytomics FC500 (Beckman Coulter, Inc., Indianapolis, IN), 400 µL binding buffer was added to each sample. Two-color analyses of apoptosis were performed, and fluorescence compensation on the flow cytometer was adjusted to minimize the overlapping of FITC and propidium iodide signals.
The TUNEL assay was performed to determine apoptosis in mouse xenografts of different treatment conditions. An in situ cell death detection kit POD (Roche Diagnostics) was used, and details of the procedure have previously been described by our laboratory.26
Quantitative Polymerase Chain Reaction (qPCR)
Details on the procedure have previously been described.26 The primers used were 5′-GTGGCCAAGCACTTCAAACC-3′ and 5′-CCCGGAGAAGGCATCCTC-3′ for ATF4, 5′-ACACCGGGAGTGTTGTCCAG-3′ and 5′-TGGGCAAGACACCCAAGTG-3′ for GADD34, and 5′-CGACCACTTTGTCAAGCTCA-3′ and 5′-AGGGGTCTACATGGCAACTG-3′ for GAPDH as an internal control.
Clinical GBM Specimens
To help translate our findings to the clinical scenario, we studied clinical specimens from 2 patients with GBM who had undergone initial surgical resection (specimens collected at this stage were denoted as treatment naive), followed by chemo-irradiation and adjuvant TMZ. Response Evaluation Criteria in solid tumors was evaluated before surgery, after surgery, and during treatment with TMZ.29 One patient (denoted as Pat. #1) showed disease recurrence after 5 cycles of adjuvant TMZ, and the second patient (Pat. #2) developed recurrence after 10 cycles of treatment. Both underwent subsequent surgery for recurrent tumor growth. The progression-free survival for Pat. #1 and Pat. #2 was 7 months and 17 months, respectively. The overall survival was 18 months and 22 months, respectively. The paired specimens (pre-TMZ and post-TMZ) from each patient were confirmed to be GBM by a specialist in neuropathology and were studied for P4HB expression. Signed informed consents were obtained from the patients or their legal guardians before sample acquisition. The study was approved by the Institution Review Board of our institution.
Immunohistochemistry
Immunohistochemical staining for P4HB in the clinical and animal tumor specimens was performed on consecutive 4-μm thick paraffin-embedded tissue sections. In brief, tissue sections were deparaffinized with xylene, and the antigen was retrieved in 10 mM sodium citrate (pH, 6.0). After peroxidase blocking, the specimens were further blocked in 5% normal goat serum (Dako, Glostrup, Denmark) for 1 h, followed by incubation with rabbit monoclonal anti-P4HB antibody (1:100 dilution; Cell Signaling Technology, Inc.) at 4°C overnight. After washing 3 times with phosphate-buffered saline, sections were incubated with a horseradish peroxidase–conjugated antibody against rabbit immunoglobulin G (IgG; Invitrogen) at 1:200 dilution. Signal was detected using a ready-to-use DAKO EnVision+ Kit (Dako) and subsequently counterstained with haematoxylin (Vector Laboratories, Burlingame, CA). Isotype-matched IgG control was used in each experiment.
U87 GBM Xenograft
To study the effects of P4HB deregulation in vivo, 1×106 U87 GBM cells (U87-S) and their subclones (U87-R, -shP4HB-1, -shP4HB-2, and -Scr-Ctrl) were inoculated subcutaneously into the right flanks of 4–6-week-old male BALB/c athymic mice. Tumor growth was measured twice weekly with calipers, and tumor volume was calculated as volume = length × width2/2, as previously described.30 After the tumor volume reached 50 mm3, mice were oral-gavaged with Ora-plus (the control group; Paddock Laboratories, Minneapolis, MN) or TMZ suspended in Ora-plus (the treatment group). TMZ was given at 55 mg/kg/day for 5 days per week for 2 cycles. Mice were observed daily and euthanized after 2 weeks of treatment. Tumors were excised for histopathological examination and Western blot analysis. The study was approved by the Committee on the Use of Live Animal for Teaching and Research, The University of Hong Kong, Hong Kong.
Results
Establishment of TMZ-Resistant U87 GBM Cell Subclones by Chronic Exposure to TMZ
We have previously identified an association between P4HB up-regulation and the acquisition of TMZ resistance in D54 GBM cell subclones by proteomics analysis.26 To facilitate subsequent in vitro and in vivo analysis, we also generated chemoresistant subclones from another GBM cell line, U87-MG. Both TMZ-resistant D54-R and U87-R subclones exhibited reduced clonogenicity when compared with the parental and passage controls.31 U87-R generated after chronic exposure to TMZ for 5 months (also termed as U87-P5) demonstrated resistance to TMZ by exhibiting a ∼4-fold increase in IC50 (U87-P5: 2176.8 µM) when compared with the parental (U87-C0: 491.72 µM) and passage control (U87-C5: 636.04 µM) (Supplementary Fig. S1A). P4HB was highly expressed in U87-P5 relative to U87-C0 and U87-C5 (Supplementary Fig. S1B). Similar to what had been described for D54-R cells, U87-P5 cells exhibited a cell structure and morphology that differed from chemosensitive cells, by adopting an elongated, spindle-like structure and shrinkage of the cytoplasm (Supplementary Fig. S1C).
P4HB Is Up-Regulated in Tumor Xenografts Developed from U87-R
As U87-MG GBM cells demonstrated higher tumorigenic capacity, compared with D54; U87-S (TMZ-sensitive) and U87-R (TMZ-resistant) were used in our in vivo model. We found that U87-R xenografts had less treatment response to TMZ, compared with U87-S cells (Fig. 1A). Tumor progression during the 14 days of TMZ treatment is shown in Fig. 1B. The U87-R group showed tumor progression, and the U87-S group showed regression (Fig. 1B). At 14 days after treatment, there was a significant difference in tumor volume (P < .01) between the U87-S-TMZ (32.32 µM) and U87-R-TMZ (273.06 µM) groups, thus confirming our in vitro findings that we had successfully established the TMZ-resistant U87 cell subclone. Moreover, mice carrying U87-S cells after TMZ treatment showed a statistically significant (P = .0002) delay in tumorigenesis and prolonged overall survival in all mice (n = 4), in comparison with those with U87-R cells (Fig. 1C). After H&E staining, a relative increase in vascularity was noted in the ex vivo xenografts developed from U87-R cells, compared with U87-S cells (Fig. 1D). Of more importance, P4HB expression was relatively up-regulated in the ex vivo xenografts from U87-R cells, confirming our in vitro findings that P4HB was associated with TMZ resistance in GBM (Fig. 1E).
Fig. 1.

In vivo tumor mouse model of TMZ-resistant GBM cells shows enhanced P4HB expression. (A) Mouse tumor xenografts (n = 4) developed from TMZ-sensitive (U87-S) and TMZ-resistant (U87-R) GBM cells were treated with Ora Plus (Ctrl) or TMZ in Ora Plus (TMZ) for 2 cycles when the tumor volume reached 50 mm3. Tumor xenografts isolated from U87-R group substantiate resistance to TMZ after 2 cycles of treatment. (B) In vivo tumor growth curves in response to TMZ treatment. Tumor volume (mm3) were expressed as: tumor volume (mm3) = (tumor length (mm) × tumor width (mm)2)/2. Each data point represents the mean ± SD of 4 animals. **P = .01. (C) Kaplan-Meier survival curves were defined by end-point of tumor volume reaching four times (200 mm3) the initial implanted tumor volume (50 mm3). Prolonged survival was seen in animals implanted with U87-R cells after treatment with TMZ when compared to those with U87-S cells (P = .0002). (D) H&E staining of xenografts from mice developed from U87-R cells shows enhanced vascular densities. Scale bar, 20 µm. Original magnification: 100×; 400× (insets). (E) Western blots show increased P4HB levels in xenografts from mice developed from U87-R cells (M1: mouse 1; M2: mouse 2).
Clinical Significance of P4HB in Recurrent GBM with Acquired TMZ Resistance
We studied paired tumor specimens before and after TMZ treatment in 2 patients with recurrent diseases. Immunohistochemical staining showed that P4HB expression was localized in the tumor periphery (Fig. 2A). No staining was found in the brain sections of normal cadaver control (Fig. 2A). Moreover, immunoblots verified our immunohistochemistry observations that P4HB expression was higher in recurrent than in treatment-naive tumors (Fig. 2B). Expression was comparatively higher in Pat. #1, who developed tumor recurrence after only 5 cycles of TMZ treatment, compared with Pat. #2, whose disease recurred after 10 cycles, suggesting that P4HB over-expression may be related to poorer tumor control.
Fig. 2.
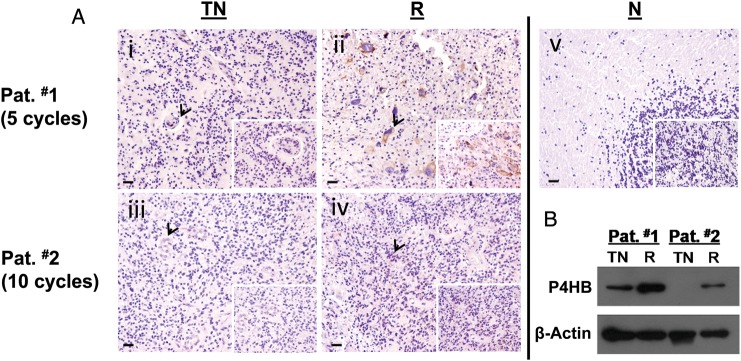
P4HB overexpressed in tumor specimens from patients with recurrence due to acquired TMZ resistance. (A) Representative pictures of immunohistochemical staining of P4HB in human GBM specimens. P4HB stained intensely in (ii) recurrent (R) GBM paraffin sections, but not in (i) treatment naive (TN) sections from patient #1 (Pat. #1) who developed recurrence after 5 cycles of treatment. Similarly, P4HB was stained positively in (iv) recurrent (R) GBM paraffin sections, but not in (iii) treatment naive (TN) sections from patient #2 (Pat. #2). (v) No staining was observed in normal brain paraffin sections obtained from cadaver donor as control. Scale bar, 10 µm. Original magnification: 100×; 400× (insets). (B) Western blot shows P4HB was highly expressed in tissues from patients developed TMZ resistance after a favorable treatment response.
Over-Expression of P4HB Enhances TMZ Resistance in GBM Cells In Vitro
Next, we investigated whether P4HB over-expression could reduce tumor response to TMZ in vitro. P4HB was transiently over-expressed in both D54-S and U87-S cells. Transfection was effective after 48 h and continued to 120 h (Fig. 3A). Chemotoxicity assay showed that P4HB over-expression was associated with significantly increased cell viability against different TMZ doses (D54-S: 250–2000 µM; U87-S: 500–1000 µM). The effect was prominent at 500 µM of TMZ, showing increased survival in both D54-S-P4HB (>20%) and U87-S-P4HB (>10%) cells relative to their parental wild-type controls (Fig. 3B). There was a ∼3-fold increase in the IC50 of D54-S-P4HB (1556.2 µM), in comparison with D54-S-WT (496.5 µM) and D54-S-Vec (607.1 µM), and a ∼2-fold increase in U87-S-P4HB cells (1009.8 µM), in comparison with U87-S-WT (538.4 µM) and U87-S-Vec (576.4 µM) (Fig. 3C). Our results showed that over-expression of P4HB in GBM cells protected TMZ-mediated cell death in vitro.
Fig. 3.

Overexpression of P4HB enhances GBM cell survival against TMZ treatment. (A) Western blot shows increased P4HB expression in D54-S and U87-S cells after transfection with P4HB cDNA clone (D54-S-P4HB and U87-S-P4HB) from 48 h (48 h) to 120 h (120 h). (B) Cell viability assay shows increased P4HB expression can significantly enhance cell survival against TMZ treatment when compared to the wild-type (WT) parental (D54-S and U87-S) and vector control (D54-S-Vec and U87-S-Vec) GBM cells. *P = .05; **P = .01. (C) Table shows increased IC50 in both D54-S-P4HB and U87-S-P4HB cells, compared with D54-S-WT and U87-S-WT, D54-S-Vec, and U87-S-Vec GBM cells.
P4HB Inhibition Resensitizes TMZ-Resistant GBM Cells to TMZ In Vitro
To evaluate the effect of P4HB inhibition as a chemosensitizer in TMZ-resistant GBM, P4HB was depleted by siRNA in both the D54-R and the U87-R cells. Both siP4HB_1 and siP4HB_2 were found to inhibit P4HB expression effectively after 48 h of transfection and up to 120 h (Fig. 4A). siCtrl- or siP4HB-transfected D54-R and U87-R cells were then examined for viability under different concentrations of TMZ (250, 500, 1000, and 2000 µM) for 72 h (Fig. 4B). Overall, P4HB knockdown alone could induce cytotoxicity in both D54-R and U87-R GBM cells (>20% relative to siCtrl alone). When combined with TMZ treatment (250–1000 µM), P4HB knockdown resulted in >40% growth inhibition relative to the siCtrl (no treatment) control. The effect was to a similar extent when compared with TMZ alone. No significant growth inhibition was observed at or beyond 2000 µM of TMZ. In clonogenic survival assay, combined treatment with siP4HB and TMZ at IC50 showed synergistic effects in TMZ-mediated cell death (Fig. 4C). The associated P4HB knockdown after 14 days of colony formation was confirmed by Western blotting (Fig. 4C).
Fig. 4.


Inhibition of P4HB sensitizes GBM cells to TMZ-mediated cell death. (A) Western blot confirmed siP4HB knockdown after 48 h and 120 h of transfection in D54-R and U87-R cells. β-Actin was used as a reference control. Two different siRNAs against different exons of P4HB were tested (ie, siP4HB_1 and siP4HB_2), and a nonspecific siRNA (siCtrl) was used in parallel. (B) Decrease in cell viability after knockdown of P4HB in D54-R and U87-R cells when compared to the siCtrl. This trend of effect is augmented in TMZ dose-dependent manner (250, 500, 1000 and 2000 µM). *P = .05; **P = .01. (C) Colony formation assay shows a decrease in cell growth after P4HB knockdown in TMZ-treated D54-R and U87-R cells (upper panel). Western blot confirmed P4HB expression was still inhibited after the incubation period of 14 days (lower panel). (D) P4HB inhibitor (bacitracin, BAC) decreased the percentage of GBM cell viability. Effect is significantly augmented at TMZ concentrations of 500 and 1000 μM. (E) Representative clonogenic pictures showing few colonies of D54-R and U87-R formed after treatment with BAC and TMZ at 500 and 1000 µM.
The chemosensitizing effect of P4HB inhibition was further demonstrated using the P4HB inhibitor, bacitracin. Bacitracin alone did not show significant inhibitory effects in D54-R (125 and 250 µM) and U87-R (250 and 500 µM) cells after 72 h of treatment. However, when combined with TMZ at 500 and 1000 µM, significant growth inhibition was observed (D54-R: >20% and >30%; U87-R: >30% and >35%, respectively) (Fig. 4D). The finding was confirmed by clonogenic survival assay (Fig. 4E). Our results suggested that inhibition of P4HB could attenuate TMZ resistance in GBM.
shP4HB Enhances TMZ Chemosensitivity In Vivo
To facilitate the in vivo and subsequent in vitro experiments, stable chemoresistant cell lines (D54-R: Scr-Ctrl, shP4HB-1 and shP4HB-2; U87-R: Scr-Ctrl, shP4HB-1, and shP4HB-2) were established from single-cell subclones after transfection with either the scrambled-control shRNA (Scr-Ctrl) or shRNAs targeting different exons of P4HB (shP4HB-1 and shP4HB-2). P4HB knockdown efficiency varied in different subclones of D54-R-shP4HB and U87-R-shP4HB. The overall inhibition was significantly more effective in U87-R-shP4HB than in D54-R-shP4HB cells at both the protein and the mRNA levels (Fig. 5A).
Fig. 5.

Permanent knockdown of P4HB in U87-R cells enhances TMZ chemosensitivity in in vivo mouse tumor xenografts. (A) D54-R and U87-R cells were transfected with 2 independent P4HB shRNA vectors (shP4HB-1 and shP4HB-2) or shRNA scrambled-control (Scr-Ctrl). The efficiency of P4HB knockdown was demonstrated by Western blot (left panel) and qPCR (right panel). **P = .01. GFP-positive populations of U87-R cells were enriched by puromycin selection, and injected subcutaneously into nude mice (shP4HB-1, shP4HB-2 and Scr-Ctrl; n = 4 per group). Xenograft growth was measured over a 14-day period after treatment with TMZ from day 0 of tumor size reached 50 mm3. Representative xenograft pictures from the treated and non-treated group as measured by (B) GFP signal and (C) tumor volume. (D) Tumor sections stained with H&E show a significant difference in cell morphology between shP4HB-1 and -2 with Scr-Ctrl. Scale bar, 20 µm. Original magnification: 100×; 400× (insets). (E) TUNEL assay was performed to quantify apoptosis on paraffin sections of xenografts. Positively stained cells were noticeable in xenografts with shP4HB-1 and -2 after TMZ treatment. (F) Xenograft growth curves show delay in tumor growth of mice implanted with both U87-R shP4HB-1 and shP4HB-2 when compared to the Scr-Ctrl. *P = .05; **P = .01. (G) Expression of P4HB in xenografts after 14 days of treatment, as demonstrated by Western blot analysis.
To investigate whether P4HB would potentiate GBM cell survival in vivo under TMZ challenge, P4HB-silenced clones and scrambled-control U87-R cells were injected subcutaneously (1 × 106 cells per animal) into the right flank region of nude mice (n = 4 in each group). Tumor growth in each group after TMZ treatment was assessed by imaging the tumors for GFP fluorescence twice weekly. Only representative images from each group (U87-R-Scr-Ctrl, U87-R-shP4HB-1, and U87-R-shP4HB-2) were showed after 14 days of TMZ treatment (Fig. 5B–D). Overall, mice implanted with U87-R-shP4HB-1 and U87-R-shP4HB-2 cells were more responsive to TMZ treatment by showing reduced fluorescence, when compared with U87-R-Scr-Ctrl (Fig. 5B). Tumor xenografts removed after animal sacrifice showed similar treatment responses by having smaller tumors (Fig. 5C). Histopathological examination of ex vivo xenografts showed significant differences in cell morphology between U87-R-Scr-Ctrl and U87-R-shP4HB. Tumors from U87-R-shP4HB-1 and U87-R-shP4HB-2 showed clear cytoplasm without shrinkage and nuclear displacement and reduced hypervascularity and geographic necrosis, when compared with the Scr-Ctrl (Fig. 5D). TUNEL assay for apoptosis was performed for the ex vivo xenografts and showed that P4HB inhibition enhanced TMZ-mediated cell death through apoptosis (Fig. 5E). Fig. 5F illustrates the tumor growth patterns under different treatment combinations. Percentage of growth in tumor volume was indexed to the initial tumor volume at day 0 before treatment. We found that mice implanted with U87-R-shP4HB-1 and U87-R-shP4HB-2 demonstrated significantly greater response to TMZ, compared with U87-R-Scr-Ctrl. A significant reduction in tumor volume was identified in both the U87-R-shP4HB-1 (TMZ; 27.90% relative to U87-R-shP4HB-1 [Ctrl]) and U87-R-shP4HB-2 (TMZ; 15.11% relative to U87-R-shP4HB-2 [Ctrl]), but not in U87-R-Scr-Ctrl (TMZ; 6.75% relative to U87-R-Scr-Ctrl [Ctrl]) at day 14 after treatment (P = .05; P = .01). Western blot confirmed the expression of P4HB in the ex vivo xenografts at the end of the study from 2 random animals (Fig. 5G).
P4HB Knockdown Triggers ER Stress and Mitochondrion-Mediated Apoptosis under TMZ
First, we studied the effect of P4HB depletion on apoptotic cell death in TMZ-resistant GBM cells. Fig. 6A demonstrates the results from flow cytometric analysis, showing an induction in the percentage of apoptotic cells after P4HB knockdown alone (D54-R: siCtrl, =2.7% and siP4HB, =3.6%; U87-R: siCtrl, =1.5% and siP4HB, =3.3%). When combined with TMZ treatment, the effect was dose-dependent and augmented, compared with untreated siCtrl (D54-R at 500 µM: siCtrl, 2.9% and siP4HB, 4.4%; U87-R at 500 µM: siCtrl, 1.9% and siP4HB, 4.0% and D54-R at 1000 µM: siCtrl, 2.9% and siP4HB, 8.9%; U87-R at 1000 µM: siCtrl, 3.8% and siP4HB, 11.3%). Our results suggest that P4HB inhibition-mediated apoptosis contributed significantly to cell death during the late phase (at 72 h) of TMZ treatment.
Fig. 6.


Chemosensitization to TMZ by P4HB inhibition activates ER stress-induced apoptosis via UPR signaling. (A) P4HB inhibition induced apoptosis. Flow cytometry analysis performed by double staining of annexin V and propidium iodide shows increased apoptotic response in D54-R and U87-R cells after P4HB inhibition by siRNA and/or TMZ treatment at 500 and 1000 µM. A representative experiment of 3 performed was shown. (B) Caspase-3/7 activity was measured by the Caspase-Glo 3/7 assay kit. Compared with the untreated and negative control groups, an increase in caspase-3/7 activity was observed in the shP4HB group and shP4HB + TMZ group. *P = .05; **P = .01. Data are means of triplicate experiments. Western blot depicting alterations in (C) the expression of apoptosis-related proteins in whole-cell lysates and (D) mitochondrial and cytosolic fraction of the release of cytochrome c (Cyto-c). COX IV was used as loading control for mitochondrial fraction and β-actin for cytosolic fraction. (E) Activation of UPR signaling was illustrated by representative blots of altered ER chaperones (GRP78) and stressor (PERK) expression prepared from cells of different groups (Scr-Ctrl + TMZ; D54-R: 0, 300 and 600 µM; U87-R: 0, 500 and 1000 µM) and shP4HB + TMZ (D54-R: 0, 300 and 600 µM; U87-R: 0, 500 and 1000 µM) for 72 h. COX IV was used as loading control. (F) Induction of ER stress markers (ATF4 and GADD34) was quantified by qPCR markedly in both D54-R shP4HB and U87-R shP4HB after TMZ treatment at our previously defined optimal concentration (300 and 600 µM; 500 and 1000 µM, respectively). Column, mean of 3 replicates ± SD. *P = .05; **P = .01.
To further characterize whether P4HB inhibition provoked any apoptosis through the executioner caspases, a homogeneous luminescent assay was used to measure caspase-3/7 activities. We found significant induction (P = .05; P = .01) of caspase-3/7 activities in shP4HB cells, compared with the scrambled no TMZ treatment control (Fig. 6B). The effect was stronger with the combination of P4HB knockdown and TMZ (D54-R at 300 µM and U87-R at 500 µM) than by P4HB knockdown. The result was confirmed by caspase cleavage analysis, because their fragments were indicative of apoptosis. As shown in Fig. 6C, the ER-associated caspase-7 and the executioner caspase-3 were substantially more activated by the treatment combinations than by individual P4HB knockdown. Overall, the effect was saturated at 600 µM (D54-R) and 1000 µM (U87-R) of TMZ concentrations.
Because activation of other apoptotic pathways, such as the mitochondrial apoptotic pathway, may also be involved under ER stress, we also examined the Bax:Bcl-2 ratio. Overall, we found a high Bax:Bcl-2 ratio and enhanced cytoplasmic cytochrome c release in TMZ-resistant GBM cells with P4HB knockdown. Similar to the activities of the caspase, the effect was synergistically increased with combined TMZ treatment in a TMZ dose-dependent manner (Fig. 6C and D). These results indicated that mitochondrial apoptotic pathway was also involved in P4HB inhibition-mediated cell death.
Suppression of P4HB Activates ER Stress-Induced UPR Signaling in Attenuating TMZ Resistance
To establish whether the chemosensitizing effect of P4HB inhibition was mediated by ER stress-induced apoptosis through the UPR signaling pathway, we examined the expression patterns of several molecular indicators. Western blot analysis indicated knockdown of P4HB alone could activate GRP78 (ER stress chaperone) and PERK (ER stress sensor) in both D54-R-shP4HB and U87-R-shP4HB cells, as demonstrated by the enhanced expressions of GRP78 and phosphorylation of PERK at Thr981 (Fig. 6E). Similarly, the effect was substantial with additional TMZ treatment (D54-R at 300 µM and U87-R at 500 µM) that became saturated at a high concentration (D54-R at 600 µM and U87-R at 1000 µM).
Two ER stress markers ATF4 and GADD34 were tested for their transcriptional activities in response to TMZ after P4HB knockdown. GADD34 is the apoptotic gene mediated by ATF4-CHOP. Overall, by qPCR analysis, we found a significant (P = .05; P = .01) induction of ATF4 and GADD34 mRNA in both D54-R-shP4HB and U87-R-shP4HB relative to the Scr-Ctrl (Fig. 6F). We tested for the optimal concentration (data not shown) and found that the synergistic effect was most significant (P = .05; P = .01) at TMZ concentrations of 300 µM (D54-R) and 500 µM (U87-R).
Overall, our findings suggested a close correlation among ER stress induction, activation of the ER stress-associated caspases, and the mitochondrial apoptotic pathway, indicating that the P4HB suppression can abrogate the ERSR-induced protective mechanism in TMZ-resistant GBM cells predominantly through proapoptotic mechanisms.
Discussion
TMZ resistance in GBM is complex and involves different cellular pathways. Although some of the potential mechanisms have been uncovered, many of them are proofs-of-principle rather than a generalizable paradigm. A significant challenge is to successfully translate these growing bodies of knowledge into robust clinical trials. Our study provided further information on the role of P4HB, an ER chaperone, in conferring TMZ resistance in GBM. This is a continuation of our previous findings that P4HB has been identified to be dysregulated in chronically established TMZ-resistant GBM cells by proteomic profiling.26,31
In the past, chaperones in the ER were thought to be responsible only for activities required for protein folding, assembly, and transportation of membrane proteins, but increasing evidence suggests that ER chaperones may play other roles in oncogenic signaling that regulates cancer cell survival, progression, and angiogenesis.32,33 Up-regulation of these chaperones has been reported to be correlated with therapeutic resistance, including chemotoxic, anti-hormonal, DNA damaging, and anti-angiogenic agents by bypassing the cell death mechanisms.34,35 Family members of ER chaperones include glucose-regulated proteins (GRPs), heat shock proteins (HSPs), lectin, and PDI. Among these subclasses, the most abundant ER chaperone, GRP78, has been characterized meticulously with regard to its potential roles in tumorigensis and therapeutic resistance.36 In the central nervous system, GRP78 has been reported to exert a neuroprotective effect in various neurodegenerative diseases and neurotrauma and in glioma's resistance against tunicamycin- and glutamate-induced cell death.37,38 Of most importance, knockdown of GRP78 augmented the effectiveness of TMZ in malignant glioma, thus making this chaperone a possible target for antineoplastic treatment.39 Our study has successfully identified another ER chaperone, P4HB, which was dysregulated in GBM with acquired TMZ-resistance. P4HB is an enzyme catalyst of posttranslational disulfide bond formation and protein folding.40 The pro-oncogenic role of P4HB has previously been reported in glioma in vitro and in vivo.41 Similar to GRP78, P4HB represents the pro-survival arm of UPR, which during ER stress, may help in ameliorating misfolded proteins and result in barring stress-induced apoptosis. Targeting P4HB by its inhibitor, bacitracin, in combination with the chemotherapeutic drugs, fenretinide and velcade, has previously been demonstrated in melanoma to enhance tumor cell death.42 Its therapeutic effect in glioma has not been reported. Bacitracin has been widely accepted as a P4HB inhibitor for protein-folding catalyst; however, it's specificity of action has not been determined in vitro or in vivo.43 In addition, its mechanism of inhibition remains largely unknown. Therefore, other P4HB inhibitors and specific antibodies are urgently needed to exploit the underlying protective effect of this ER-resident protein catalyst in glioma.
In fact, ERSR has recently received much attention as a molecular pathway that can be modulated to induce glioma cell cytotoxicity. The pathway represents a cellular yin-yang process: at low level, it is potentially cell protective, but at high levels, it is cytotoxic through apoptosis or autophagy.17 The pivotal role of ERSR in protecting tumor cells under chronic stress conditions suggests that it may also determine chemo-sensitivity and may be exploited as a therapeutic opportunity in overcoming chemoresistance. Glioma has recently been shown to be sensitive to agents that interfere with ERSR.44 Different pharmacologic agents aimed at manipulating the ERSR are becoming available to be used alone or in combination with chemotherapeutic drugs. These are mainly small molecules affecting the ER Ca2+ homeostasis, such as THAP45 and flavonoids,44 or by interfering with the appropriate folding of proteins, such as nonsteroidal anti-inflammatory drugs.46 The ideal drugs will only elicit a tumor-specific effect by activating the pro-death arm in tumor cells but not in normal cells. Unfortunately, few of these agents showed selective tumor-targeting effect. In fact, some demonstrated unfavorable CNS bioavailability and adverse effects at dosages required to elicit apoptosis.47 Recently, a phase I clinical trial reported that the combination of ERSR-inducing agents, BOR, and TMZ could successfully control tumor progression in malignant glioma, suggesting that ERSR induction in glioma may provide a therapeutic alternative for the development of novel and effective anti-neoplastic agents.48
Our study represents the first report to our knowledge that up-regulation of P4HB is associated with acquired TMZ resistance in GBM through the protective mechanism of UPR. Thus, P4HB may be used as a target to sensitize GBM to TMZ treatment. The anti-tumor effect, however, was cell line– and dose-dependent, with D54-R cells being more susceptible to P4HB knockdown alone or its combination with TMZ. This observation has previously been reported in that P4HB knockdown alone induced different levels of cytotoxicity that was cell line–dependent.49 We also demonstrated that, although knockdown of P4HB alone could induce cytotoxicity both in vitro and in vivo, combined treatment with TMZ was even more effective. Although many reports have suggested that P4HB acted mainly as a catalyst for thiol-disulfide exchange reactions, its substrate specificities and mechanism of cooperation with other catalysts and chaperones in the cell are incompletely understood.40 On the basis of our findings, we conjecture that, under TMZ challenge, P4HB present at basal level in TMZ-sensitive GBM cells would perturb ER homeostasis via the activation of ER stress sensor PERK (Fig. 7). The release of PERK from P4HB will initiate the UPR signaling cascade to regulate the expression of different target genes of other ER chaperones, folding enzymes, and pro-apoptotic proteins, aiming to restore the ER homeostatic balance. Prolonged and intense ER stress would induce cell death via CHOP. In TMZ-resistant GBM cells, however, there is an excess of P4HB in the ER lumen sufficient enough to correct the unfolded proteins during the rechallenge with TMZ without activating the ER sensor. Inhibition of P4HB in TMZ-resistant GBM cells will have the clinically beneficial effect of reversing the cell back to the typical state of TMZ-sensitive cells, thus enhancing their response to TMZ. Future work aims to focus on the delivery of anti-P4HB therapies, such as the siRNA targeting P4HB or compounds that specifically suppress P4HB biological activities. The feasibility of delivering siRNA into brain by means of Ommaya reservoirs may potentially minimize or avoid systemic exposure and the potential of hepatotoxicity.50
Fig. 7.

Schematic model put forward the potential role of P4HB at different GBM cell states to TMZ treatment. ER stress is induced during the treatment of TMZ to TMZ-sensitive GBM cells and activates the proapopotic UPR signaling pathway. We hypothesize that P4HB as an ER chaperone will be dissociated from one of the ER stressor PERK, so as to enhance binding to the unfolded proteins in the ER lumen. Dissociation of P4HB from the ER stress transducer will trigger the PERK-eIF2α pathway by inducing the transcription of ATF4 which in turn enhancing the expression of proapoptotic CCAAT/enhancer-binding protein-homologous protein (CHOP). CHOP will subsequently activate the apoptotic machinery consisting of Bax, caspase-3 and -7, leading to cell death. In TMZ-resistant GBM cells, the presence of high level of P4HB is sufficiently bound to the unfolded proteins, therefore no induction of proapoptotic UPR signaling by ER stress. Nevertheless, P4HB inhibition can alternatively resensitize TMZ-resistant GBM cells to TMZ because of the insufficient P4HB binding to misfolded proteins. After prolonged ER stress, UPR induced apoptosis will be activated.
The present study has certain limitations that need to be acknowledged. GBM is known to belong to a heterogeneous group of neoplasms with differing cellular lineages, genetic changes, biological behaviors, and responses to chemotherapeutics. Our model involves only 2 cell lines and their TMZ-resistant subclones, and they have already revealed varying degrees of responses to our treatments. In vivo subcutaneous xenograft mouse model is immunosuppressed and is therefore not reproducing the tumor-host immune interactions to which human brain tumor would have been exposed. The number of clinical specimens involved in the present study was low, because paired clinical GBM specimens from patients with treatment-naive and recurrent GBM are very rare. Our aim was to provide illustrative rather that conclusive data with our 2 paired clinical specimens.
In conclusion, our study provides evidence that P4HB protects GBM cells and endows them with resistance to TMZ. Down-regulation of P4HB enhances chemosensitivity via the UPR signaling pathway. P4HB alone or in combination with TMZ may represent a novel approach to treat recurrent GBM with acquired TMZ resistance.
Supplementary Material
Acknowledgments
We thank the Philip Wong Foundation for providing general funding support.
Conflict of interest statement. No potential conflict of interest to disclose.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Krex D, Klink B, Hartmann C, et al. Long-term survival with glioblastoma multiforme. Brain. 2007;130:2596–2606. doi: 10.1093/brain/awm204. [DOI] [PubMed] [Google Scholar]
- 3.Holland EC. Glioblastoma multiforme: the terminator. Proc Natl Acad Sci USA. 2000;97:6242–6244. doi: 10.1073/pnas.97.12.6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oliva CR, Nozell SE, Diers A, et al. Acquisition of temozolomide chemoresistance in gliomas leads to remodeling of mitochondrial electron transport chain. J Biol Chem. 2010;285:39759–39767. doi: 10.1074/jbc.M110.147504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le Calve B, Rynkowski M, Le Mercier M, et al. Long-term in vitro treatment of human glioblastoma cells with temozolomide increases resistance in vivo through up-regulation of GLUT transporter and aldo-keto reductase enzyme AKR1C expression. Neoplasia. 2010;12:727–739. doi: 10.1593/neo.10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 7.Liu X, Han EK, Anderson M, et al. Acquired resistance to combination treatment with temozolomide and ABT-888 is mediated by both base excision repair and homologous recombination DNA repair pathways. Mol Cancer Res. 2009;7:1686–1692. doi: 10.1158/1541-7786.MCR-09-0299. [DOI] [PubMed] [Google Scholar]
- 8.Curtin NJ, Wang LZ, Yiakouvaki A, et al. Novel poly(ADP-ribose) polymerase-1 inhibitor, AG14361, restores sensitivity to temozolomide in mismatch repair-deficient cells. Clin Cancer Res. 2004;10:881–889. doi: 10.1158/1078-0432.ccr-1144-3. [DOI] [PubMed] [Google Scholar]
- 9.Lefranc F, Brotchi J, Kiss R. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J Clin Oncol. 2005;23:2411–2422. doi: 10.1200/JCO.2005.03.089. [DOI] [PubMed] [Google Scholar]
- 10.Huang H, Lin H, Zhang X, Li J. Resveratrol reverses temozolomide resistance by downregulation of MGMT in T98G glioblastoma cells by the NF-kappaB-dependent pathway. Oncol Rep. 2012;27:2050–2056. doi: 10.3892/or.2012.1715. [DOI] [PubMed] [Google Scholar]
- 11.Chen L, Han L, Shi Z, et al. LY294002 enhances cytotoxicity of temozolomide in glioma by down-regulation of the PI3K/Akt pathway. Mol Med Report. 2012;5:575–579. doi: 10.3892/mmr.2011.674. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Chen L, Bao Z, et al. Inhibition of STAT3 reverses alkylator resistance through modulation of the AKT and beta-catenin signaling pathways. Oncol Rep. 2011;26:1173–1180. doi: 10.3892/or.2011.1396. [DOI] [PubMed] [Google Scholar]
- 13.Happold C, Roth P, Wick W, et al. Distinct molecular mechanisms of acquired resistance to temozolomide in glioblastoma cells. J Neurochem. 2012;122:444–455. doi: 10.1111/j.1471-4159.2012.07781.x. [DOI] [PubMed] [Google Scholar]
- 14.Sathornsumetee S, Rich JN. New treatment strategies for malignant gliomas. Expert Rev Anticancer Ther. 2006;6:1087–1104. doi: 10.1586/14737140.6.7.1087. [DOI] [PubMed] [Google Scholar]
- 15.Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2012 doi: 10.1038/onc.2012.130. [published online ahead of print April 16, 2012] doi:10.1038/onc.2012.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higa A, Chevet E. Redox signaling loops in the unfolded protein response. Cell Signal. 2012;24:1548–1555. doi: 10.1016/j.cellsig.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 17.Schonthal AH, Chen TC, Hofman FM, Louie SG, Petasis NA. Preclinical development of novel anti-glioma drugs targeting the endoplasmic reticulum stress response. Curr Pharm Des. 2011;17:2428–2438. doi: 10.2174/138161211797249242. [DOI] [PubMed] [Google Scholar]
- 18.Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318:1351–1365. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- 19.Yorimitsu T, Klionsky DJ. Endoplasmic reticulum stress: a new pathway to induce autophagy. Autophagy. 2007;3:160–162. doi: 10.4161/auto.3653. [DOI] [PubMed] [Google Scholar]
- 20.Schonthal AH. Targeting endoplasmic reticulum stress for cancer therapy. Front Biosci (Schol Ed) 2012;4:412–431. doi: 10.2741/s276. [DOI] [PubMed] [Google Scholar]
- 21.Oliver L, Olivier C, Marhuenda FB, Campone M, Vallette FM. Hypoxia and the malignant glioma microenvironment: regulation and implications for therapy. Curr Mol Pharmacol. 2009;2:263–284. doi: 10.2174/1874467210902030263. [DOI] [PubMed] [Google Scholar]
- 22.Ciechomska IA, Gabrusiewicz K, Szczepankiewicz AA, Kaminska B. Endoplasmic reticulum stress triggers autophagy in malignant glioma cells undergoing cyclosporine A-induced cell death. Oncogene. 2012 doi: 10.1038/onc.2012.174. [published online ahead of print May 14, 2012] doi:10.1038/onc.2012.174. [DOI] [PubMed] [Google Scholar]
- 23.Zhong J, Kong X, Zhang H, et al. Inhibition of CLIC4 Enhances Autophagy and Triggers Mitochondrial and ER Stress-Induced Apoptosis in Human Glioma U251 Cells under Starvation. PLoS One. 2012;7:e39378. doi: 10.1371/journal.pone.0039378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang H, White EJ, Conrad C, Gomez-Manzano C, Fueyo J. Autophagy pathways in glioblastoma. Methods Enzymol. 2009;453:273–286. doi: 10.1016/S0076-6879(08)04013-5. [DOI] [PubMed] [Google Scholar]
- 25.Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11:448–457. doi: 10.1038/sj.cdd.4401359. [DOI] [PubMed] [Google Scholar]
- 26.Sun S, Wong TS, Zhang XQ, et al. Protein alterations associated with temozolomide resistance in subclones of human glioblastoma cell lines. J Neurooncol. 2012;107:89–100. doi: 10.1007/s11060-011-0729-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noiva R. Protein disulfide isomerase: the multifunctional redox chaperone of the endoplasmic reticulum. Semin Cell Dev Biol. 1999;10:481–493. doi: 10.1006/scdb.1999.0319. [DOI] [PubMed] [Google Scholar]
- 28.Sun S, Poon RT, Lee NP, et al. Proteomics of hepatocellular carcinoma: serum vimentin as a surrogate marker for small tumors (<or=2 cm) J Proteome Res. 2010;9:1923–1930. doi: 10.1021/pr901085z. [DOI] [PubMed] [Google Scholar]
- 29.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 30.Zhou YH, Wu X, Tan F, et al. PAX6 suppresses growth of human glioblastoma cells. J Neurooncol. 2005;71:223–229. doi: 10.1007/s11060-004-1720-4. [DOI] [PubMed] [Google Scholar]
- 31.Sun S, Lee D, Lee NP, et al. Hyperoxia resensitizes chemoresistant human glioblastoma cells to temozolomide. J Neurooncol. 2012;109:467–475. doi: 10.1007/s11060-012-0923-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581:3641–3651. doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kern J, Untergasser G, Zenzmaier C, et al. GRP-78 secreted by tumor cells blocks the antiangiogenic activity of bortezomib. Blood. 2009;114:3960–3967. doi: 10.1182/blood-2009-03-209668. [DOI] [PubMed] [Google Scholar]
- 34.Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem. 2003;278:20915–20924. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- 35.Dong D, Ko B, Baumeister P, et al. Vascular targeting and antiangiogenesis agents induce drug resistance effector GRP78 within the tumor microenvironment. Cancer Res. 2005;65:5785–5791. doi: 10.1158/0008-5472.CAN-05-0754. [DOI] [PubMed] [Google Scholar]
- 36.Pootrakul L, Datar RH, Shi SR, et al. Expression of stress response protein Grp78 is associated with the development of castration-resistant prostate cancer. Clin Cancer Res. 2006;12:5987–5993. doi: 10.1158/1078-0432.CCR-06-0133. [DOI] [PubMed] [Google Scholar]
- 37.Penas C, Guzman MS, Verdu E, Fores J, Navarro X, Casas C. Spinal cord injury induces endoplasmic reticulum stress with different cell-type dependent response. J Neurochem. 2007;102:1242–1255. doi: 10.1111/j.1471-4159.2007.04671.x. [DOI] [PubMed] [Google Scholar]
- 38.Suyama K, Watanabe M, Sakabe K, et al. Overexpression of GRP78 protects glial cells from endoplasmic reticulum stress. Neurosci Lett. 2011;504:271–276. doi: 10.1016/j.neulet.2011.09.045. [DOI] [PubMed] [Google Scholar]
- 39.Pyrko P, Schonthal AH, Hofman FM, Chen TC, Lee AS. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007;67:9809–9816. doi: 10.1158/0008-5472.CAN-07-0625. [DOI] [PubMed] [Google Scholar]
- 40.Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11:2807–2850. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- 41.Goplen D, Wang J, Enger PO, et al. Protein disulfide isomerase expression is related to the invasive properties of malignant glioma. Cancer Res. 2006;66:9895–9902. doi: 10.1158/0008-5472.CAN-05-4589. [DOI] [PubMed] [Google Scholar]
- 42.Lovat PE, Corazzari M, Armstrong JL, et al. Increasing melanoma cell death using inhibitors of protein disulfide isomerases to abrogate survival responses to endoplasmic reticulum stress. Cancer Res. 2008;68:5363–5369. doi: 10.1158/0008-5472.CAN-08-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karala AR, Ruddock LW. Bacitracin is not a specific inhibitor of protein disulfide isomerase. FEBS J. 2010;277:2454–2462. doi: 10.1111/j.1742-4658.2010.07660.x. [DOI] [PubMed] [Google Scholar]
- 44.Das A, Banik NL, Ray SK. Flavonoids activated caspases for apoptosis in human glioblastoma T98G and U87MG cells but not in human normal astrocytes. Cancer. 2010;116:164–176. doi: 10.1002/cncr.24699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen LY, Chiang AS, Hung JJ, Hung HI, Lai YK. Thapsigargin-induced grp78 expression is mediated by the increase of cytosolic free calcium in 9L rat brain tumor cells. J Cell Biochem. 2000;78:404–416. doi: 10.1002/1097-4644(20000901)78:3<404::aid-jcb6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 46.Sareddy GR, Geeviman K, Ramulu C, Babu PP. The nonsteroidal anti-inflammatory drug celecoxib suppresses the growth and induces apoptosis of human glioblastoma cells via the NF-kappaB pathway. J Neurooncol. 2012;106:99–109. doi: 10.1007/s11060-011-0662-x. [DOI] [PubMed] [Google Scholar]
- 47.Johnson GG, White MC, Grimaldi M. Stressed to death: targeting endoplasmic reticulum stress response induced apoptosis in gliomas. Curr Pharm Des. 2011;17:284–292. doi: 10.2174/138161211795049660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kubicek GJ, Werner-Wasik M, Machtay M, et al. Phase I trial using proteasome inhibitor bortezomib and concurrent temozolomide and radiotherapy for central nervous system malignancies. Int J Radiat Oncol Biol Phys. 2009;74:433–439. doi: 10.1016/j.ijrobp.2008.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hashida T, Kotake Y, Ohta S. Protein disulfide isomerase knockdown-induced cell death is cell-line-dependent and involves apoptosis in MCF-7 cells. J Toxicol Sci. 2011;36:1–7. doi: 10.2131/jts.36.1. [DOI] [PubMed] [Google Scholar]
- 50.Thakker DR, Natt F, Husken D, et al. Neurochemical and behavioral consequences of widespread gene knockdown in the adult mouse brain by using nonviral RNA interference. Proc Natl Acad Sci USA. 2004;101:17270–17275. doi: 10.1073/pnas.0406214101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.