

Abstract
Dopamine D2 receptor antagonism is a unifying property of all antipsychotic drugs in clinical use for schizophrenia. While often effective at ameliorating psychosis, these drugs are largely ineffective at treating negative and cognitive symptoms. Increasing attention is being focused on the complex genetics of the illness and the signaling pathways implicated in its pathophysiology. We review targeted approaches for pharmacotherapy involving the glutamatergic, GABAergic and cholinergic pathways. We also describe a number of the major genetic findings that identify signaling pathways representing potential targets for novel pharmacological intervention. These include genes in the 22q11 locus, DISC1, neuregulin/ERB4, and components of the Akt/GSK-3 pathway.
Schizophrenia, signaling and drug development
Schizophrenia is a debilitating psychiatric disorder that affects 1% of the worldwide population. It occurs both as a sporadic and as a heritable disease, typically presenting in adolescence or early adulthood and leads to great disability and distress. The clinical characteristics include positive symptoms (delusions, hallucinations, and disorganized thought, speech, and/or behavior), negative symptoms (amotivation, social withdrawal, poor relatedness, and a reduction in affective expression) and cognitive deficits (poor working memory and deficits in attention, processing speed and executive function). Patients with schizophrenia also suffer disproportionately from mood symptoms and substance abuse, and approximately 10% die from suicide1.
Schizophrenia is increasingly being understood as a neurodevelopmental disorder, with a clear genetic risk and subtle neuropathology. Although the symptoms that establish the diagnosis are usually not present until young adulthood, prodromal symptoms and endophenotypic features of cognitive and social deficits can precede psychotic illness and manifest in unaffected relatives. Treatments remain palliative and no diagnostic tests are yet available despite recognized trends in patients, including ventricular enlargement, reduced medial temporal lobe volume, and increased striatal dopamine storage and release1,2.
The advent of antipsychotic medications acting at dopamine (DA) D2 receptors (Figure 1) revolutionized the treatment of schizophrenia primarily by alleviating positive symptoms. Based on these drugs’ anti-dopaminergic properties, a DA hypothesis proposed that the positive symptoms of schizophrenia are due to an excess of DA signaling in the striatal and/or mesolimbic areas of the brain3. In contrast, negative symptoms are thought to be related to deficits in prefrontal cortical DA signaling, likely through D1 receptors4,5. The DA D2 receptor couples to Gi/o proteins to inhibit adenylate cyclase and also to modulate voltage-gated K+ and Ca2+ channels. More recently, it also has been shown to signal via an arrestin-mediated, G-protein-independent pathway6 (Figure 1). Remarkably, the mechanisms by which D2 receptor blockers exert their therapeutic actions are unknown, and the specific downstream effector molecule or molecules that must be targeted for therapeutic efficacy remain to be determined.
Figure 1. Dopamine D2receptor antagonism as a unifying property of all antipsychotic drugs in clinical use.
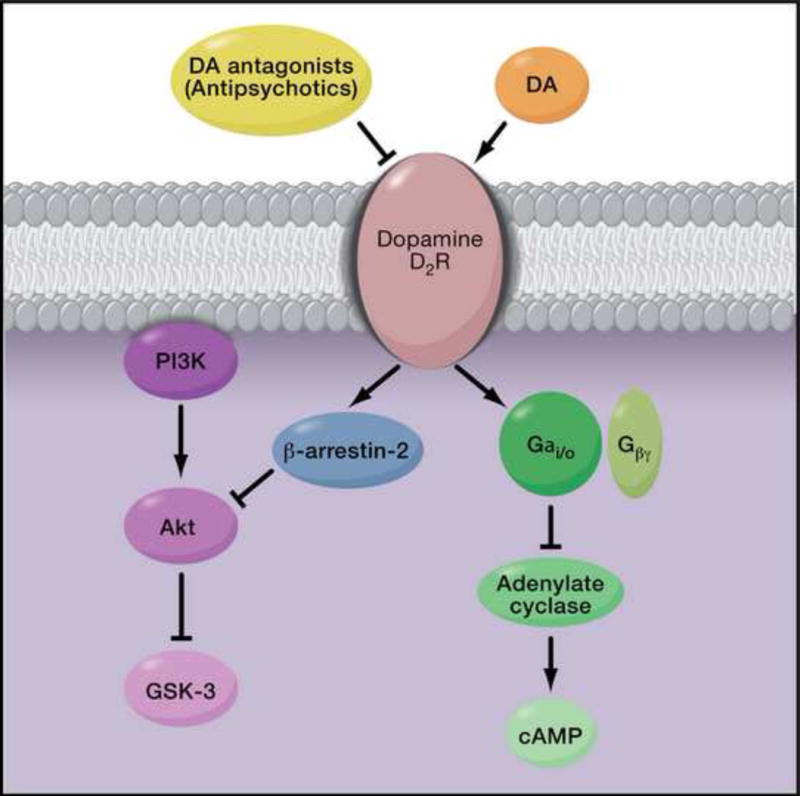
Current antipsychotic medications are thought to alleviate symptoms by blocking dopamine (DA) D2 receptor (D2R) activation and blunting dopaminergic signaling. Binding of DA to D2R results in G-protein dependent and G-protein-independent signaling. The DA D2R couples to Gi/o G-proteins to inhibit adenylate cyclase and also to modulate voltage-gated K+ and Ca2+ channels. DA binding also inhibits Akt activity in a G-protein-independent manner by recruitment of the scaffolding protein β-arrestin-2, which in turn recruits Akt and the phosphatase, PP2A. PP2A dephosphorylates Akt, leading to its inactivation and enhanced activity of the downstream kinase GSK-3.
While D2 receptor antagonism is a unifying property of all antipsychotic drugs in clinical use, these compounds have limited effectiveness against cognitive and negative symptoms. Current research efforts, which we will review below, are focused on designing drugs that target other neurotransmitter signaling pathways. Although it is not yet possible to integrate these findings into a unified pathophysiological mechanism, as these pathways are better defined, it should become increasingly possible to develop mechanistically novel and more efficacious medications.
Glutamatergic signaling
NMDA antagonists (such as phencyclidine (PCP) or ketamine) exacerbate symptoms in people with schizophrenia, and even a single exposure can mimic symptoms of schizophrenia in both healthy controls and in animal models4. Although direct NMDA agonists cannot be used clinically, allosteric enhancers such as glycine, D-serine, or D-alanine have been used with mixed results5. The glycine transporter modulates the amount of glycine available to the NMDA receptor and thus, when blocked, may provide a better glycine reserve for the receptor than a direct glycinergic agonist6 (Figure 2). Consistent with this, sarcosine, a glycine transporter antagonist, may be effective as monotherapy for positive and negative symptoms, though further work needs to be done7.
Figure 2. Glutamaergic and GABAergic Signaling.
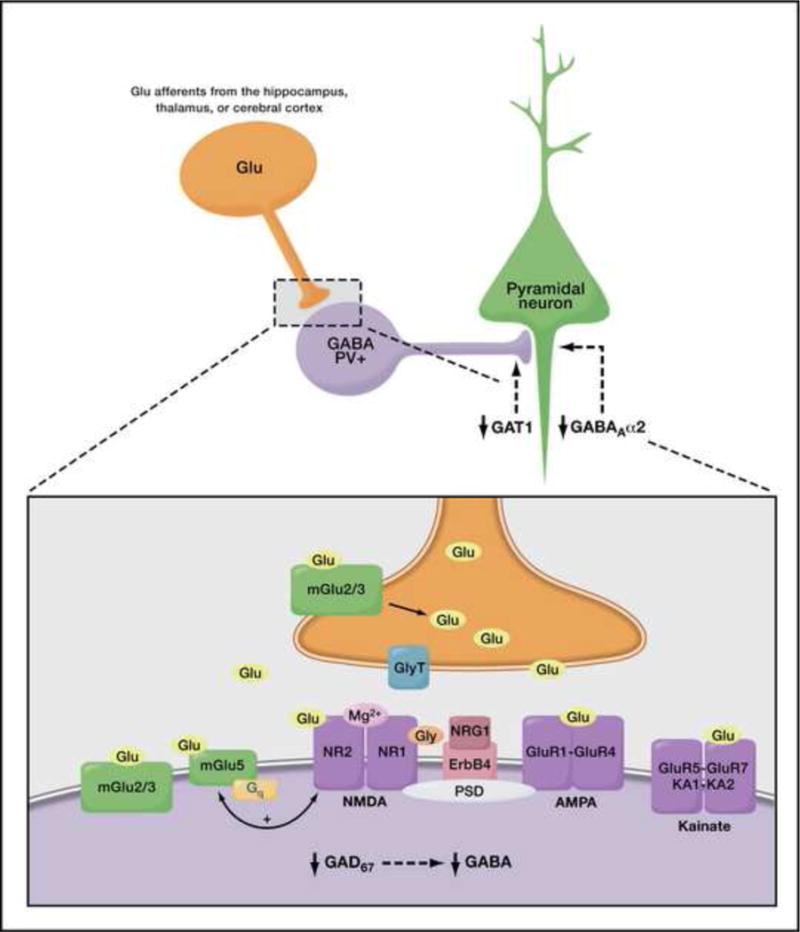
GABA receptors mediate activity in the dorsolateral prefrontal cortex (DLPFC), which plays an important role in working memory. GABA production is controlled by glutamate decarboxylase GAD67, the expression of which is decreased in patients with schizophrenia. Altered expression patterns of GABA transporter (GAT1) and the GABAA receptor alpha 2 subunit (GABAAα2) have also been observed, and α2-positive allosteric modulators are being explored for therapeutic benefits. Decreased GABA contributes to worsening of the synchronization of pyramidal cells, which is thought to contribute to deficits in working memory. Deficits in glutamatergic signaling have also been implicated in schizophrenia. Blocking the glycine transporter (GlyT) can increase the amount of the allosteric potentiator glycine that is available to the NMDA receptor (NR1/2) and enhance NMDA neurotransmission, as can D-serine, and D-cycloserine. A type II (2/3) mGluR agonist has shown initial promise in decreasing both positive and negative symptoms, although this has not been replicated. Positive allosteric modulators of mGluR5 have shown promise in animal models. Adapted with permission from Lewis and Moghaddam, Archives of Neurology 63, 1372–1376, copyright © 2006 American Medical Association. All rights reserved.
Other glutamatergic targets are also active areas of research (Figure 2). Agonism of the metabotropic glutamate receptor (mGluR) was shown to reverse PCP’s effects on locomotor activity, stereotyped movements and extracellular glutamate levels in the rat8. Because of these and other findings9, including reversal of some ketamine effects in humans10, mGluR2/3 agonists suitable for clinical use were developed and, in a preliminary study, found to improve both positive and negative symptoms11. However, these findings have not been replicated.
Other mGluRs are also increasingly the focus of research, including 5 and 8. MGluR5 may play a role in the regulation of the NMDA receptor, particularly in the forebrain. Rats with a knockout of mGluR5 show deficits in prepulse inhibition (PPI; a process that correlates with sustained attention and is a common endophenotype of schizophrenia), similar to those caused by PCP12. Similarly, rats pretreated with MPEP (a selective mGluR5 antagonist) show more significant cognitive deficits and increased hyperlocomotor behavior compared to rats administered PCP alone13. Subtype-specific allosteric modulators of mGluRs are being developed and studied preclinically13.
A mGluR5 specific positive allosteric modulator, CDPPB, was tested in vivo in rats and found to block amphetamine-induced locomotor activity14. ADX47273, another mGluR5 modulator, has also shown initial promise in rodents and may ultimately be studied in humans15. Biphenyl-indanone A (BINA), an mGluR2 allosteric modulator, inhibits PCP-induced locomotion in rats but does not alter amphetamine-induced locomotion16.
GABAergic signaling
Postmortem and imaging studies of people with schizophrenia have identified abnormalities in GABA neurotransmission that are associated with poor cognitive functioning. GABAA receptors play an important role in mediating activity in the dorsolateral prefrontal cortex (DLPFC), which is critical for working memory17. GABA production is controlled by the enzyme GAD67 (67 kD isoform of the glutamic acid decarboxylase), the expression of which is decreased in parvalbumin (PV) expressing neurons, leading to lower levels of GABA in the DLPFC18 (Figure 2). Decreased GABA contributes to impaired synchronization of pyramidal cells and is thus theorized to be related to deficits in working memory19. To remedy this deficiency, MK-0777, an allosteric modulator of the GABAA α2 receptor, was developed with the aim of increasing GABA transmission from chandelier neurons, leading to enhanced neuronal synchronization across the DLPFC19. Focusing on this α2 GABAA subunit may more specifically target the PV+ neurons and decrease the risk for further disruption of synchronization and potentially worsening cognition, such as can occur with conventional benzodiazepines. In an initial double-blind, placebo-controlled trial of MK-0777, schizophrenia patients on stable doses of antipsychotic medications improved in three working memory tasks, although the study was underpowered for statistical significance20; additional studies are ongoing, with at least one larger study failing to replicate these results21. Baclofen, a GABAB agonist, has also been shown to reverse PCP effects on PPI in rats in a dose-dependent fashion22.
Cholinergic signaling
The cholinergic system has also gained attention as a potential target for treating negative and cognitive symptoms because cholinergic neurons innervate anatomical structures implicated in schizophrenia and participate in processes that are altered in patients such as attention, working memory, and motivated behaviors23. Relevant cholinergic nuclei are found in the (1) nucleus basalis of Meynert and medial septum, which innervate the prefrontal cortex and hippocampus, respectively, (2) laterodorsal tegmental area, which innervates the ventral tegmental area, and (3) cholinergic interneurons in the nucleus accumbens (Figure 3). Curiously, about 80% of individuals with schizophrenia smoke, in contrast to a substantially smaller fraction of the general population, and this has been interpreted to reflect a potential self-medication mechanism to ameliorate deficits in sensory filtering and cognitive processes24. Studies in animal models and postmortem tissue, as well as recent clinical studies suggest a direct role for the alpha7 nicotinic (α7) and muscarinic M1 receptor subtypes in the production of these symptoms25,26, although a role for other nicotinic (α4β2) and muscarinic (M4 and M5) receptors has not been ruled out (Figure 3).
Figure 3. Cholinergic Signaling.
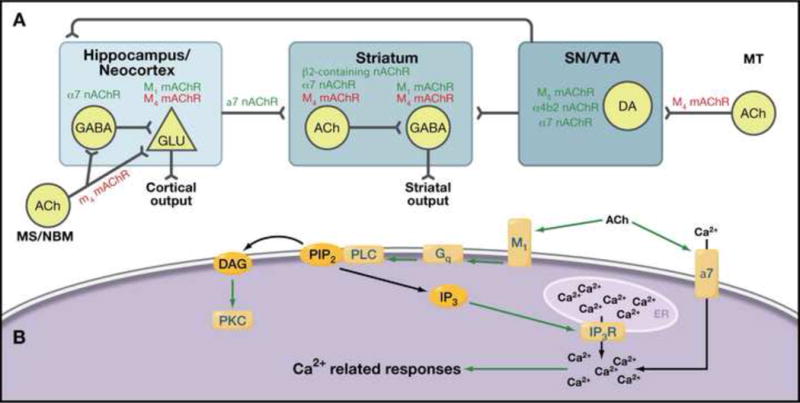
(A) Expression profile of nicotinic and muscarinic receptors within structures associated with schizophrenia (squares) and areas projecting to these structures. Green: excitatory receptors; Red: inhibitory receptor; MS: medial septum; NBM: nucleus basalis of meynert; MT: mesopontine tegmentum; SN substantia nigra pars compacta; VTA: ventral tegmental area. Although it has been very difficult to target specific muscarinic receptors with drugs, the recent identification of highly specific allosteric modulators of multiple receptor subtypes will make it possible to explore targeted pharmacotherapies. (B) Intracellular signaling associated with nicotinic receptor alpha 7 (nAChR α7) and the G-protein-coupled muscarinic receptor M1. nAChR α7 has high permeability for Ca2+ ions and channel opening promotes Ca2+ influx from the extracellular milieu and subsequent activation of Ca2+-associated signals. M1 signal is mediated by the Gq/11-PLC pathway, which includes activation of PKC and Ca2+-associated signaling. α7 agonists have shown potential promise in treating negative and cognitive symptoms, but rapid receptor desensitization makes this a difficult target.
Alpha7 receptors
In patients with schizophrenia, a reduction in α7 nicotinic receptor levels in the prefrontal cortex and hippocampus has been reported, with no significant change in the number of α7-positive neurons. In rodents, reduction in hippocampal α7 levels or activation correlates with deficits in PPI. In addition, an autosomal dominant polymorphism of the α7 gene (15q14) has been linked to PPI deficits in humans24,25.
Activation of α7 by AZD0328 has been shown to enhance cortical DA release and improve learning and attention processes in rodents27. In clinical studies, α7 agonists have been assessed for effects on cognitive symptoms. In a three-arm crossover-designed study, DMXB-A (a partial agonist selective for α7 and an α4β2 antagonist) showed improvement in negative symptoms28. Improvements in working memory and attention were observed at the end of the first arm, but these were obscured by a potential practice effect at the end of the study. Recently, varenicline (a partial agonist for α4β2 and a full agonist for α7) was evaluated in a small open-label study29. Although mood-associated side effects have been reported30–32, varenicline treatment demonstrated a significant improvement in verbal but not spatial memory, and a decrease in smoking.
M1 muscarinic receptors
Reduced levels of M1 receptors have been observed in cortical areas of schizophrenia patients, with a profound reduction in M1 levels in the DLPFC in a subpopulation of patients (75% reduction in 25% of the patients). In addition, studies in M1 knockout mice have shown deficits in working memory-related tasks but normal hippocampal-related memory33. These results suggest that impairment of cortical M1 signaling induces cortical dysfunction or information exchange deficits between cortex and hippocampus that could lead to the working memory deficits observed in schizophrenia. In a small pilot study, xanomeline (a M1/M4 agonist and M5 antagonist) produced a significant improvement in cognitive function, especially in verbal memory and working memory34; positive and negative symptoms also improved. Altogether, evidence tends to suggest that improvements in working memory are related to M1 activation in cortical areas, especially in the DLPFC, although the antipsychotic effects of xanomeline could be associated with activation of M1 or M4 receptors in striatal neurons or inhibition of M5 receptors in dopaminergic projections of the ventral tegmental area. Furthermore, since xanomeline also binds to certain serotonin receptors as well as the dopamine D3 receptor35, its mechanism of action is difficult to ascertain.
Genetics and animal models
Schizophrenia, like other common diseases, is multifactorial in nature with contributions from multiple susceptibility genes in conjunction with epigenetic, stochastic, and environmental factors36. Numerous family, twin and adoption studies have shown that genetic factors play a major role in the development of schizophrenia — a monozygotic twin of a person diagnosed with schizophrenia has ~50% likelihood to develop the disorder, as opposed to the ~1% prevalence in the general population37. Support for a gene-environment interaction comes from epidemiological data focusing on the pre-/perinatal period and adolescence. Maternal factors that increase risk include prenatal infections and malnutrition38. Perinatal hypoxia appears to be a key component for the association of preterm birth and obstetric complications with increased risk39,40. Cannabis exposure, urban living, and stress during adolescence, a period of ongoing brain development in humans, also increase the risk of schizophrenia38.
A number of meta-analyses have examined the results of over 1000 genetic association studies attempting to link candidate genes to schizophrenia41,42. The elucidation of genetic mechanisms has been challenging: both common allelic variants, or single nucleotide polymorphisms (SNPs), even when replicated, cause only a slight increase in disease risk (Odds Ratios generally <1.5). Recent genome wide association studies have led to the identification of several rare copy number variations (CNVs), which are more highly penetrant and appear more strongly associated with psychiatric disease43. The genetic architecture underlying disease susceptibility is characterized by both the frequency and penetrance of risk alleles. The common disease-common allele hypothesis emphasizes the importance of relatively common alleles, each of small effect, acting together to increase disease risk. Conversely, the common disease-rare allele hypothesis emphasizes the impact of individually rare yet highly penetrant alleles. These models are not mutually exclusive and there may be interaction between common and rare alleles at the functional level43.
The wealth of genetic information points to multiple disease pathways, similar to findings in other complex polygenic disorders such as inflammatory bowel disease, diabetes, and hypertension. However, the same alleles have been associated with disparate neuropsychiatric disorders (schizophrenia, bipolar and unipolar depression, mental retardation, seizures, autism), making it difficult to develop a comprehensive mechanistic framework based on current genetic data. Furthermore, the nonspecificity of the genetic findings also makes it difficult to design a pharmacological intervention that will treat all the symptoms of schizophrenia and makes it more likely that novel therapies inspired by genetic findings will target symptoms that might be shared by a number of psychiatric disorders, e.g., cognitive dysfunction. Consistent with this notion, genetic research has led to the development of a number of animal models of cognitive endophenotypes of psychiatric disease44–46. Below we describe a number of the major genetic findings that implicate signaling pathways representing potential targets for the development of novel pharmacological agents.
22q11 Deletion Syndrome & Schizophrenia Candidate Genes
The 22q11 Deletion Syndrome (22q11DS) (Figure 4) is a congenital malformation syndrome caused by 22q11.2 microdeletions, which occurs in one of every 6000 births47. It has been estimated that ~25–30% of all children with 22q11.2 microdeletions go on to develop schizophrenia48. Conversely, 22q11.2 microdeletions account for up to 1–2% of non-familial (sporadic) cases of schizophrenia49,50. To date, the bidirectional association for this CNV has not been demonstrated for any other chromosomal locus or schizophrenia candidate gene. Knock-out mice with a deletion of the region that corresponds with the 22q11.2 locus have decreased density of dendritic spines and glutamatergic synapses, impaired dendritic growth51, as well as impaired connectivity between the hippocampus and prefrontal cortex52.
Figure 4. The 22q11 locus.
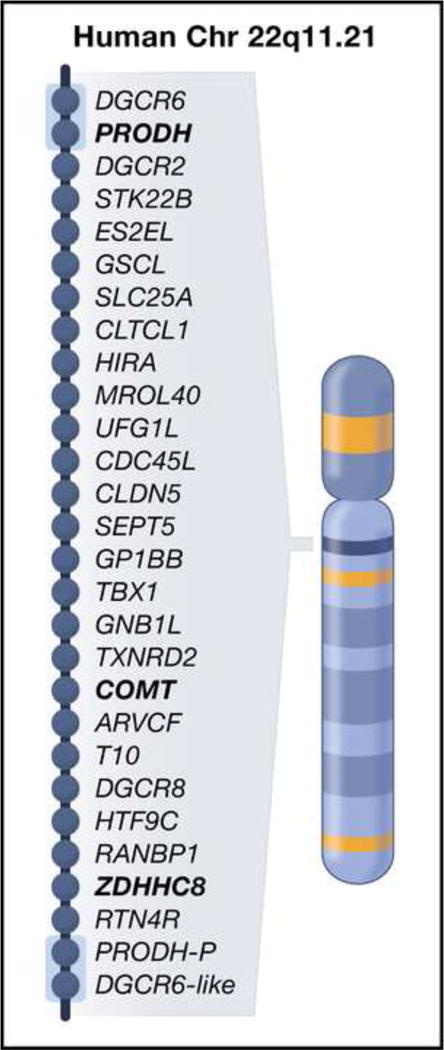
This 1.5 megabase region is flanked by low-copy repeat sequences (light blue boxes) making it prone to nonhomologous recombination. Approximately 30% of all individuals with 22q11.3 microdeletions develop symptoms of schizophrenia. Three schizophrenia candidate genes in this region are highlighted in bold font: proline dehydrogenase, PRODH, catechol-O-methyltransferase, COMT, and a palmitoyl-transferase, ZDHHC8. PRODH-P and DGRC6-like are pseudogenes. Adapted with permission from Dr. Alexander Arguello and Dr. Joseph Gogos, Department of Physiology and Cellular Biophysics, Columbia University, New York, NY.
Proline dehydrogenase (PRODH), located at the 22q11.2 locus, has been implicated in the development of schizophrenia in patients with 22q11 DS53,54. Several subsequent studies have provided additional evidence that PRODH may play a role in the pathogenesis of schizophrenia in other patient populations55. There are at least two mechanisms by which decreased activity of PRODH can disturb neuronal function and affect disease susceptibility. First, L-proline itself may function as a direct modulator of glutamatergic transmission in the brain56,57. Second, several lines of independent research have also implicated PRODH in the initiation of apoptosis58,59 as proline oxidation supports the generation of reactive oxygen species by donating reducing potential to an electron transport chain. Prodh-deficient mice present with regional alterations of GABA, glutamate, and dopamine in the brain accompanied by deficits in sensorimotor gating57. Furthermore, measurements of serum and brain L-proline levels revealed an increase in mice homozygous for this mutation. These elevated proline levels are comparable to those observed in some individuals with the 22q11 microdeletion and in certain carriers of PRODH rare variants.
ZDHHC8, another schizophrenia candidate gene located at the 22q11 locus, encodes a putative palmitoyl-transferase (PAT)60,61. Protein palmitoylation involves the reversible post-translational attachment of the 16-carbon saturated fatty acid palmitate to specific cysteine residues and is critical for membrane targeting62,63. In addition to patients with 22q11.2 microdeletions, several other patient populations with schizophrenia were found to harbor a genetic variant of ZDHHC864. Mukai et al. demonstrated that re-introduction of enzymatically active ZDHHC8 protein to the 22q11 mouse model prevents the dendritic defects caused by the deletion51. Zdhhc8-deficient mice also have similar alterations in hippocampal neurons, as well as both cognitive and behavioral deficits. ZDHHC8 can palmitoylate postsynaptic density-95 (PSD95), an adaptor molecule known to modulate the number of dendritic spines62, and possibly dendritic branches65.
COMT, which encodes for catechol-O-methyltransferase, is also located in the 22q11 locus66. COMT metabolizes catecholamines, including dopamine, and variation in COMT activity may have effects specific to the prefrontal cortex. The contribution of COMT to the development of schizophrenia is not clear. Both high and low activity of this enzyme might contribute to schizophrenia susceptibility, depending on the genetic context48. Furthermore, COMT appears to have a functionally complex allelic architecture with certain alleles (Val158Met) affecting the stability of the protein while other alleles determine the level of expression67.
Transcriptional profiling and pharmacological manipulations revealed a transcriptional and behavioral interaction between PRODH and COMT that may modulate the risk and/or the expression of the 22q11-associated psychiatric phenotypes56. COMT upregulation has been hypothesized to be one of the mechanisms that compensates for the enhanced dopaminergic signaling induced by PRODH deficiency in the frontal cortex. Therefore, individuals with schizophrenia who have a 22q11.2 deletion may be at a particular disadvantage because they are hemizygous for both these genes and perhaps unable to compensate for the cortical dopaminergic dysregulation induced by PRODH deficiency.
Neuregulin/ERB4
More than 80 SNPS within the NRG1/ErbB4 receptor/ligand pair (Figure 5) have been associated with schizophrenia43. Although this association has been negative in some studies, there are multiple reports of NRG1 association with endophenotypes in patients: decreased PPI68 , reduced white matter integrity69,70, hypofrontality, age of onset of psychosis, and premorbid IQ71. The gene produces multiple isoforms through alternate promoters and splicing72.The majority of schizophrenia-associated genetic polymorphisms in NRG1 are found in the 5’ region73, possibly leading to alterations of NRG1 protein expression and/or function.
Figure 5. Neuregulin (NRG1) Isoforms and ErbB (EGF receptor tyrosine kinase) signaling pathways.
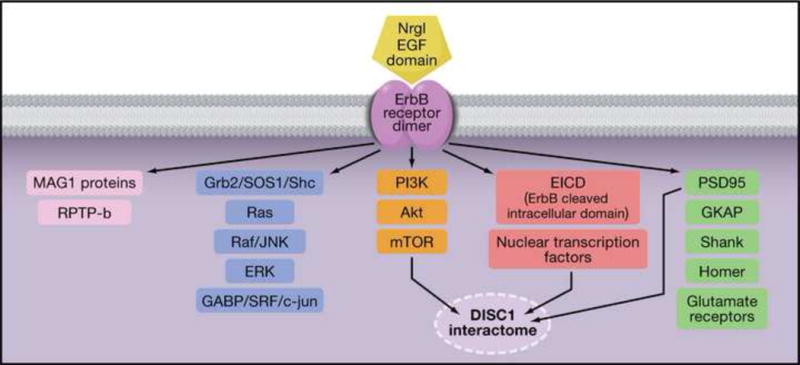
More than 80 SNPs within the Erb4/NRG1 receptor/ligand pair have been associated with schizophrenia. All NRG1 isoforms signal through an EGF domain, either through paracrine, juxtacrine, or autocrine mechanisms. ErbB2, ErbB3, and ErbB4 form dimers and heterodimers, which signal through multiple pathways. Classical signals are transduced through the Grb2 and Shc adaptor molecules and the Raf–MEK–ERK, or JUN kinase cascade to activate transcription factors such as GABP, SRF, and c-jun. Also activated are the PI3K, AKT, mTOR pathway. Brain-specific receptor-type proteintyrosine phosphatase, RPTP-beta, modulates ErbB4 signals through an interaction with MAGI proteins. Additionally, ErbB4 colocalizes with PSD95 and may modulate NMDA, AMPA, and metabotropic glutamate receptor function. The non-cannonical ErbB signals are transmitted by a cleaved ErbB intracellular domain (EICD), which translocates to the nucleus and interacts with nuclear transcription factors. Three ErbB signaling pathways overlap with the disrupted in Schizophrenia 1, DISC1, interactome, namely, AKT-mTOR, EICD, and PSD95 mediated signals.
NRG1 plays a role in numerous processes implicated in schizophrenia, including myelination, glial cell development, migration of radial glial cells during cortical development, neuronal plasticity via NMDA receptor function, development of GABAergic interneurons, and dopamine and serotonin receptor and monoamine transporter expression72. Recent work has highlighted the modulating role of NRG1 in dendritic spine formation74 and maintenance of electrophysiological gamma oscillations, which are critical for information processing in the hippocampus75.
Complete disruption of Nrg1 through traditional knockout approaches is lethal. Animals with heterozygous disruption of Nrg1 or of ErbB4 receptor function are hyperactive and show PPI deficits76, altered social behavior and anxiety77, memory deficits, reduced inhibitory interneurons and increased ventricular volume, all consistent with schizophrenia74. Interestingly, NRG1 has been shown to increase the expression of α7 nicotinic receptors in ventral hippocampal projections and interneurons, and the expression of α7 receptors is reduced in type III NRG1 deficient animals78. Recent findings support the idea that altered NRG1-mediated signaling changes the nicotinic receptor profile in presynaptic inputs, leading to deficits in PPI by altering glutamatergic transmission from ventral hippocampus to nucleus accumbens. Additionally, the Nrg1/ErbB signaling pathway connects to an extensive array of partners, many of which may be suitable future drug targets, such as PI3 kinases, the PTPRZ1-RPTPβ phosphatase, the BACE secretase and more distantly, the DISC1 pathway79,80 (Figure 5).
Disc1
Disrupted in schizophrenia (DISC1) (Figure 6) is a gene locus originally identified in a Scottish family, many of whom carried a balanced translocation between chromosomes 1 and 1181. Of 37 individuals with this translocation, 29 had a psychiatric diagnosis including schizophrenia (7), bipolar disorder (1), and recurrent major depression (10). Linkage and association studies have also supported a role for the DISC1 locus in schizophrenia82. Recent studies have shown abnormalities in the expression of DISC1 splice variants in schizophrenia83, whereas postmortem findings of DISC1 expression have been limited and preliminary, showing no clear patterns of alteration82.
Figure 6. Disrupted in Schizophrenia1 (DISC1).
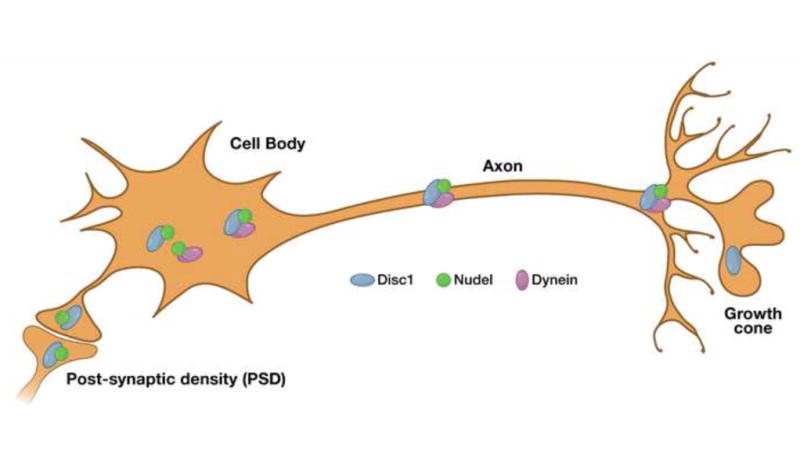
Disrupted in schizophrenia (DISC1) is a gene locus originally identified in a Scottish family, many of whom carried a balanced translocation between chromosomes 1 and 11 and who had a high frequency of psychiatric disorders, including schizophrenia, bipolar disorder, and recurrent major depression. Linkage and association studies have also supported a role for the DISC1 locus in schizophrenia. DISC1 associates with important cellular components, including the centrosome, microtubules, terminals and neurite growth cones, enabling it to play a role in various cellular functions, such as neuronal migration and axonal elongation, as well as microtubule transport and organization. Note that DISC1 can also inhibit GSK-3 activity, and thus potentially overlaps with dopaminergic signaling and NRG1/ERB4 signaling. Adapted with permission from Macmillan Publishers Ltd: Molecular Psychiatry (Chubb et al. [2008] Mol. Psych. 13, 36–64), copyright 2008.
Alterations or suppression of DISC1 in cell cultures and in mice cause impaired neurite outgrowth, abnormal neuronal migration, and abnormal pyramidal neuronal orientation and development of the cerebral cortex, similar to the observed pathology of schizophrenia84. The product of DISC1 also interacts with NUDEL, important in the transport of microtubules and cellular migration, microtubule-associated proteins and the dynein motor protein complex, and promotes microtubule organization in the cell84–86 (Figure 6). Additionally, findings that the DISC1 protein localizes to the synapse, as well as the potential importance of DISC1 for neurite development, suggest a significant role for DISC1 at the synapse87. Finally, alterations of DISC1 in mice produce deficits in working memory88.
These data on DISC1 suggest the possibility that a microtubule-stabilizing and synapse-reinforcing agent might be beneficial in schizophrenia. One such agent is NAP, an eight amino acid neuroprotective peptide89 that can cross the cell membrane, bind to glial tubulin, and promote microtubule reorganization90. NAP also increases synaptogenesis in both hippocampal and cortical regions and promotes neurite outgrowth in cells from rat hippocampal regions91. NAP may affect synaptic plasticity by promoting synaptic reconnections after injury in the mature brain91. Most importantly, cognitive/memory enhancing effects after NAP administration have been observed in animal models of cholinotoxicity, Apo-E deficiency, Alzheimer’s disease, and middle age89,92. Therefore, the targeted treatment of neurocognition in individuals with schizophrenia with a microtubule-stabilizing agent such as NAP may be possible.
Akt1/GSK-3
Akt is a protein kinase involved in a variety of cellular functions including metabolism, cell stress, and cell-cycle regulation. Akt also plays a role in regulating neuronal cell size, survival and possibly synaptic plasticity93–95. While three isoforms of Akt have been identified (Akt1, Akt2 and Akt3)96, Akt1 has been the primary focus in almost all studies examining roles for Akt in schizophrenia. Akt1 haplotypes cosegregate with schizophrenia, suggesting that the AKT1 gene may be a schizophrenia susceptibility gene97. Similarly, genetic studies demonstrated association of AKT1 gene polymorphisms with schizophrenia in diverse populations98,99. In lymphocytes derived from people with schizophrenia, there was a 68% reduction in Akt1 levels relative to control subjects97. Additionally, in brain, relative to control subjects, reductions in Akt1 were also found in the hippocampus and frontal cortex using post-mortem brain samples from those with schizophrenia97.
Phosphatidylinositol 3-kinase (PI3K) promotes Akt recruitment to the plasma membrane, where it is activated through sequential phosphorylation94,100 (Figure 1). Consistent with the relevance of this pathway for schizophrenia, polymorphisms in the promoter region for PI3K gene, PI3KC3, have been found to increase risk for schizophrenia101,102. Once active, Akt phosphorylates numerous molecules including glycogen synthase kinase-3 (GSK-3)103, a protein that is also regulated by DISC1104. Besides mediating glucose metabolism, GSK-3 may also function to regulate synaptic plasticity105. Whereas Akt is activated by phosphorylation, GSK-3 is inactivated when phosphorylated by Akt103. Consistent with reduced Akt1, levels of the phosphorylated GSK-3 isoform, GSK-3β, were shown to be diminished in the frontal cortex of people with schizophrenia97. Moreover, Akt is inactivated by the PP2A phosphatase in an arrestin-dependent complex promoted by D2 receptor activation, thereby increasing GSK-3 activity106. By blocking DA D2 receptor activation, antipsychotic medications would be expected to enhance Akt activity, and this has been postulated to be part of their mechanism of therapeutic efficacy107.
Conclusion
Our current criteria for the diagnosis of schizophrenia evolved to group patients with shared symptoms and course of illness. The general effectiveness of D2 receptor antagonists in ameliorating psychosis suggested the possibility of a common pathobiology. However, current antipsychotics have multiple side effects with the potential to induce profound morbidity and are ineffective in treating cognitive deficits and negative symptoms. The complex genetics of the syndrome we call schizophrenia suggests a much greater heterogeneity of its etiology and pathophysiology. To date clinical trials of nondopaminergic therapies have met with at best limited success, and the challenge of developing new drugs with efficacy in schizophrenia is daunting. As we enter the era of personalized medicine, in which treatments may be more selectively matched to specific patient subtypes, developing novel agents directed at pathophysiologic or etiologic targets will be necessary. Grouping patients based on genetics, neurochemical, structural and functional imaging, electrophysiologic assessment and other methods of identifying endophenotypic features may increase the power of clinical trials to validate novel targeted treatments. For example, comparing treatment response to α7 nicotinic receptor drugs based on NRG1 genotype, or response to drugs that work through glutamatergic or GABAergic mechanisms based on impaired Gamma oscillations, may lead to more effective treatments of specific deficits in distinct patient populations.
Cognitive symptoms are most predictive of outcome in schizophrenia, and any therapeutic advance in this area would be of great importance. New drugs might replace dopaminergic drugs altogether or might be used as adjunctive agents to improve cognitive and/or negative symptoms. There are immense challenges in developing newer more effective medications, especially since the targets haven’t been elucidated fully, nor have efforts in evaluating these targets been exhausted. Yet, there are signs of hope. The recent identification of novel subtype-selective allosteric modulators of acetylcholine receptors, for example, provides an opportunity for the first time to evaluate cholinergic therapies in a way not contaminated by lack of selectivity and off target effects. Given the high cost and level of risk in developing drugs with new mechanisms of action, it will be important to test them in rigorous and innovative trials that will yield clear and reliable results.
Acknowledgments
The co-first authors are supported by NIMH Schizophrenia Training Grant T32 MH018870. This work also was supported in part by a NARSAD Young Investigator Award (NMB), by NIH grants DA022413 and MH54137 (JAJ) and by the Lieber Center for Schizophrenia Research and Treatment.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lewis DA, Lieberman JA. Catching up on schizophrenia: natural history and neurobiology. Neuron. 2000;28:325–334. doi: 10.1016/s0896-6273(00)00111-2. [DOI] [PubMed] [Google Scholar]
- 2.Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III--the final common pathway. Schizophr Bull. 2009;35:549–562. doi: 10.1093/schbul/sbp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carlsson A. The current status of the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1988;1:179–186. doi: 10.1016/0893-133x(88)90012-7. [DOI] [PubMed] [Google Scholar]
- 4.Steinpreis RE. The behavioral and neurochemical effects of phencyclidine in humans and animals: some implications for modeling psychosis. Behav Brain Res. 1996;74:45–55. doi: 10.1016/0166-4328(95)00162-x. [DOI] [PubMed] [Google Scholar]
- 5.Buchanan RW, et al. The Cognitive and Negative Symptoms in Schizophrenia Trial (CONSIST): the efficacy of glutamatergic agents for negative symptoms and cognitive impairments. Am J Psychiatry. 2007;164:1593–1602. doi: 10.1176/appi.ajp.2007.06081358. [DOI] [PubMed] [Google Scholar]
- 6.Javitt DC. Glycine transport inhibitors and the treatment of schizophrenia. Biol Psychiatry. 2008;63:6–8. doi: 10.1016/j.biopsych.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 7.Lane HY, et al. Sarcosine (N-methylglycine) treatment for acute schizophrenia: a randomized, double-blind study. Biol Psychiatry. 2008;63:9–12. doi: 10.1016/j.biopsych.2007.04.038. [DOI] [PubMed] [Google Scholar]
- 8.Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281:1349–1352. doi: 10.1126/science.281.5381.1349. [DOI] [PubMed] [Google Scholar]
- 9.Moghaddam B. Targeting metabotropic glutamate receptors for treatment of the cognitive symptoms of schizophrenia. Psychopharmacology. 2004;174:39–44. doi: 10.1007/s00213-004-1792-z. [DOI] [PubMed] [Google Scholar]
- 10.Krystal JH, et al. Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology (Berl) 2005;179:303–309. doi: 10.1007/s00213-004-1982-8. [DOI] [PubMed] [Google Scholar]
- 11.Patil ST, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- 12.Kinney GG, et al. Metabotropic glutamate subtype 5 receptors modulate locomotor activity and sensorimotor gating in rodents. J Pharmacol Exp Ther. 2003;306:116–123. doi: 10.1124/jpet.103.048702. [DOI] [PubMed] [Google Scholar]
- 13.Conn PJ, et al. Activation of metabotropic glutamate receptors as a novel approach for the treatment of schizophrenia. Trends Pharmacol Sci. 2009;30:25–31. doi: 10.1016/j.tips.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kinney GG, et al. A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J Pharmacol Exp Ther. 2005;313:199–206. doi: 10.1124/jpet.104.079244. [DOI] [PubMed] [Google Scholar]
- 15.Schlumberger C, et al. Comparison of the mGlu(5) receptor positive allosteric modulator ADX47273 and the mGlu(2/3) receptor agonist LY354740 in tests for antipsychotic-like activity. Eur J Pharmacol. 2009 doi: 10.1016/j.ejphar.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 16.Galici R, et al. Biphenyl-indanone A, a positive allosteric modulator of the metabotropic glutamate receptor subtype 2, has antipsychotic- and anxiolytic-like effects in mice. J Pharmacol Exp Ther. 2006;318:173–185. doi: 10.1124/jpet.106.102046. [DOI] [PubMed] [Google Scholar]
- 17.Rao SG, et al. Destruction and creation of spatial tuning by disinhibition: GABA(A) blockade of prefrontal cortical neurons engaged by working memory. J Neurosci. 2000;20:485–494. doi: 10.1523/JNEUROSCI.20-01-00485.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hashimoto T, et al. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci. 2003;23:6315–6326. doi: 10.1523/JNEUROSCI.23-15-06315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis DA, Moghaddam B. Cognitive dysfunction in schizophrenia: convergence of gamma-aminobutyric acid and glutamate alterations. Arch Neurol. 2006;63:1372–1376. doi: 10.1001/archneur.63.10.1372. [DOI] [PubMed] [Google Scholar]
- 20.Lewis DA, et al. Subunit-selective modulation of GABA type A receptor neurotransmission and cognition in schizophrenia. Am J Psychiatry. 2008;165:1585–1593. doi: 10.1176/appi.ajp.2008.08030395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buchanan RW. American College of Neuropsychopharmacology. 2009. MK0777 for the treatment of cognitive impairements in people with schizophrenia. [Google Scholar]
- 22.Fejgin K, et al. Prefrontal GABA(B) receptor activation attenuates phencyclidine-induced impairments of prepulse inhibition: involvement of nitric oxide. Neuropsychopharmacology. 2009;34:1673–1684. doi: 10.1038/npp.2008.225. [DOI] [PubMed] [Google Scholar]
- 23.Berman JA, et al. Cholinergic circuits and signaling in the pathophysiology of schizophrenia. Int Rev Neurobiol. 2007;78:193–223. doi: 10.1016/S0074-7742(06)78007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mobascher A, Winterer G. The molecular and cellular neurobiology of nicotine abuse in schizophrenia. Pharmacopsychiatry. 2008;41(Suppl 1):S51–59. doi: 10.1055/s-2008-1081463. [DOI] [PubMed] [Google Scholar]
- 25.Ochoa EL, Lasalde-Dominicci J. Cognitive deficits in schizophrenia: focus on neuronal nicotinic acetylcholine receptors and smoking. Cell Mol Neurobiol. 2007;27:609–639. doi: 10.1007/s10571-007-9149-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raedler TJ, et al. Towards a muscarinic hypothesis of schizophrenia. Mol Psychiatry. 2007;12:232–246. doi: 10.1038/sj.mp.4001924. [DOI] [PubMed] [Google Scholar]
- 27.Sydserff S, et al. Selective alpha7 nicotinic receptor activation by AZD0328 enhances cortical dopamine release and improves learning and attentional processes. Biochem Pharmacol. 2009;78:880–888. doi: 10.1016/j.bcp.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 28.Freedman R, et al. Initial phase 2 trial of a nicotinic agonist in schizophrenia. Am J Psychiatry. 2008;165:1040–1047. doi: 10.1176/appi.ajp.2008.07071135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith RC, et al. Cognitive and antismoking effects of varenicline in patients with schizophrenia or schizoaffective disorder. Schizophr Res. 2009;110:149–155. doi: 10.1016/j.schres.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 30.FDA Information for Healthcare Professionals: Varenicline (marketed as Chantix) and Bupropion (marketed as Zyban, Wellbutrin, and generics) 2009 http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm169986.htm.
- 31.Popkin MK. Exacerbation of recurrent depression as a result of treatment with varenicline. Am J Psychiatry. 2008;165:774. doi: 10.1176/appi.ajp.2008.07111735. [DOI] [PubMed] [Google Scholar]
- 32.Freedman R. Exacerbation of schizophrenia by varenicline. Am J Psychiatry. 2007;164:1269. doi: 10.1176/appi.ajp.2007.07020326. [DOI] [PubMed] [Google Scholar]
- 33.Scarr E, Dean B. Muscarinic receptors: do they have a role in the pathology and treatment of schizophrenia? J Neurochem. 2008;107:1188–1195. doi: 10.1111/j.1471-4159.2008.05711.x. [DOI] [PubMed] [Google Scholar]
- 34.Shekhar A, et al. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. 2008;165:1033–1039. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- 35.PDSP NIMH Psychoactive Drug Screening Program: Ki Database. http://pdsp.med.unc.edu/pdsp.php.
- 36.Karayiorgou M, Gogos JA. Dissecting the genetic complexity of schizophrenia. Molecular psychiatry. 1997;2:211–223. doi: 10.1038/sj.mp.4000271. [DOI] [PubMed] [Google Scholar]
- 37.Ayhan Y, et al. Animal models of gene-environment interactions in schizophrenia. Behavioural brain research. 2009;204:274–281. doi: 10.1016/j.bbr.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keshavan MS, et al. Schizophrenia, “just the facts”: what we know in 2008 Part 3: neurobiology. Schizophr Res. 2008;106:89–107. doi: 10.1016/j.schres.2008.07.020. [DOI] [PubMed] [Google Scholar]
- 39.Nicodemus KK, et al. Serious obstetric complications interact with hypoxia-regulated/vascular-expression genes to influence schizophrenia risk. Molecular psychiatry. 2008;13:873–877. doi: 10.1038/sj.mp.4002153. [DOI] [PubMed] [Google Scholar]
- 40.Clarke MC, et al. The role of obstetric events in schizophrenia. Schizophrenia bulletin. 2006;32:3–8. doi: 10.1093/schbul/sbj028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allen NC, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nature genetics. 2008;40:827–834. doi: 10.1038/ng.171. [DOI] [PubMed] [Google Scholar]
- 42.Jia P, et al. SZGR: a comprehensive schizophrenia gene resource. Mol Psychiatry. 15:453–462. doi: 10.1038/mp.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sebat J, et al. Rare structural variants in schizophrenia: one disorder, multiple mutations; one mutation, multiple disorders. Trends Genet. 2009;25:528–535. doi: 10.1016/j.tig.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ellenbroek BA, Cools AR. Animal models for the negative symptoms of schizophrenia. Behavioural pharmacology. 2000;11:223–233. doi: 10.1097/00008877-200006000-00006. [DOI] [PubMed] [Google Scholar]
- 45.Kellendonk C, et al. Modeling cognitive endophenotypes of schizophrenia in mice. Trends in neurosciences. 2009;32:347–358. doi: 10.1016/j.tins.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gottesman, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. The American journal of psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- 47.Botto LD, et al. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003;112:101–107. doi: 10.1542/peds.112.1.101. [DOI] [PubMed] [Google Scholar]
- 48.Karayiorgou M, Gogos JA. The molecular genetics of the 22q11-associated schizophrenia. Brain research. 2004;132:95–104. doi: 10.1016/j.molbrainres.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 49.Lasseter VK, et al. Follow-up report of potential linkage for schizophrenia on chromosome 22q: Part 3. American journal of medical genetics. 1995;60:172– 173. doi: 10.1002/ajmg.1320600217. [DOI] [PubMed] [Google Scholar]
- 50.Xu B, et al. Strong association of de novo copy number mutations with sporadic schizophrenia. Nature genetics. 2008;40:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 51.Mukai J, et al. Palmitoylation-dependent neurodevelopmental deficits in a mouse model of 22q11 microdeletion. Nature neuroscience. 2008;11:1302–1310. doi: 10.1038/nn.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sigurdsson T, et al. Impaired hippocampal-prefrontal synchrony in a genetic mouse model of schizophrenia. Nature. 464:763–767. doi: 10.1038/nature08855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu H, et al. Genetic variation in the 22q11 locus and susceptibility to schizophrenia. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:16859–16864. doi: 10.1073/pnas.232186099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu H, et al. Genetic variation at the 22q11 PRODH2/DGCR6 locus presents an unusual pattern and increases susceptibility to schizophrenia. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:3717–3722. doi: 10.1073/pnas.042700699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li T, et al. Evidence for association between novel polymorphisms in the PRODH gene and schizophrenia in a Chinese population. Am J Med Genet B Neuropsychiatr Genet. 2004;129B:13–15. doi: 10.1002/ajmg.b.30049. [DOI] [PubMed] [Google Scholar]
- 56.Paterlini M, et al. Transcriptional and behavioral interaction between 22q11.2 orthologs modulates schizophrenia-related phenotypes in mice. Nature neuroscience. 2005;8:1586–1594. doi: 10.1038/nn1562. [DOI] [PubMed] [Google Scholar]
- 57.Gogos JA, et al. The gene encoding proline dehydrogenase modulates sensorimotor gating in mice. Nature genetics. 1999;21:434–439. doi: 10.1038/7777. [DOI] [PubMed] [Google Scholar]
- 58.Donald SP, et al. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer research. 2001;61:1810–1815. [PubMed] [Google Scholar]
- 59.Maxwell SA, Davis GE. Differential gene expression in p53-mediated apoptosis-resistant vs. apoptosis-sensitive tumor cell lines. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:13009–13014. doi: 10.1073/pnas.230445997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fukata M, et al. Identification of PSD-95 palmitoylating enzymes. Neuron. 2004;44:987–996. doi: 10.1016/j.neuron.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 61.Roth AF, et al. The yeast DHHC cysteine-rich domain protein Akr1p is a palmitoyl transferase. The Journal of cell biology. 2002;159:23–28. doi: 10.1083/jcb.200206120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.El-Husseini AE, et al. PSD-95 involvement in maturation of excitatory synapses. Science (New York, N.Y) 2000;290:1364–1368. [PubMed] [Google Scholar]
- 63.el-Husseini Ael D, Bredt DS. Protein palmitoylation: a regulator of neuronal development and function. Nature reviews. 2002;3:791–802. doi: 10.1038/nrn940. [DOI] [PubMed] [Google Scholar]
- 64.Mukai J, et al. Evidence that the gene encoding ZDHHC8 contributes to the risk of schizophrenia. Nature genetics. 2004;36:725–731. doi: 10.1038/ng1375. [DOI] [PubMed] [Google Scholar]
- 65.Charych EI, et al. Activity-independent regulation of dendrite patterning by postsynaptic density protein PSD-95. J Neurosci. 2006;26:10164–10176. doi: 10.1523/JNEUROSCI.2379-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shifman S, et al. A highly significant association between a COMT haplotype and schizophrenia. American journal of human genetics. 2002;71:1296–1302. doi: 10.1086/344514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bray NJ, et al. A haplotype implicated in schizophrenia susceptibility is associated with reduced COMT expression in human brain. American journal of human genetics. 2003;73:152–161. doi: 10.1086/376578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hong LE, et al. Evidence of missense mutations on the neuregulin 1 gene affecting function of prepulse inhibition. Biol Psychiatry. 2008;63:17–23. doi: 10.1016/j.biopsych.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McIntosh AM, et al. The effects of a neuregulin 1 variant on white matter density and integrity. Mol Psychiatry. 2008;13:1054–1059. doi: 10.1038/sj.mp.4002103. [DOI] [PubMed] [Google Scholar]
- 70.Winterer G, et al. Association of 5′ end neuregulin-1 (NRG1) gene variation with subcortical medial frontal microstructure in humans. Neuroimage. 2008;40:712– 718. doi: 10.1016/j.neuroimage.2007.12.041. [DOI] [PubMed] [Google Scholar]
- 71.Hall J, et al. A neuregulin 1 variant associated with abnormal cortical function and psychotic symptoms. Nat Neurosci. 2006;9:1477–1478. doi: 10.1038/nn1795. [DOI] [PubMed] [Google Scholar]
- 72.Mei L, Xiong WC. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci. 2008;9:437–452. doi: 10.1038/nrn2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harrison PJ, Law AJ. Neuregulin 1 and schizophrenia: genetics, gene expression, and neurobiology. Biol Psychiatry. 2006;60:132–140. doi: 10.1016/j.biopsych.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 74.Chen YJ, et al. Type III neuregulin-1 is required for normal sensorimotor gating, memory-related behaviors, and corticostriatal circuit components. J Neurosci. 2008;28:6872–6883. doi: 10.1523/JNEUROSCI.1815-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fisahn A, et al. Neuregulin-1 modulates hippocampal gamma oscillations: implications for schizophrenia. Cereb Cortex. 2009;19:612–618. doi: 10.1093/cercor/bhn107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stefansson H, et al. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet. 2002;71:877–892. doi: 10.1086/342734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Desbonnet L, et al. Mutant models for genes associated with schizophrenia. Biochem Soc Trans. 2009;37:308–312. doi: 10.1042/BST0370308. [DOI] [PubMed] [Google Scholar]
- 78.Hancock ML, et al. Presynaptic type III neuregulin1-ErbB signaling targets {alpha}7 nicotinic acetylcholine receptors to axons. J Cell Biol. 2008;181:511–521. doi: 10.1083/jcb.200710037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jaaro-Peled H, et al. Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends Neurosci. 2009;32:485–495. doi: 10.1016/j.tins.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hoistad M, et al. Linking white and grey matter in schizophrenia: oligodendrocyte and neuron pathology in the prefrontal cortex. Front Neuroanat. 2009;3:9. doi: 10.3389/neuro.05.009.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.St Clair D, et al. Association within a family of a balanced autosomal translocation with major mental illness. Lancet. 1990;336:13–16. doi: 10.1016/0140-6736(90)91520-k. [DOI] [PubMed] [Google Scholar]
- 82.Chubb JE, et al. The DISC locus in psychiatric illness. Molecular psychiatry. 2008;13:36–64. doi: 10.1038/sj.mp.4002106. [DOI] [PubMed] [Google Scholar]
- 83.Nakata K, et al. DISC1 splice variants are upregulated in schizophrenia and associated with risk polymorphisms. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:15873–15878. doi: 10.1073/pnas.0903413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kamiya A, et al. A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nature cell biology. 2005;7:1167–1178. doi: 10.1038/ncb1328. [DOI] [PubMed] [Google Scholar]
- 85.Brandon NJ, et al. Disrupted in Schizophrenia 1 and Nudel form a neurodevelopmentally regulated protein complex: implications for schizophrenia and other major neurological disorders. Molecular and cellular neurosciences. 2004;25:42–55. doi: 10.1016/j.mcn.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 86.Morris JA, et al. DISC1 (Disrupted-In-Schizophrenia 1) is a centrosome-associated protein that interacts with MAP1A, MIPT3, ATF4/5 and NUDEL: regulation and loss of interaction with mutation. Human molecular genetics. 2003;12:1591–1608. doi: 10.1093/hmg/ddg162. [DOI] [PubMed] [Google Scholar]
- 87.Camargo LM, et al. Disrupted in Schizophrenia 1 Interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Molecular psychiatry. 2007;12:74–86. doi: 10.1038/sj.mp.4001880. [DOI] [PubMed] [Google Scholar]
- 88.Kvajo M, et al. A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:7076–7081. doi: 10.1073/pnas.0802615105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gozes I, et al. NAP: research and development of a peptide derived from activity-dependent neuroprotective protein (ADNP) CNS drug reviews. 2005;11:353–368. doi: 10.1111/j.1527-3458.2005.tb00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Divinski I, et al. A femtomolar acting octapeptide interacts with tubulin and protects astrocytes against zinc intoxication. The Journal of biological chemistry. 2004;279:28531–28538. doi: 10.1074/jbc.M403197200. [DOI] [PubMed] [Google Scholar]
- 91.Smith-Swintosky VL, et al. Activity-dependent neurotrophic factor-9 and NAP promote neurite outgrowth in rat hippocampal and cortical cultures. J Mol Neurosci. 2005;25:225–238. doi: 10.1385/JMN:25:3:225. [DOI] [PubMed] [Google Scholar]
- 92.Matsuoka Y, et al. A neuronal microtubule-interacting agent, NAPVSIPQ, reduces tau pathology and enhances cognitive function in a mouse model of Alzheimer’s disease. The Journal of pharmacology and experimental therapeutics. 2008;325:146–153. doi: 10.1124/jpet.107.130526. [DOI] [PubMed] [Google Scholar]
- 93.Lin CH, et al. A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron. 2001;31:841–851. doi: 10.1016/s0896-6273(01)00433-0. [DOI] [PubMed] [Google Scholar]
- 94.Franke TF. PI3K/Akt: getting it right matters. Oncogene. 2008;27:6473–6488. doi: 10.1038/onc.2008.313. [DOI] [PubMed] [Google Scholar]
- 95.Niizuma K, et al. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. Journal of neurochemistry. 2009;109(Suppl 1):133–138. doi: 10.1111/j.1471-4159.2009.05897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dummler B, Hemmings BA. Physiological roles of PKB/Akt isoforms in development and disease. Biochem Soc Trans. 2007;35:231–235. doi: 10.1042/BST0350231. [DOI] [PubMed] [Google Scholar]
- 97.Emamian ES, et al. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat Genet. 2004;36:131–137. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]
- 98.Xu MQ, et al. Association of AKT1 gene polymorphisms with risk of schizophrenia and with response to antipsychotics in the Chinese population. The Journal of clinical psychiatry. 2007;68:1358–1367. doi: 10.4088/jcp.v68n0906. [DOI] [PubMed] [Google Scholar]
- 99.Thiselton DL, et al. AKT1 is associated with schizophrenia across multiple symptom dimensions in the Irish study of high density schizophrenia families. Biol Psychiatry. 2008;63:449–457. doi: 10.1016/j.biopsych.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sarbassov DD, et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 101.Stopkova P, et al. Identification of PIK3C3 promoter variant associated with bipolar disorder and schizophrenia. Biol Psychiatry. 2004;55:981–988. doi: 10.1016/j.biopsych.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 102.Duan S, et al. A family-based association study of schizophrenia with polymorphisms at three candidate genes. Neurosci Lett. 2005;379:32–36. doi: 10.1016/j.neulet.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 103.Beaulieu JM, et al. The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol Sci. 2007;28:166–172. doi: 10.1016/j.tips.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 104.Mao Y, et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell. 2009;136:1017–1031. doi: 10.1016/j.cell.2008.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Peineau S, et al. The role of GSK-3 in synaptic plasticity. British journal of pharmacology. 2008;153(Suppl 1):S428–437. doi: 10.1038/bjp.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Beaulieu JM, et al. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 107.Masri B, et al. Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13656–13661. doi: 10.1073/pnas.0803522105. [DOI] [PMC free article] [PubMed] [Google Scholar]