

Abstract
BACKGROUND & AIMS
Zinc homeostasis in cells is maintained through tight regulation of zinc influx, efflux, and distribution to intracellular organelles by zinc transporters. The Zrt-Irt-like protein (ZIP) transporters facilitate zinc influx to the cytosol. Expression of the ZIP family member Zip14 can be induced by inflammatory cytokines, which also initiate liver regeneration. Hepatocyte proliferation is required for liver regeneration. Zinc regulates cell proliferation, tissue growth, and many mitogenic signaling pathways; we investigated its role in hepatocytes.
METHODS
Wild-type and Zip14−/− mice that underwent partial hepatectomy (70% of liver removed) were used as models of liver regeneration. We also analyzed AML12 hepatocytes that overexpressed Zip14. Proliferation was assessed with proliferating cell nuclear antigen, CD1, and Ki67 markers and along with assays of zinc content was related to protein tyrosine phosphatase 1B (PTP1B) and extracellular signal–regulated kinase 1/2 signaling.
RESULTS
Zip14 was up-regulated and hepatic zinc content increased during liver regeneration. Increased hepatic zinc inhibited activity of the phosphatase PTP1B and increased phosphorylation of c-Met, which promoted hepatocyte proliferation. AML12 cells that overexpressed Zip14 increased in zinc content and proliferation; PTP1B was inhibited and phosphorylation of c-Met increased. The increases in hepatic levels of zinc and hepatocyte proliferation that occurred following partial hepatectomy were not observed in Zip14−/− mice.
CONCLUSIONS
The transporter Zip14 mediates hepatic uptake of zinc during liver regeneration and for hepatocyte proliferation. These findings indicate that zinc transporter activity regulates liver tissue growth by sequestering zinc. Reagents that regulate ZIP14 activity might be developed as therapeutics to promote liver regeneration in patients with chronic liver disease.
Keywords: Micronutrient, Signal Transduction, Tissue Regeneration, Interleukin-6
Stimulation of the regenerative capacity of the liver is a target for the treatment of chronic liver diseases.1 These therapeutic approaches include partial hepatectomy (PHx), split liver transplantation, and live donor liver transplantation. Enhanced liver regeneration (LR) is vital for the survival of the recipient and also for the donor in live donor transplantation. The enhancement of LR is also required for the success of pharmacologic approaches that aim to reverse fibrosis to treat liver disease.2 Although LR offers curative options, many patients develop postoperative complications because the remnant liver or graft is too small or of poor quality to sustain sufficient organ function.1 Hence, effective LR is a major goal in such clinical situations.
Zinc has the potential to be a factor on which to develop new therapies to enhance liver LR. Zinc supplementation has been used in a few clinical trials and was shown to have a positive effect on liver function.3–7 The functional assessments in all of those trials were measurements of serum alanine aminotransferase (ALT) activity. Thus, it is not clear if zinc has any direct effect on proliferation of hepatocytes, as required for regeneration.8 Zinc is involved in cell proliferation both as a structural element9–12 and as a regulatory element in many mitogenic signaling pathways13–15; therefore, zinc could have a positive influence on the regeneration process.
Cytokines are involved in the initiation of LR.16 They prime hepatocytes to enter the cell cycle and to respond to the effect of mitogenic factors. The main participants in this cytokine network are tumor necrosis factor (TNF)-α and interleukin (IL)-6. Both TNF-α and IL-6 levels increase within the first few hours after PHx. IL-6 causes activation of STAT3 and its translocation to the nucleus in Kupffer cells and hepatocytes.17 PHx in mice lacking the TNFR1 is associated with markedly reduced activation of nuclear factor κB and STAT3 and reduced DNA synthesis; however, injection of IL-6 before PHx restored DNA synthesis.18 IL-6 knockout mice also show an impaired regenerative response that is corrected by IL-6 injection.19 Zinc homeostasis has numerous components that are induced by proinflammatory cytokines, including metallothionein and the zinc transporter Zip14.20 These factors may up-regulate during LR as a consequence of their regulation by these immune mediators.
Our hypothesis is that zinc and one or more zinc transporters may have an important role in LR. To investigate this hypothesis, we have examined zinc transporter expression during LR and explored a role for zinc in regulation of the hepatocyte growth factor (HGF) pathway during hepatocyte proliferation.
Materials and Methods
Animals and PHx
Young adult (8–12 weeks) male C57BL/6 mice were from Charles River Breeding Laboratories (Wilmington, MA). Zip14−/+ and Zip14+/+ founder mice were obtained through a contract with the Mutant Mouse Regional Resource Center at the University of California, Davis. A targeted mutation in Zip14 gene (exons 3–5) was generated in strain 129/SvEvBrd-derived embryonic stem cells. The chimeric mice were bred to C57BL/6J albino mice to generate Zip14+/− mice. Zip14−/− mice were obtained through further breeding of founder Zip14+/− mice at the University of Florida. Mice were given free access to tap water and received commercial rodent diets (Harlan Teklad 7912, Indianapolis, IN). Genomic DNA was extracted from mouse tail samples for genotyping. Treatments with purified diets (AIN76a) were low zinc (ZnD, <0.5 mg Zn/kg diet), zinc adequate (ZnA, 30 mg Zn/kg diet), and high zinc (ZnH, 180 mg Zn/kg diet).21 Mice were fed a ZnA diet for 4 days and then underwent either 70% PHx or a sham operation. Other mice were randomly assigned to be fed ZnA, ZnD, or ZnH diets for 1 week before PHx or a sham operation. A rat 70% PHx method22 modified for the mouse23,24 was used for excision of the left and median lobes. Mice were killed up to 48 hours after PHx. Isoflurane anesthetic was used for all procedures. Buprenorphine (0.1 mg/kg subcutaneously) was used for analgesia. Protocols were approved by the University of Florida Institutional Animal Care and Use Committee.
Cell Culture and Treatments
The AML12 hepatocyte cell line was established from a transgenic mouse where human transforming growth factor α is constitutively produced.25 Transforming growth factor α regulates normal growth through binding to the epidermal growth factor receptors; therefore, at serum starvation and for subsequent steps, the medium was supplemented with epidermal growth factor receptor inhibitor (Calbiochem, San Diego, CA). The hepatocytes were maintained in Dulbecco’s modified Eagle medium/F-12. The cells were serum starved for 20 hours and either pretreated with pyrithione and zinc or macrophage-conditioned medium (CM) and then HGF (Imgenex, San Diego, CA) for either 30 minutes or 48 hours. RAW264.7 mouse macrophages were used to produce the CM.
Biochemical Analyses
IL-6 level was measured in macrophage-conditioned medium by enzyme-linked immunosorbent assay. Serum ALT level was measured by a colorimetric end point method. Blood was collected by cardiac puncture under anesthesia. Serum was obtained by 2-stage centrifugation. Liver tissue and cells were digested in HNO3. Zinc concentrations were measured by flame atomic absorption spectrophotometry and were normalized for tissue weight or for total protein concentration.
RNA Isolation and Quantitative Polymerase Chain Reaction
Liver tissue was collected in RNAlater (Ambion, Austin, TX) and homogenized (Polytron) in TRIzol reagent (Ambion). Cells were placed directly in TRIzol reagent. Total RNA samples were treated with Turbo DNA-free reagents (Ambion). Primer/probe sequences for the polymerase chain reactions (PCRs) are provided in Supplementary Tables 1–3. Assays were one-step reverse-transcriptase reactions (Applied Biosystems, Foster City, CA), and relative quantitation used TATA binding protein messenger RNA (mRNA) as the normalizer.
Immunoblotting
Polyclonal rabbit antibodies against Zip6, Zip14, and zinc transporter (ZnT) 8 were raised in-house as described previously26 to the peptides listed in Supplementary Table 4. Immunoglobulin G fractions were affinity purified (Pierce, Rockford, IL). Liver tissue samples were flash frozen in liquid nitrogen at collection. Frozen liver tissue was homogenized in lysis buffer (Santa Cruz Biotechnology, Santa Cruz, CA) containing protease inhibitor cocktail (Santa Cruz Biotechnology) and phosphatase inhibitor (Santa Cruz Biotechnology). AML12 hepatocytes were washed and harvested into ice-cold phosphate-buffered saline that contained protease inhibitors. Proteins were separated by sodium dodecyl sulfate/polyacrylamide gel electrophoresis. Transfer to nitrocellulose membrane was confirmed by Ponceau Red staining. Immunoblot conditions are described in Supplementary Table 5. Immunoreactivity was visualized by enhanced chemiluminescence.
Zip14 Overexpression and Small Interfering RNA Knockdown
AML12 hepatocytes were reverse transfected either with pCMV-SportZip14 vector by using Effectene transfection reagent (Qiagen, Germantown, MD) and 1 μg of DNA or Zip14 small interfering RNA by using HiPerFect transfection reagent (Qiagen) and 10 nmol/L of small interfering RNA. Secondary treatments were conducted between 24 and 96 hours of transfection.
Bromodeoxyuridine Assay
A bromodeoxyuridine (BrdU) proliferation assay (Calbiochem) was conducted after AML12 hepatocytes were serum starved and pretreated with pyrithione and zinc. Cells were washed twice and then 40 ng/mL HGF was added. For Zip14-overexpressing hepatocytes (as previously described), after 48 hours in culture, cells were serum starved for 20 hours and then 40 ng/mL HGF was added.
Protein Tyrosine Phosphatase 1B Assay
The AML12 hepatocytes were serum starved for 20 hours and then treated with either pyrithione and zinc or CM for 2 hours. The nonspecific phosphatase inhibitor sodium orthovanadate (Sigma, St Louis, MO) and protein tyrosine phosphatase 1B (PTP1B)-specific inhibitors, 3-(3,5-dibromo-4-hydroxy-benzoyl)-2-ethyl-benzofuran-6-sulfonicacid-(4-(thiazol-2-ylsulfamyl)-phenyl)-amide (Calbiochem) and potassium bisperoxo (1,10-phenanthroline) oxovanadate (Santa Cruz Biotechnology), were included as positive controls for inhibition of phosphatase activity. Membrane proteins were isolated (BioVision, Mountain View, CA) from the cells using a Dounce homogenizer ice-cold homogenizing buffer with protease inhibitor cocktail (BioVision). Membrane protein pellets were dissolved in HEPES buffer and incubated with phosphopeptide substrate (ELEF-pY-MDYE-NH2) (AnaSpec, Fremont, CA). Inorganic phosphate release was measured using a colorimetric phosphate assay (Biovision).
Details of experimental procedures are presented in Supplementary Materials and Methods.
Statistical Analysis
Data are expressed as means ± SD. Comparisons among groups used analysis of variance (ANOVA) with Tukey–Kramer multiple comparisons test or t test with Welch correction. Longitudinal data were analyzed by repeated-measures ANOVA. Significance was set at P < .05.
Results
Increases in Hepatic Zinc Concentration in Response to PHx
Hepatocytes are the first cells of the liver that enter the G1 phase of the cell cycle after PHx.8 In mice, proliferation peaks between 24 and 48 hours, depending on the strain16; therefore, we concentrated on the first 48 hours after PHx. The expression of CD1 as a G1-phase and proliferating cell nuclear antigen (PCNA) as an S-phase marker of the 2 stages of the cell cycle was measured. Both CD1 and PCNA were increased after PHx (Figure 1A). We hypothesized that changes in liver zinc concentration might start earlier, because induction of hepatic zinc accumulation after proinflammatory stimuli is rapid. There were significant decreases (P < .05) in serum and an increase (P < .05) in liver zinc concentrations, peaking at 10 hours after PHx (Figure 1B), showing that zinc metabolism is altered early in the LR process.
Figure 1.

Changes in proliferation markers, Zip14 and hepatic zinc in response to PHx over 48 hours. (A) Liver was collected from PHx and sham-operated mice. Quantitative PCR and Western blot analysis of CD1 and PCNA expression. (B) Total zinc concentrations in liver and serum. (C) Quantitative PCR analysis of ZIP transporters. (D) Quantitative PCR and Western blot analysis showing Zip14 expression after PHx. Values are means ± SD (n = 5 for PHx group and n = 3 for sham-operated group). *P < .05, **P < .01, ***P < .0001.
Zinc Transporters Are Differentially Expressed in Response to PHx
Changes in liver zinc content in response to PHx suggest that zinc transporter activity might have a role in the LR process. Because changes were observed at 10 hours after PHx, expression of the full panel of 14 Zip and 10 ZnT zinc transporters was measured at that time point (Figure 1C and Supplementary Figures 1 and 2). Zip1, Zip3, Zip6, Zip7, Zip10, Zip14, ZnT7, and ZnT8 were up-regulated, and Zip8 was down-regulated. Among all the ZIP transporters, Zip6 and Zip14 mRNAs had the most significant increase (P < .001) in comparison to the sham control (Figure 1C). Zip14 had the highest expression at 10 hours after PHx (Figure 1C). Zip14 may play a major role in facilitating zinc mobilization to hepatocytes. Zip14 up-regulation started as early as 2 hours (P < .01 at 2 hours, P < .0001 at 10 hours), and at 24 hours expression had returned to that of the sham control (Figure 1D). This result supported our hypothesis that zinc transporter activity helps augment the additional zinc required for proliferation after PHx. In support of this is that Zip14 is regulated by IL-620 and LR is initiated through a cytokine-dependent (IL-6 and TNF-α) pathway.16
Zinc Supplementation Enhances LR
A dietary zinc study was conducted to test the effect of zinc supplementation in the mouse PHx model (Figure 2A). At the end of the feeding period, PHx or sham operations were conducted. Serum and hepatic zinc concentrations were lower in ZnD-fed mice and were higher in ZnH-fed mice (Supplementary Figure 3). The ZnH-fed mice had significantly (P < .001) lower serum ALT levels 24 hours after PHx than ZnA-fed mice, suggesting that ZnH-fed mice had improved liver function (Figure 2B). Expression of the proliferation marker PCNA was significantly (P < .001) higher in the ZnH-fed mice in comparison to ZnA-fed mice (Figure 2C). Furthermore, ZnH-fed mice had a significantly (P < .004) higher liver/body weight ratio when compared with ZnA-fed mice (Figure 2D). Taken together, these 2 latter measures point toward enhanced proliferation.
Figure 2.

Dietary zinc content influences serum ALT activity and liver PCNA expression. (A) Serum ALT activity for 48 hours after PHx. (B) Serum ALT activity of mice fed a low (ZnD), adequate (ZnA), or high (ZnH) level of dietary zinc for 1 week with blood obtained 24 hours after PHx. (C) PCNA expression as measured by quantitative PCR and Western blot analysis in liver from mice in B. (D) Liver/body weight ratio measurements. Values are means ± SD (n = 5 for PHx and n =3 for sham groups).
To further investigate the mechanism through which zinc may play a role in LR, in vitro experiments were conducted with AML12 murine hepatocytes. To test the direct effect of zinc, the cells were treated with 2 and 8 μmol/L zinc along with the ionophore pyrithione to chelate zinc to enhance cellular uptake and then with HGF. Following zinc and pyrithione treatment, there was a 3-fold increase (P < .001) in cellular zinc concentration, assuring that intracellular zinc concentration was increased (Figure 3A). Next, the effect of zinc on hepatocyte proliferation was tested by the measurement of PCNA (Figure 3B and C). Zinc-pretreated (8 μmol/L) hepatocytes had significantly (P < .001) higher PCNA expression than HGF-treated hepatocytes without zinc. Further confirmation of the zinc effect on proliferation was obtained from a BrdU incorporation assay, a measure of DNA replication (Figure 3D). The almost identical patterns of PCNA expression and BrdU incorporation strongly suggest that hepatocyte proliferation was enhanced by zinc.
Figure 3.

Zinc treatment of AML12 hepatocytes influences proliferation in response to HGF stimulation. (A) Following 20 hours of serum starvation, AML12 hepatocytes were pre-treated with a combination of pyrithione and 8 μmol/L zinc and then total zinc concentrations were measured. (B–D) Cells were incubated with zinc before addition of HGF. After 48 hours, cells were harvested and (B and C) PCNA expression and (D) BrdU incorporation were measured. Values are means ± SD (n = 3–5).
For a better simulation of the in vivo initiation step of LR, we used a model in which CM, obtained from either control (CM) or lipopolysaccharide (LPS)-treated (LPS-CM) RAW cells, was used to treat AML12 hepatocytes. IL-6 production by these cells was confirmed by quantitative PCR and enzyme-linked immunosorbent assay (Supplementary Figure 4). Zip14 expression in LPS-CM–treated hepatocytes was substantially increased (P < .01) (Figure 4A). To further confirm the IL-6 regulation of Zip14, recombinant IL-6 was added to the LPS-CM, which further enhanced Zip14 expression (P < .001) (Figure 4A). Furthermore, depletion of IL-6 in the LPS-CM, using neutralizing IL-6 antibody, diminished Zip14 expression to the control level (Supplementary Figures 5 and 6), confirming that IL-6 –regulated Zip14 up-regulation was similar to that produced by LR initiation in vivo.
Figure 4.

Comparison of Zip14 up-regulation with the intracellular zinc concentration and proliferation of AML12 hepatocytes. Macrophage CM was obtained from LPS-treated RAW cells. (A) AML12 hepatocytes were incubated with CM for 2 hours. Cells were harvested, and Zip14 expression was measured by quantitative PCR and Western blot analysis. The same samples were used to measure (B) metallothionein mRNA and (C) PCNA mRNA and protein. (B) Total zinc concentration of AML12 hepatocytes was measured. Values are means ± SD (n =3–9). Values with a different superscript are statistically different (P < .05 to .001).
To investigate potential physiologic consequences of Zip14 up-regulation, total zinc concentration and a zinc-regulated gene, metallothionein, expression in the hepatocytes were measured. Both were significantly higher (P < .001) in combined LPS-CM and IL-6 –treated hepatocytes than cells treated with CM or IL-6 alone (Figure 4B). To test the effect of Zip14-facilitated zinc transport on hepatocyte proliferation, after macrophage-CM pre-treatment, the cells were treated with HGF. PCNA expression was significantly higher in cells treated with LPS-CM alone (P < .05) or LPS-CM and IL-6 (P < .001) (Figure 4C).
Zinc Inhibits PTP1B Activity in AML12 Hepatocytes
c-Met receptor kinase phosphorylation sites are dephosphorylated by PTP1B.27 Because it has also been shown that zinc inhibits PTP1B enzymatic activity,28 we hypothesized that augmentation of hepatocyte proliferation by zinc might be through inhibition of PTP1B activity during LR. To test PTP1B enzymatic activity, phosphate ion release from a phosphopeptide substrate was measured. There was no difference between zinc-pre-treated and control cell lysates in activity (Supplementary Figure 7). Incubation of the hepatocytes with zinc (2–32 μmol/L) and pyrithione produced a dose-dependent inhibition of PTP1B activity in both total and membrane fractions (Supplementary Figure 8). PTP1B is a membrane-bound protein; thus, further experiments were conducted with only membrane fractions. PTP1B enzymatic activity was diminished 60% in 8 μmol/L zinc-pretreated cells (Figure 5A). The amount of inhibition by zinc was similar to the amount of inhibition by nonspecific sodium orthovanadate and PTP-1B–specific phosphatase inhibitors (Figure 5B). A 40% decrease in PTP1B activity was also detected in LPS-CM– and IL-6 –treated cells. These results collectively show that zinc inhibits PTP1B activity.
Figure 5.
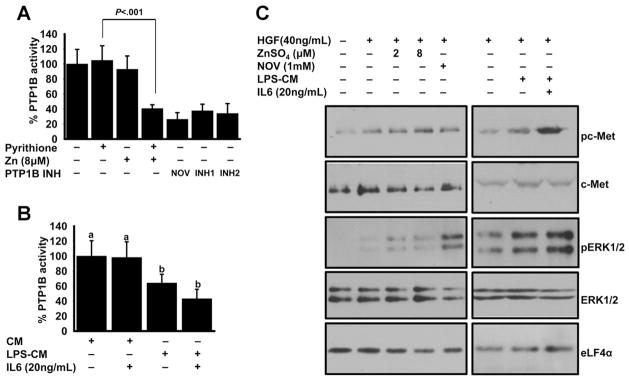
Effect of zinc on PTP1B activity and c-Met pathway. AML12 hepatocytes were treated with either pyrithione or 8 μmol/L zinc, along with (A) phosphatase inhibitors for 30 minutes or (B) macrophage CM for 2 hours. Cells were harvested and total membrane fractions were incubated with phosphosubstrate and inorganic phosphate release was measured. (C) AML12 hepatocytes were treated with either 2 different concentrations of zinc or CM for 2 hours. Cells were harvested and total cell lysates were used for the detection of indicated proteins by Western blot analysis. Values are means ± SD (n = 3) (B). Values with a different superscript are statistically different (P < .05 to .001).
Zinc Enhances c-Met Phosphorylation in AML12 Hepatocytes
To test the hypothesis that inhibition of PTP1B may result in enhanced c-Met phosphorylation through zinc or combined pretreatment of LPS-CM and IL-6, cells were treated with HGF and lysates were tested using phosphospecific antibodies. As shown, there was a greater amount of phosphorylation of c-Met with zinc or LPS-CM and IL-6 pretreatment (Figure 5C). c-Met protein was used as a loading control, and there were no changes in the band intensities between treatment groups. c-Met phosphorylation activates many downstream signaling pathways. Among them, extracellular signal–regulated kinase (ERK) 1/2 was shown to be the main target during LR.29 In agreement, ERK1/2 phosphorylation was higher in the zinc- or LPS-CM– and IL-6 –treated conditions (Figure 5C). No changes in band intensities were found for the loading controls, ERK1/2, and eukaryotic initiation factor 4α (elf4α) proteins. These results further proved a role of zinc in the c-Met–activated hepatocyte proliferation pathway during the LR process.
Overexpressed Zip14 Enhances Hepatocyte Proliferation
AML12 hepatocytes were transiently transfected with either pCMV-Sport6-Zip14 or pCMV-Sport6 empty vector for 96 hours. Overexpression of Zip14 was confirmed by quantitative PCR (P < .001) and Western blot analyses (Figure 6A). During the transfection, hepatocytes were serum starved and incubated with HGF for 48 hours. Higher PCNA expression was detected in the Zip14-over-expressing cells when compared with the empty-vector transfected control cells (Figure 6A). In addition, significantly higher (P < .001) BrdU incorporation was found in Zip14-overexpressing cells (Figure 6B). These results suggested that Zip14 overexpression enhanced hepatocyte proliferation.
Figure 6.

Zip14 overexpression in AML12 hepatocytes influences proliferation and the c-Met signaling pathway. AML12 hepatocytes were transfected with either pCMV Sport-Zip14 or Zip14 small interfering RNA and then serum starved for 20 hours. Next, cells were incubated either with HGF and BrdU or HGF alone for 24 hours. (A, B, and E) Cells were harvested and used to measure Zip14 and PCNA expression or BrdU incorporation. (C) After 96 hours of Zip14 transient transfection, total cell membranes were isolated and used for assay of phosphatase activity. (D and F) Cells were serum starved in the last 20 hours of transfection and then were incubated with HGF for 30 minutes. Total cell lysates were used for Western blot analysis. Values are means ± SD (n = 3–5).
We hypothesized that the overexpressed Zip14 facilitates an increase in intracellular zinc concentration that may have a role in producing the PTP1B inhibition of the HGF– c-Met pathway during proliferation. Membrane PTP1B phosphatase activity was decreased 40% in the Zip14-overexpressing cells (Figure 6C). To investigate the role of Zip14 in the c-MET–ERK1/2 pathway, Zip14 was either overexpressed or silenced in AML12 hepatocytes. Following HGF treatment, the hepatocytes were harvested and proteins were analyzed by sodium dodecyl sulfate/polyacrylamide gel electrophoresis. Enhanced phosphorylation of both the c-Met kinase domain and ERK1/2 was detected when Zip14 was overexpressed (Figure 6D), whereas decreased phosphorylation of both the c-Met kinase domain and ERK1/2 was detected when Zip14 was silenced (Figure 6E and F).
Zip14−/− Causes Decreased Hepatocyte Proliferation
In addition to genotyping, the Zip14−/− was confirmed at both the protein and mRNA level (Supplementary Figure 9). Intestine and liver tissues were chosen for the confirmation because these 2 tissues abundantly express Zip14.20,30 PHx surgeries were conducted with Zip14−/− and wild-type (WT) mice. In these sets of experiments, comparisons were made between pre-PHx and post-PHx conditions. At 24 hours after PHx, Zip14 was up-regulated at the mRNA (not shown) and protein level in WT mice (Figure 7A). In contrast, Zip14 was not detectable in post-PHx liver of the Zip14−/− mice. Total hepatic zinc level was significantly (P < .001) increased in the WT mice in response to PHx (Figure 7B). However, that increase in hepatic zinc levels was not present in Zip14−/− mice, strongly suggesting that Zip14 is the main zinc transporter that facilitates zinc mobilization to the liver during LR. CD1 and PCNA expression, as measures of cell proliferation, examined in liver tissue at 24 hours and 48 hours after PHx, was lower in Zip14−/− mice than WT mice (Figure 7C). Similar results were observed in Ki67 detection by immunohistochemistry (Figure 7C). Ki67-positive cells indicative of cell proliferation were measured by digital imaging and were significantly lower in Zip14−/− mice than WT mice (P < .009) (Figure 7C). Zip14−/− mice had significantly (P < .04) higher serum ALT levels when compared with WT mice (Figure 7D). Furthermore, the liver/body weight ratio was measured at 1, 2, 5, and 7 days after PHx. Zip14−/− mice had a significantly lower liver/body weight ratio at days 5 and 7 after PHx (P < .02 and P < .03) when compared with WT mice (Figure 7E).
Figure 7.

Zip14−/− reduces hepatic zinc accumulation and proliferation in response to PHx. The PHx was performed on WT and Zip14−/− mice. (A) Zip14 protein expression and (B) liver zinc concentration were measured. (C) Liver CD1, PCNA protein, and Ki67 staining were measured by Western blot and immunohistochemistry, respectively. Quantification of Ki67-positive cells was performed by digital imaging. (D) Serum ALT measurements. (E) Liver/body weight ratio measurements. Values are means ± SD (n = 4). (F) Schematic representation of involvement of zinc and Zip14 in LR. Kupffer and innate immune system cells produce IL-6, which up-regulates Zip14 at the priming step of LR. Zip14 up-regulation leads to an increase in hepatic zinc concentration. Increased hepatic zinc inhibits PTP1B activity and thus enhances hepatocyte proliferation by increased c-Met–ERK1/2 phosphorylation.
Discussion
The aim of this research was to gain a better understanding of the involvement of zinc and zinc transport in the LR process. Both in vivo and in vitro experiments revealed increased hepatic zinc uptake during LR and enhanced hepatocyte proliferation. Our previous data have shown that Zip14 is localized to the plasma membrane of hepatocytes and that it is a Zn2+ ion transporter.20 Here we report that an increase in hepatic zinc uptake during LR was mainly facilitated by the most up-regulated zinc transporter, Zip14, as confirmed with the novel use of a Zip14−/− mouse. Zip14 up-regulation was mainly caused by IL-6; however, the presence of other nonparenchymal cell–produced factors was required as shown with hepatocytes cultured with CM. PTP1B is the enzyme responsible for dephosphorylation of the c-Met kinase domain. The inhibition of PTP1B by zinc via Zip14 activity leads to enhanced c-Met–ERK1/2 phosphorylation and results in enhanced hepatocyte proliferation. A model integrating these events in LR is presented (Figure 7F).
The priming step of LR resembles an acute phase response during inflammation.9 Hypozincemia is one of the hallmarks of inflammation,31 and redistribution of hepatic zinc during inflammation has been reported.32 Taken together, these observations suggest that hepatic zinc content changes during LR. An increase in hepatic zinc level and proliferation of hepatocytes were previously shown during LR at 24 hours after PHx in mice.33 This suggests that any change mediated by zinc that affects proliferation should occur earlier than 24 hours after PHx. We observed that hepatic zinc level started to increase as early as 2 hours after PHx and stayed elevated for up to 48 hours. This novel finding is important in terms of the requirement of zinc in early steps of hepatocyte proliferation during LR in the mouse.
In an effort to generate an in vitro model for the simulation of the initiation step of LR, macrophage CM was used to pretreat AML12 hepatocytes. IL-6 was a most important component because of both its role in the priming step of LR and its regulatory role in Zip14 expression. The comparison of CM with recombinant IL-6 treatment relates to the controversy over the consistency of an IL-6 effect on hepatocyte proliferation in vitro. Both inhibitory34 and enhancing35 effects of IL-6 on proliferation have been reported. These differences were explained by Sun et al,36 who showed that the presence of nonparenchymal cell components was required for IL-6 to have a positive effect on hepatocyte proliferation. They also showed that IL-6 influenced the HGF pathway. When cocultured hepatocytes were pretreated with c-Met antibody and then IL-6, they detected a decrease in hepatocyte proliferation. Therefore, macrophage CM was used along with HGF in the in vitro model in our experiments.
A major challenge in these experiments was to connect the enhancing effect of zinc on LR to a mechanism of action. Mounting evidence suggests that zinc ions influence signaling pathways through inhibition of phosphatase activity.26,28 Because HGF activates c-Met through phosphorylation at the active residues Tyr-1234 and Tyr-1235, we focused on PTP1B, which regulates this pathway and influences proliferation through phosphorylation of ERK1/2. Assays were conducted to evaluate the effect of zinc on PTP1B phosphatase activity. Total cellular membrane fractions were used as a source of endogenous PTP1B because this enzyme is membrane bound. However, this approach did not eliminate the possibility that there could be other tyrosine phosphatases in the lysate. To circumvent this potential complication, we used a highly specific consensus peptide, ELEFpYMDYE, as a PTP1B-specific substrate. The consensus peptide sequence was obtained from studies of PTP1B that used kinetic,37 structural,38 and alanine scanning39 approaches. PTP1B inhibition by zinc was further proven by the increased phosphorylation of the physiological substrate of PTP1B, c-Met kinase domain phosphotyrosines, as a result of zinc pretreatment (Figure 5C).
The possible mechanism for the dose-dependent zinc inhibition of PTP1B activity was not addressed in the current experiments. We speculate that zinc may bind to the cysteine residue of the PTP1B active site and cause inhibition of enzymatic activity. This idea was supported by the fact that the inhibitory effect of zinc was diminished when the reducing reagent, dithiothreitol, was included in the reaction (data not shown). Dithiothreitol increases the redox potential, thus causing PTP1B to return to the active form. Such interplay between zinc and redox was shown previously with regard to sequestration and release of zinc ions by cysteine-rich metallothioneins.40
We have shown that the inhibition of PTP1B activity caused increased phosphorylation of the c-Met receptor at the kinase domain. Other studies have shown that downstream c-Met signaling produces activation of Ras/ERK/MAPK, PI3K/Akt, Rac/Pak, and Crk/Rap1 pathways.41 Among these pathways, phosphorylation of ERK1/2 was not detected in the regenerating liver of the conditional Met mutant mice.29 This suggested that c-Met signaling contributes significantly to ERK1/2 activation in the regenerating liver. Therefore, we chose to investigate ERK1/2 phosphorylation to evaluate the effect of zinc on the downstream signaling of c-Met activation. Zinc inhibition of MAPK phosphatases resulted in increased phosphorylation of ERK1/2 in neural cells,42 raising the possibility that the increase in ERK1/2 phosphorylation could be independent from upstream c-Met activation. However, this possibility was eliminated by further investigation of ERK1/2 phosphorylation in hepatocytes treated with zinc (without HGF). In these cells, there was no significant change in ERK1/2 phosphorylation between control and zinc-pretreated cell preparation (data not shown). Therefore, it was concluded that the increase in ERK1/2 phosphorylation was a downstream effect of HGF/c-Met activation after pretreatment with zinc.
It is most relevant that Zip14−/− mice exhibit decreased proliferation following PHx. This decrease possibly resulted from impaired hepatic zinc transport because, unlike WT mice, no increase was observed in hepatic zinc level in Zip14−/− mice after PHx. In addition, hepatic zinc concentrations were the same in WT and Zip14−/− mice before PHx, suggesting that the Zip14−/− genotype does not result in reduced liver zinc availability. A likely explanation for maintenance of normal liver zinc levels is through simultaneous activity of other ZIP transporters, because Zip1, Zip3, Zip6, Zip7, and Zip10 in addition to Zip14 were significantly up-regulated during LR. Even though Zip14−/− did not produce zinc deficiency in liver, proliferation was impaired, supporting the idea that Zip14 may have a role in zinc transport function during LR and inflammation. One of those possible roles for Zip14 could be related to adipogenesis during LR. Markedly increased hepatocellular fat was detected at 12 to 24 hours in post-PHx mice.43 Interestingly, Zip14 participates in zinc uptake during adipogenesis.44 In addition, Zip14 is up-regulated in the early stages of adipocyte differentiation in 3T3-L1 cells. The time of Zip14 up-regulation (10 hours after PHx) during LR slightly precedes the time of the hepatocellular fat accumulation (12–24 hours after PHx); thus, Zip14 could participate in early stages of adipogenesis during LR. Another possible role for Zip14 in LR could be to influence metalloprotease activity. Hepatic biomatrix degradation by metalloproteases is essential for the initiation of LR.2 Zip14 belongs to the LZT subfamily of the ZIP transporter family of proteins.30 The LZT subfamily proteins share the conserved HEXPHE motif. This motif is similar to the consensus sequence for the catalytic zinc-binding site of metalloproteases (HEXXH).
Zinc supplementation has been shown in several clinical trials to have a positive effect on liver function based on serum ALT levels.3–7 In this report, zinc and Zip14 are proposed as potential therapeutic targets to treat specific liver diseases because enhancement of LR by zinc was shown in response to PHx.
Supplementary Material
Acknowledgments
Funding
Supported by National Institutes of Health grants R01DK31127 and R01DK94244 and the Boston Family Endowment of the University of Florida Foundation (to R.J.C.).
Abbreviations used in this paper
- ANOVA
analysis of variance
- BrdU
bromodeoxyuridine
- CM
conditioned medium
- ERK
extracellular signal–regulated kinase
- HGF
hepatocyte growth factor
- IL
interleukin
- LPS
lipopolysaccharide
- LR
liver regeneration
- PCNA
proliferating cell nuclear antigen
- PCR
polymerase chain reaction
- PHx
partial hepatectomy
- PTP1B
protein tyrosine phosphatase 1B
- TNF
tumor necrosis factor
- WT
wild-type
- ZnA
zinc adequate
- ZnD
zinc deficient
- ZnH
zinc supplement
- ZnT
zinc transporter
Footnotes
Conflicts of interest
The authors disclose no conflicts.
To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at http:/dx.doi.org/10.1053/j.gastro.2012.02.046.
References
- 1.Clavien PA, Oberkofler CE, Raptis DA, et al. What is critical for liver surgery and partial liver transplantation: size or quality? Hepatology. 2010;52:715–729. doi: 10.1002/hep.23713. [DOI] [PubMed] [Google Scholar]
- 2.Schuppan D, Popov Y. Rationale and targets for antifibrotic therapies. Gastroenterol Clin Biol. 2009;33:949–957. doi: 10.1016/j.gcb.2009.07.021. [DOI] [PubMed] [Google Scholar]
- 3.Weismann K, Christensen E, Dreyer V. Zinc supplementation in alcoholic cirrhosis. A double-blind clinical trial. Acta Med Scand. 1979;205:361–366. doi: 10.1111/j.0954-6820.1979.tb06065.x. [DOI] [PubMed] [Google Scholar]
- 4.Murakami Y, Koyabu T, Kawashima A, et al. Zinc supplementation prevents the increase of transaminase in chronic hepatitis C patients during combination therapy with pegylated interferon alpha-2b and ribavirin. J Nutr Sci Vitaminol (Tokyo) 2007;53:213–221. doi: 10.3177/jnsv.53.213. [DOI] [PubMed] [Google Scholar]
- 5.Takagi H, Nagamine T, Abe T, et al. Zinc supplementation enhances the response to interferon therapy in patients with chronic hepatitis C. J Viral Hepat. 2001;8:367–337. doi: 10.1046/j.1365-2893.2001.00311.x. [DOI] [PubMed] [Google Scholar]
- 6.Matsuoka S, Matsumura H, Nakamura H, et al. Zinc supplementation improves the outcome of chronic hepatitis C and liver cirrhosis. J Clin Biochem Nutr. 2009;45:292–303. doi: 10.3164/jcbn.08-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagamine T, Takagi H, Takayama H, et al. Preliminary study of combination therapy with interferon-alpha and zinc in chronic hepatitis C patients with genotype 1b. Biol Trace Elem Res. 2000;75:53–63. doi: 10.1385/BTER:75:1-3:53. [DOI] [PubMed] [Google Scholar]
- 8.Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007;213:286–300. doi: 10.1002/jcp.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jirakulaporn T, Muslin AJ. Cation diffusion facilitator proteins modulate Raf-1 activity. J Biol Chem. 2004;279:27807–27815. doi: 10.1074/jbc.M401210200. [DOI] [PubMed] [Google Scholar]
- 10.Chesters JK, Petrie L, Vint H. Specificity and timing of the Zn2+ requirement for DNA synthesis by 3T3 cells. Exp Cell Res. 1989;184:499–508. doi: 10.1016/0014-4827(89)90347-9. [DOI] [PubMed] [Google Scholar]
- 11.Berg JM, Shi Y. The galvanization of biology: a growing appreciation for the roles of zinc. Science. 1996;271:1081–1085. doi: 10.1126/science.271.5252.1081. [DOI] [PubMed] [Google Scholar]
- 12.King J, Cousins RJ. Zinc. In: Shils ME, Shike M, Ross AC, et al., editors. Modern nutrition in health and disease. 10. Baltimore, MD: Lippincott Williams and Wilkins; 2005. pp. 271–285. [Google Scholar]
- 13.Beyersmann D, Haase H. Functions of zinc in signaling, proliferation and differentiation of mammalian cells. Biometals. 2001;14:331–341. doi: 10.1023/a:1012905406548. [DOI] [PubMed] [Google Scholar]
- 14.Siemes C, Quast T, Klein E, et al. Normalized proliferation of normal and psoriatic keratinocytes by suppression of sAPPalpha-release. J Invest Dermatol. 2004;123:556–563. doi: 10.1111/j.0022-202X.2004.23320.x. [DOI] [PubMed] [Google Scholar]
- 15.Kang X, Song Z, McClain JC, et al. Zinc supplementation enhances hepatic regeneration by preserving hepatocyte nuclear factor-4α in mice subjected to long-term ethanol administration. Am J Pathol. 2008;172:916–925. doi: 10.2353/ajpath.2008.070631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fausto N, Riehle KJ. Mechanisms of liver regeneration and their clinical implications. J Hepatobiliary Pancreat Surg. 2005;12:181–189. doi: 10.1007/s00534-005-0979-y. [DOI] [PubMed] [Google Scholar]
- 17.Taub R, Greenbaum LE, Peng Y. Transcriptional regulatory signals define cytokine-dependent and -independent pathways in liver regeneration. Semin Liver Dis. 1999;19:117–127. doi: 10.1055/s-2007-1007104. [DOI] [PubMed] [Google Scholar]
- 18.Yamada Y, Kirillova I, Peschon JJ, et al. Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci U S A. 1997;94:1441–1446. doi: 10.1073/pnas.94.4.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cressman DE, Greenbaum LE, DeAngelis RA, et al. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996;274:1379–1383. doi: 10.1126/science.274.5291.1379. [DOI] [PubMed] [Google Scholar]
- 20.Liuzzi JP, Lichten LA, Rivera S, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to the hypozincemia of the acute-phase response. Proc Natl Acad Sci U S A. 2005;102:6843–6848. doi: 10.1073/pnas.0502257102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liuzzi JP, Bobo JA, Lichten LA, et al. Responsive transporter genes within the murine intestinal-pancreatic axis form a basis of zinc homeostasis. Proc Natl Acad Sci U S A. 2004;101:14355–14360. doi: 10.1073/pnas.0406216101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Higgins GM, Anderson RM. Experimental pathology of the liver. Arch Pathol Lab Med. 1931;12:186–202. [Google Scholar]
- 23.Greene AK, Puder M. Partial hepatectomy in the mouse: technique and perioperative management. J Invest Surg. 2003;16:99–102. [PubMed] [Google Scholar]
- 24.Boyce S, Harrison D. A detailed methodology of partial hepatectomy in the mouse. Lab Anim (NY) 2008;37:529–532. doi: 10.1038/laban1108-529. [DOI] [PubMed] [Google Scholar]
- 25.Wu JC, Merlino G, Fausto N. Establishment and characterization of differentiated, nontransformed hepatocyte cell lines derived from mice transgenic for transforming growth factor alpha. Proc Natl Acad Sci U S A. 1994;91:674–678. doi: 10.1073/pnas.91.2.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aydemir TB, Liuzzi JP, McClellan S, et al. Zinc transporter ZIP8 (SLC39A8) and zinc influence IFN-gamma expression in activated human T cells. J Leukoc Biol. 2009;86:337–348. doi: 10.1189/jlb.1208759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sangwan V, Paliouras GN, Abella JV, et al. Regulation of the Met receptor-tyrosine kinase by the protein-tyrosine phosphatase 1B and T-cell phosphatase. J Biol Chem. 2008;283:34374–34383. doi: 10.1074/jbc.M805916200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haase H, Maret W. Intracellular zinc fluctuations modulate protein tyrosine phosphatase activity in insulin/insulin-like growth factor-1 signaling. Exp Cell Res. 2003;291:289–298. doi: 10.1016/s0014-4827(03)00406-3. [DOI] [PubMed] [Google Scholar]
- 29.Borowiak M, Garratt AN, Wüstefeld T, et al. Met provides essential signals for liver regeneration. Proc Natl Acad Sci U S A. 2004;101:10608–10613. doi: 10.1073/pnas.0403412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor KM, Morgan HE, Johnson A, et al. Structure-function analysis of a novel member of the LIV-1 subfamily of zinc transporters, ZIP14. FEBS Lett. 2005;579:427–432. doi: 10.1016/j.febslet.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 31.Moshage H. Cytokines and the hepatic acute phase response. J Pathol. 1997;181:257–266. doi: 10.1002/(SICI)1096-9896(199703)181:3<257::AID-PATH756>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 32.Cousins RJ, Leinart AS. Tissue-specific regulation of zinc metabolism and metallothionein genes by interleukin 1. FASEB J. 1988;2:2884–2890. doi: 10.1096/fasebj.2.13.2458983. [DOI] [PubMed] [Google Scholar]
- 33.Milin C, Tota M, Domitrovic R, et al. Metal tissue kinetics in regenerating liver, thymus, spleen, and submandibular gland after partial hepatectomy in mice. Biol Trace Elem Res. 2005;108:225–243. doi: 10.1385/BTER:108:1-3:225. [DOI] [PubMed] [Google Scholar]
- 34.Moran DM, Mayes N, Koniaris LG, et al. Interleukin-6 inhibits cell proliferation in a rat model of hepatocellular carcinoma. Liver Int. 2005;25:445–457. doi: 10.1111/j.1478-3231.2005.01083.x. [DOI] [PubMed] [Google Scholar]
- 35.Ohira H, Miyata M, Kuroda M, et al. Interleukin-6 induces proliferation of rat hepatocytes in vivo. J Hepatol. 1996;25:941–947. doi: 10.1016/s0168-8278(96)80300-x. [DOI] [PubMed] [Google Scholar]
- 36.Sun R, Jaruga B, Kulkarni S, et al. IL-6 modulates hepatocyte proliferation via induction of HGF/p21cip1: regulation by SOCS3. Biochem Biophys Res Commun. 2005;338:1943–1949. doi: 10.1016/j.bbrc.2005.10.171. [DOI] [PubMed] [Google Scholar]
- 37.Zhang ZY, Thieme-Sefler AM, Maclean D, et al. Substrate specificity of the protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 1993;90:4446–4450. doi: 10.1073/pnas.90.10.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jia Z, Barford D, Flint AJ, et al. Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science. 1995;268:1754–1758. doi: 10.1126/science.7540771. [DOI] [PubMed] [Google Scholar]
- 39.Vetter SW, Keng YF, Lawrence DS, et al. Assessment of protein-tyrosine phosphatase 1B substrate specificity using “inverse alanine scanning”. J Biol Chem. 2000;275:2265–2268. doi: 10.1074/jbc.275.4.2265. [DOI] [PubMed] [Google Scholar]
- 40.Maret W, Krezel A. Cellular zinc and redox buffering capacity of metallothionein/thionein in health and disease. Mol Med. 2007;13:371–375. doi: 10.2119/2007-00036.Maret. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Birchmeier C, Birchmeier W, Gherardi E, et al. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 42.Ho Y, Samarasinghe R, Knoch ME, et al. Selective inhibition of mitogen-activated protein kinase phosphatases by zinc accounts for extracellular signal-regulated kinase 1/2-dependent oxidative neuronal cell death. Mol Pharmacol. 2008;74:1141–1151. doi: 10.1124/mol.108.049064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shteyern E, Liao Y, Muglia LJ, et al. Disruption of hepatic adipogenesis is associated with impaired liver regeneration in mice. Hepatology. 2004;40:1322–1332. doi: 10.1002/hep.20462. [DOI] [PubMed] [Google Scholar]
- 44.Tominaga K, Kagata T, Johmura Y, et al. SLC39A14, a LZT protein, is induced in adipogenesis and transports zinc. FEBS J. 2005;272:1590–1599. doi: 10.1111/j.1742-4658.2005.04580.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.