

Abstract
The CSF is a complex fluid with a dynamically varying proteome throughout development and in adulthood. During embryonic development, the nascent CSF differentiates from the amniotic fluid upon closure of the anterior neural tube. CSF volume then increases over subsequent days as the neuroepithelial progenitor cells lining the ventricles and the choroid plexus generate CSF. The embryonic CSF contacts the apical, ventricular surface of the neural stem cells of the developing brain and spinal cord. CSF provides crucial fluid pressure for the expansion of the developing brain and distributes important growth promoting factors to neural progenitor cells in a temporally-specific manner. To investigate the function of the CSF, it is important to isolate pure samples of embryonic CSF without contamination from blood or the developing telencephalic tissue. Here, we describe a technique to isolate relatively pure samples of ventricular embryonic CSF that can be used for a wide range of experimental assays including mass spectrometry, protein electrophoresis, and cell and primary explant culture. We demonstrate how to dissect and culture cortical explants on porous polycarbonate membranes in order to grow developing cortical tissue with reduced volumes of media or CSF. With this method, experiments can be performed using CSF from varying ages or conditions to investigate the biological activity of the CSF proteome on target cells.
Keywords: Neuroscience, Issue 73, Neurobiology, Developmental Biology, Anatomy, Physiology, Stem Cell Biology, Cellular Biology, Biomedical Engineering, Medicine, Surgery, Neural Stem Cells (NSCs), stem cells, Cerebral Cortex, Cerebrospinal Fluid, CSF, ventricular embryonic CSF, Isolation, Brain, Cerebral Cortical Explant, tissue, culture, mouse, animal model
Introduction
The CSF is a complex fluid that bathes the developing neuroepithelium 1-6 and provides essential pressure 7 and growth promoting cues for the developing brain 8-12. To study the CSF over the course of brain development, we developed techniques to isolate ventricular CSF from developing rat or mouse embryos during various stages of development 6,9. Previous methods of isolation included using a glass micro-needle and isolating the CSF using a micro-injector 1,2. Our method utilizes a glass micro-capillary pipette whose tip has been pulled to create an ultrafine point for improved tissue penetration. The glass micro-capillary pipette is connected to an aspirator so that ventricular CSF collection can be controlled with gentle changes in pressure. To investigate the stem cell influences of CSF signals, we dissect cerebral cortical explants, place them on polycarbonate membranes, and float them on appropriate culture medium supplemented with CSF samples 9. With this technique, reduced volumes of media are sufficient to culture the tissue, allowing for an efficient use of CSF 9.
Protocol
1. Embryo Isolation/Preparation
This technique can be used for mouse or rat. In this protocol we demonstrate the CSF collection technique and cerebral cortical explant dissection with mouse embryonic brain. We will comment on any important differences for rats versus mice that exist within the general techniques. For the embryonic age staging system, E1 is classified as the day of the plug for rats, and E0.5 is classified as the day of the plug for mice.
Prepare a micro-dissection dish with Sylgard Silicone Elastomer. Sylgard 184 Silicone Elastomer is supplied as a two part liquid component, Part A and Part B. Part A and Part B are mixed in a 10:1 ratio by weight or volume. After the liquid components are mixed, pour the sylgard elastomer into a Petri dish to cover the entire surface of the dish. Some air bubbles may be present that will dissipate during the curing process. Place the Petri dish lid on and allow the elastomer to cure. The elastomer can be cured at room temperature for 24 hr, or at higher temperatures (e.g. 37 °C) for faster curing. Once the liquid components solidify, the dissection dish is ready to use. The dish can be used repeatedly for multiple experiments, provided that it is cleaned well following the procedure.
Preparation of the micro-capillary pipette (needle). Micro-capillary pipettes are prepared by applying heat and pull using a Narishige PC-10 vertical micropipette puller with the following settings: One step pull; Heater #2 set to 58; 100 g pull weight. The fine tip of the micropipette is carefully snapped off using fine #55 forceps. The resulting average inner diameter of the needle is 85 μm.
Prepare the aspirator assembly for CSF aspiration. Insert micro-plunger provided with micro-capillary pipettes into the capillary needle. Alternatively, attach a plastic disposable filter to the end of the aspirator tube assembly that is connected to the micro-capillary pipette. Push the needle through the gasket into position on the opposite end of the aspirator assembly.
Transfer embryo isolated from the litter to a micro-dissection dish prepared with Sylgard.
Remove the extra-embryonic membranes and tissues so that the embryo is clearly exposed. Each tissue layer-first the uterine wall and then the decidua-can be dissected using fine iridectomy scissors (i.e. Fine Science Tools # 15000-02). At each implantation site, first the uterine wall, then the decidua can be incised parallel to the long axis of the uterus, and the incision can then be opened further using fine forceps. The decidua can be removed after a similar incision, exposing the fetal membranes. Care should be exercised so that the fetal membranes are not incised or punctured.
Wash with sterile Hanks balanced salt solution (HBSS) and remove excess fluid from the surrounding embryo using a pipette as well as a kimwipe or filter paper cut into triangles.
2. Ventricular CSF Collection
Visualize embryo under the dissection microscope: for mouse, the embryo should be laying on its side, such that one has a sagittal view of the developing embryo. With rat embryos E16 or older, position the embryo on its dorsal spinal column, along its longest planar dimension, from a cranial to caudal direction, as if the embryo is lying on its back. In this manner the CSF can be collected from both right and left lateral ventricle.
Steadily insert the micro-capillary pipette into the lateral ventricle, mesencephalic ventricle, or cisterna magna, attempting not to contact the neuroepithelial cells once the pipette has been inserted. For mouse embryos E14.5 or older, the CSF can be aspirated from the right ventricle and then the needle removed and inserted into the left ventricle. Because of the patency of the ventricles and the neural tube in embryos younger than E14.5, the entire CSF volume can often be aspirated from the lateral ventricles with a single needle insertion. However, this is not always the case as developmental times and patency of the connecting ventricles may vary slightly, and therefore, the micro-capillary pipette can also be inserted into the cisterna magna to collect the maximal CSF volume.
Once the micro-capillary pipette is inserted into the lateral ventricle, mesencephalic ventricle, or cisterna magna, carefully and gently begin aspirating the CSF into the pipette, using either the micro-plunger to create negative pressure and aspirate the CSF, or by providing a gentle negative pressure created by mouth such that CSF starts to gently flow into the micro-capillary pipette in a slow and controlled fashion. We recommend that the viewer checks with their own local officials regarding institutional policies on these approaches.
Continue to apply negative pressure and collect the CSF into the micro-capillary pipette. In both mouse and rat embryos of E16.5/E17 or younger, it is possible to observe the ventricle collapse slightly by the appearance of a divot forming on the side of the head that the micro-capillary pipette is in. In older embryos, due to the larger size of the brain, it may not be possible to observe the head collapsing.
Stop applying pressure and gently remove the micro-capillary pipette from the lateral ventricle.
Gently expel the CSF sample into an Eppendorf tube that has been chilled on ice.
Continue collecting the CSF from an entire litter of animals and pool the samples into the same tube.
Centrifuge at 10,000 x g at 4 °C for 10 min to remove any contaminating cells.
Analyze the samples for any signs of contaminating neuroepithelial cells or red blood cells. Signs of contamination are usually indicated by the fluid appearing cloudy or red/pink, or the presence of a pellet with a blood tinged spot. After centrifugation, the CSF can be analyzed microscopically for any evidence of cells or cellular debris. If there are signs of contamination the fluid should be discarded. As an additional control, the pellet content may also be stained for cells. A clean CSF sample can be used for cell culture, cortical culture, spectrometry, western blot, ELISA, and other assays. The clear CSF should be transferred to a new sterile Eppendorf tube. The sample can now be used for analysis, pooled with other samples, or snap frozen with liquid nitrogen and stored at -80 °C. The freezing and thawing of small samples of fluid may result in a small volume decrease and changes in protein concentration. This can be avoided by freeze drying the samples.
3. Cortical Explant Dissection
Transfer the E14.5 embryo isolated from the litter onto a micro-dissection dish prepared with Sylgard.
Remove embryo from extra-embryonic membranes and tissues as described in Step 1.5.
Wash with sterile Hanks balanced salt solution (HBSS) and remove excess fluid from the surrounding embryo using a pipette.
Dissect through the cervical region separating the head from the rest of the embryo (Figure 1A).
Using a fine scalpel (ophthalmic knife), cut down the midline of the scalp. Grasp each side of this incision with fine forceps and remove the skin. Next, use iridectomy scissors to make an incision that runs the length of midline of the developing cranium. Subsequently, make two additional incisions, approximately 1/3 of the distance from the anterior and posterior ends of the developing skull. Use fine forceps to grasp the "flaps" of cranium that result from this part of the dissection, and remove the developing skull tissue, exposing the cortex (Figure 1B).
Using the scalpel bisect the brain along the midline in the mid-sagital plane, separating the right and left cortical hemispheres (Figures 1B, C). The pia and arachnoid are left attached to the cortex, and the dura is removed together with the developing skull.
Prepare each cortical hemisphere separately. Place the hemisphere medial side up so that you can see the ganglionic eminence and the developing cortex (Figures 1C, D).
Using the scalpel, make a coronal incision through the cortical neuroepithelium caudal to the developing olfactory bulb. This incision should begin at the anterior boundary of the lateral and medial ganglionic eminences, and extend through the anterior cingulate region of the developing cortical rudiment (Figure 1E).
Make another coronal incision just caudal to the posterior boundary of the lateral ganglionic eminence (Figure 1F). This incision should also extend from the lateral boundary of the ganglionic eminence to the medial wall of the developing cortical rudiment.
Retract the cortical "flap" created by the two incisions made in steps 3.7 and 3.8, using a scalpel or gentle pressure from a stream of HBSS delivered with a 200 μl pipettor to avoid mechanical damage to the cortex. Then make a transverse incision along the boundary separating the lateral ganglionic eminence from the developing cortex (Figures 1G, H). Then, dissect away the developing hippocampus and cortical hem of the medial telencephalon, at the apex of the neocortex where the lateral cortical surface meets the interhemispheric wall.
Make a transverse incision along the boundary separating the lateral ganglionic eminence from the developing cortex (Figure 1H).
Remove any extra dissected tissue that is on the Sylgard plate from the dissected cortical explant. One can use gentle pipetting with fresh HBSS buffer to pipette away any extra dissected tissue. The dissected cortex is now with meningeal side facing down (Figure 1I).
4. Transferring of Cortical Explant
Prepare a platinum wire loop connected to a glass pipette. Heat the glass pipette with a folded platinum wire inserted in the end of the glass pipette. By twisting the end of the platinum wire loop, the size of the loop can be increased or decreased, and the loop can be shaped to fit the size of the explant desired. (This can be prepared beforehand and reused.)
Heat the end of the platinum wire to sterilize the loop.
Isolate the dissected cortical explant and remove the extra HBSS media by gentle pipetting, leaving a small amount to use with the wire loop.
Place a 1 cm polycarbonate membrane in a 4 cm diameter imaging dish.
Using the side of the platinum wire loop, gently flip the cortex so that the meningeal side is facing up.
Lift the cortical explant off the dissection dish, by placing the explant in the middle of the loop and using the intrinsic hydrostatic forces to lift the explant on a flat plane.
Gently place the explant on the polycarbonate membrane, and lift the platinum wire loop away such that the explant remains on the membrane on a flat plane with the ventricular surface making contact with membrane.
Gently lift the membrane and pipette 50 μl of CSF or other media under the membrane.
Cover the imaging dish, place it in a humidified secondary container, and incubate at 37 °C in 5% CO2 for 24 hr to support continued cell proliferation and explant growth. Figure 2 depicts a schematic of the final cortical explant dish preparation.
Representative Results
The CSF collection should yield a clear, transparent fluid. There should be no evidence of contamination from blood, as demonstrated by a red or yellow tinged fluid in the aspirate and in the Eppendorf tube. There should also be no evidence of tissue in the aspirate and Eppendorf tube. When the CSF is centrifuged, one can also assess the CSF microscopically to ensure that there is no contamination. If there are signs of contamination, the CSF should be discarded. From one E14.5 mouse litter, one can anticipate collecting 10-15 μl CSF. Table 1 depicts average volumes of CSF collected from average litter sizes in both mice and rats from various embryonic ages. When pure CSF samples have been collected, the CSF can be analyzed using a number of different techniques. Figure 3 shows a representative silver stained gel using 2ug of mouse E14.5 CSF.
Cerebral cortical explants can be grown with varying volumes of CSF plus a basal medium if necessary so that the total volume is 50 μl. Figure 4 shows representative explants grown with 50 μl of 100% embryonic CSF for 24 hr. The explants have been shown to survive and proliferate with 100% CSF and have tissue histology similar to rodent embryos at the same gestational age 9 , as indicated by immunoreactivity to phospho-histone H3 (PH3), a marker of cell division, along the ventricular surface, BrdU, a marker of DNA synthesis, incorporation along the ventricular zone, and Tuj1, a neuronal marker, in the developing cortical plate.
| Sprague Dawley litter Embryonic Age | Volume per litter | CD1 litter Embryonic Age | Volume per litter |
| E13 | 30-50 μl | E10.5 | 15-20 μl |
| E14 | 40-75 μl | E12.5 | 15-20 μl |
| E16 | 50-90 μl | E14.5 | 10-15 μl |
| E18 | 40-75 μl | E16.5 | 5-10 μl |
Table 1. Average Volumes of CSF Obtained From Standard Sized Litters.
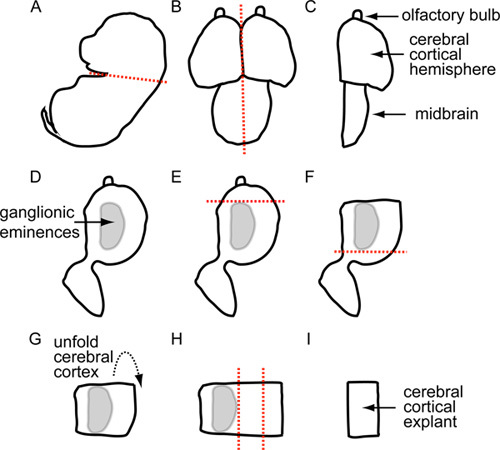 Figure 1. Schematic Outlining Cerebral Cortical Explant Dissection.
Figure 1. Schematic Outlining Cerebral Cortical Explant Dissection.
 Figure 2. Schematic Diagram of Final Dish Preparation.
Figure 2. Schematic Diagram of Final Dish Preparation.
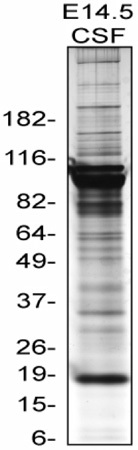 Figure 3. Silver stain of mouse E14.5 CSF. This is a representative Silver stain of 2ug mouse E14.5 CSF protein run on 4-12% Bis-Tris NuPAGE gradient gel from Invitrogen. Silver stain done with SilverQuest Silver staining kit developed for 5 min 40 sec.
Figure 3. Silver stain of mouse E14.5 CSF. This is a representative Silver stain of 2ug mouse E14.5 CSF protein run on 4-12% Bis-Tris NuPAGE gradient gel from Invitrogen. Silver stain done with SilverQuest Silver staining kit developed for 5 min 40 sec.
 Figure 4. E15 rat cortical explants grown for 24 hr. These are representative images of cerebral cortical explants grown for 24 hr in 100% embryonic CSF. These explants have been fixed by Carnoy's method, paraffin sectioned, and prepared for immunostaining. PH3 (red), and Tuj1 (green), BrdU (blue).
Figure 4. E15 rat cortical explants grown for 24 hr. These are representative images of cerebral cortical explants grown for 24 hr in 100% embryonic CSF. These explants have been fixed by Carnoy's method, paraffin sectioned, and prepared for immunostaining. PH3 (red), and Tuj1 (green), BrdU (blue).
Discussion
The described method for ventricular CSF collection has yielded relatively pure samples of embryonic CSF with stable protein composition and consistent activity in a number of cellular assays 9 . With a good collection technique and litter size of ten E14.5 mice, one can expect to collect 10-15 μl of CSF, and from a litter of E16 rats, one can expect to collect about 50-90 μl of CSF. This collection technique minimizes contamination from blood and tissue, by careful observation, centrifugation, and discarding of samples with any evidence of cellular debris or blood tinge. We have analyzed the fluid microscopically to assess for cellular debris or cells within the CSF after centrifugation, and found that samples that have no pellet after centrifugation are free from cellular contamination. In contrast to the proteome of the developing brain tissue or to samples of contaminated CSF (data not shown), using this method of CSF extraction, the CSF proteome did not contain any mitochondrial proteins when analyzed with a mass spectrometer 6 .
Based on the physical localization of the ventricular CSF within the telencephalic vesicles and neural tube, the only method of isolation is to pierce through the developing skin, skull, and telencephalon. Therefore, needle insertion through these tissues is a source for possible contamination. The tip of the needle should be as slender as possible, such that the needle can be inserted smoothly through the tissue and into the ventricle. The micro-capillary pipette may collect tissue within the bore of the needle during insertion of the needle into the ventricle. Depending on the age of the embryo, and if the choroid plexus has developed within the ventricle, it is important to not aspirate contents of the choroid plexus into the pipette. However, it is important to create sufficient negative pressure such that the CSF collects into the pipette, without disturbing the surrounding developing brain tissue or choroid plexus. The presence of tissue contamination can be visualized under the dissecting microscope, by pipetting CSF onto a glass slide with a glass cover slip and directly visualizing for cellular debris.
The methods we describe for CSF isolation provide relatively pure samples of CSF as we have determined to date. When known contaminated CSF samples have been analyzed using mass spectrometry, they have revealed the presence of mitochondrial proteins (data not shown). An inherent flaw in the technique is that the CSF is obtained by puncturing the developing skull and cortex, and therefore cells and tissue may be present in the CSF. By centrifuging the CSF, assaying for a pellet, and microscopically analyzing the CSF under the microscope, we have optimized a method for CSF isolation that yields relatively pure samples of CSF.
The technique for ventricular embryonic CSF collection and cerebral cortical explants has been described and shown in the movie for an embryonic mouse. However, this technique can be used for ventricular CSF collection and cerebral cortical explants from all embryonic rodents and has been applied to both rats and mice. The method described in this protocol was designed specifically for the isolation of ventricular CSF, and is not intended for collection of CSF in the subarachnoid space.
The technique for cerebral cortical explants was created to be able to grow explants with a reduced volume of media, given the limited quantities of embryonic CSF available for individual experiments. The smallest volume of medium used to grow explants reliably for 24 hr was 50 μl. Occasionally, explants were cultured for longer than 24 hr, with the longest period being 72 hr. With longer experiments, it is best to supplement the growth media every 24 hr. Once the explants are cultured for the desired period of time, the explants can be used for a number of various assays including immunofluoresence, generating neurospheres, cell death, or RNA extraction.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We are grateful for support from the NIH (Award numbers R00 NS072192 to M.K.L., HD029178 to A-S.L., and 2 RO1 NS032457 to C.A.W.). M.K.L. is the recipient of the Children's Hospital Boston Career Development Fellowship/Harvard Medical School Shore Fellowship and a Fellow of the Alfred P. Sloan Foundation. C.A.W. is an Investigator of the Howard Hughes Medical Institute.
References
- Dziegielewska KM, Evans CA, Lai PC, Lorscheider FL, Malinowska DH, Mollgard K, Saunders NR. Proteins in cerebrospinal fluid and plasma of fetal rats during development. Developmental Biology. 1981;83(1):193–200. doi: 10.1016/s0012-1606(81)80024-3. [DOI] [PubMed] [Google Scholar]
- Gato A, Martin P, Alonso MI, Martin C, Pulgar MA, Moro JA. Analysis of cerebro-spinal fluid protein composition in early developmental stages in chick embryos. Journal of Experimental Zoology. Part A, Comparative Experimental Biology. 2004;301(4):280–289. doi: 10.1002/jez.a.20035. [DOI] [PubMed] [Google Scholar]
- Gato A, Moro JA, Alonso MI, Bueno D, Mano DL, Martin C. Embryonic cerebrospinal fluid regulates neuroepithelial survival, proliferation, and neurogenesis in chick embryos. The Anatomical Record. Part A, Discoveries in Molecular, Cellular, and Evolutionary Biology. 2005;284(1):475–484. doi: 10.1002/ar.a.20185. [DOI] [PubMed] [Google Scholar]
- Parada C, Gato A, Aparicio M, Bueno D. Proteome analysis of chick embryonic cerebrospinal fluid. Proteomics. 2006;6(1):312–320. doi: 10.1002/pmic.200500085. [DOI] [PubMed] [Google Scholar]
- Parada C, Gato A, Bueno D. Mammalian embryonic cerebrospinal fluid proteome has greater apolipoprotein and enzyme pattern complexity than the avian proteome. Journal of Proteome Research. 2005;4(6):2420–2428. doi: 10.1021/pr050213t. [DOI] [PubMed] [Google Scholar]
- Zappaterra MD, Lisgo SN, Lindsay S, Gygi SP, Walsh CA, Ballif BA. A comparative proteomic analysis of human and rat embryonic cerebrospinal fluid. Journal of Proteome Research. 2007;6(9):3537–3548. doi: 10.1021/pr070247w. [DOI] [PubMed] [Google Scholar]
- Desmond ME, Jacobson AG. Embryonic brain enlargement requires cerebrospinal fluid pressure. Developmental Biology. 1977;57(1):188–198. doi: 10.1016/0012-1606(77)90364-5. [DOI] [PubMed] [Google Scholar]
- Lehtinen MK, Walsh CA. Neurogenesis at the brain-cerebrospinal fluid interface. Annual Review of Cell and Developmental Biology. 2011;27:653–679. doi: 10.1146/annurev-cellbio-092910-154026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Zappaterra MW, Chen X, Yang YJ, Hill AD, Lun M, Maynard T, Gonzalez D, Kim S, Ye P, et al. The cerebrospinal fluid provides a proliferative niche for neural progenitor cells. Neuron. 2011;69(5):893–905. doi: 10.1016/j.neuron.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C, Alonso MI, Santiago C, Moro JA, De la Mano A, Carretero R, Gato A. Early embryonic brain development in rats requires the trophic influence of cerebrospinal fluid. International Journal of Developmental Neuroscience: The Official Journal of the International Society for Developmental Neuroscience. 2009;27(7):733–7340. doi: 10.1016/j.ijdevneu.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Martin C, Bueno D, Alonso MI, Moro JA, Callejo S, Parada C, Martin P, Carnicero E, Gato A. FGF2 plays a key role in embryonic cerebrospinal fluid trophic properties over chick embryo neuroepithelial stem cells. Developmental Biology. 2006;297(2):402–416. doi: 10.1016/j.ydbio.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Zappaterra MW, Lehtinen MK. The cerebrospinal fluid: regulator of neurogenesis, behavior, and beyond. Cellular and Molecular Life Sciences: CMLS. 2012;69(17):2863–2878. doi: 10.1007/s00018-012-0957-x. [DOI] [PMC free article] [PubMed] [Google Scholar]