

Abstract
Aims
To characterize the population pharmacokinetics of ranitidine in critically ill children and to determine the influence of various clinical and demographic factors on its disposition.
Methods
Data were collected prospectively from 78 paediatric patients (n = 248 plasma samples) who received oral or intravenous ranitidine for prophylaxis against stress ulcers, gastrointestinal bleeding or the treatment of gastro-oesophageal reflux. Plasma samples were analysed using high-performance liquid chromatography, and the data were subjected to population pharmacokinetic analysis using nonlinear mixed-effects modelling.
Results
A one-compartment model best described the plasma concentration profile, with an exponential structure for interindividual errors and a proportional structure for intra-individual error. After backward stepwise elimination, the final model showed a significant decrease in objective function value (−12.618; P < 0.001) compared with the weight-corrected base model. Final parameter estimates for the population were 32.1 l h−1 for total clearance and 285 l for volume of distribution, both allometrically modelled for a 70 kg adult. Final estimates for absorption rate constant and bioavailability were 1.31 h−1 and 27.5%, respectively. No significant relationship was found between age and weight-corrected ranitidine pharmacokinetic parameters in the final model, with the covariate for cardiac failure or surgery being shown to reduce clearance significantly by a factor of 0.46.
Conclusions
Currently, ranitidine dose recommendations are based on children's weights. However, our findings suggest that a dosing scheme that takes into consideration both weight and cardiac failure/surgery would be more appropriate in order to avoid administration of higher or more frequent doses than necessary.
Keywords: gastro-oesophageal reflux, nonlinear mixed-effects modelling, population pharmacokinetics, ranitidine, stress ulcers
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Ranitidine is routinely prescribed in paediatric intensive care units as a prophylactic treatment for stress ulcers, gastrointestinal bleeding or to negate the harmful effects of gastro-oesophageal reflux and gastric aspiration.
Despite the widespread use of ranitidine, little is known about its pharmacokinetics in paediatric patients.
WHAT THIS STUDY ADDS
A population pharmacokinetic model has been developed to evaluate the pharmacokinetics of ranitidine in paediatric patients who received the drug as part of their therapy in the intensive care unit.
The model showed that in addition to weight, cardiac failure/surgery was a significant covariate that affected ranitidine clearance (reduced its value by a factor of 0.463).
Introduction
Stress ulcers and upper gastrointestinal bleeding are frequent complications of critical illness in children admitted to paediatric intensive care units [1, 2], of whom up to 25% develop gastrointestinal bleeding or perforation [3–5].
Gastro-oesophageal reflux (GOR) is also an important disorder in children [6, 7]. During the first year of life, when the oesophageal sphincter is still developing, GOR is common and may cause oesophagitis [8]. In hospitalized paediatric patients, GOR may complicate other underlying conditions and increase the risk of life-threatening respiratory symptoms associated with gastric aspiration [9, 10].
A gastric pH below 2.5 is one of the risk factors for development of stress ulcers and gastrointestinal bleeding [11]. Ranitidine is therefore prescribed routinely in paediatric intensive care units for prevention of stress ulcers [2, 12–14] and to negate harmful effects of GOR/gastric aspiration or the erosive side-effects of certain drugs (e.g. corticosteroids). Despite widespread use of ranitidine, oral administration is still unlicensed in children under 3 years of age and parenteral administration is unlicensed in children <6 months. Furthermore, pharmacokinetic-based dosage guidelines are lacking in these patient groups.
The aim of the present study was to use sparse data to investigate the population pharmacokinetic profile of intravenous and oral ranitidine in critically ill children and characterize potentially important factors that could lead to variability in ranitidine concentrations.
Methods
Patients and data collection
Ninety-one children who received ranitidine as part of their normal treatment while in the paediatric intensive care unit at either the Royal Belfast Hospital for Sick Children, Belfast or the Alder Hey Children's Hospital, Liverpool participated in the study, which was approved by the Research Ethics Committee, Queen's University Belfast. Ranitidine was administered orally and/or intravenously (bolus doses). Blood samples were taken opportunistically when blood sampling was required as part of routine clinical care. In addition to information on dosing and times of sampling, the following data were collected for each child: age, gender, weight, laboratory test results, concomitant drug therapies and concomitant illness.
Blood samples (0.5 ml) were collected in EDTA collection tubes and centrifuged at 1800g for 10 min to separate the plasma component. Plasma samples were stored at −20°C prior to analysis.
Ranitidine assay
Plasma concentrations of ranitidine were determined by a selective, reversed phase high-performance liquid chromatography assay that was developed and validated in our laboratory using nizatidine as an internal standard. The method utilized 200 μl of plasma, and sample preparation involved solid phase extraction using Oasis® HLB cartridges (1 ml/30 g; Waters®, Dublin, Ireland). The method was found to be linear over the concentration range 25–2000 ng ml−1 and was validated over that range. The limit of quantification (LOQ) was 25 ng ml−1. The intra- and interday variability ranged from 0.2 to 6.2% and from 1.3 to 1.9%, respectively. For pharmacokinetic analysis, concentrations that were detectable, i.e. peak present in chromatogram, but below the LOQ (n = 26, i.e. 10.4% of the final data set) were replaced with LOQ/2 (12.5 ng ml−1) as suggested by Hing et al. [15].
Population pharmacokinetic modelling
Population pharmacokinetic analysis was performed by means of nonlinear mixed-effect modelling using NONMEM® (version VI, level 1.0) 1989–2006 installed on a personal computer in conjunction with DIGITAL Visual Fortran compiler (version 5.0.A) in combination with WFN (Wings For NONMEM®, version 601) [17]. Census® (version 1.0) was used for management of data analysis [18] and Xpose® [19] for graphical visualization of results. The first-order conditional estimation method with interaction was used to estimate population mean parameters, interindividual variability (IIV) in these parameters and residual variability between measured and predicted ranitidine concentrations.
The complete data set was used for development of the pharmacokinetic model. Potential pharmacokinetic models considered were linear one- and two-compartment models with first order absorption. The IIV for clearance (CL) and volume of distribution (V) parameters was modelled using an exponential scale because they must be constrained to be greater than zero and their distribution is often right skewed; however, the IIVs on both bioavailability and absorption rate constant had to be removed for the model to minimize successfully.
The statistical residual variability model considered both the constant coefficient of variation (proportional) and additive error models throughout the model development, according to the following equation:
where Cij is the measured and Cpred,ij is the model predicted concentration of the ith individual at the jth sampling time and εij is the residual error term, which is a random variable with zero mean and variance of σ2. Simplification of the residual error model was considered during model building by removing the residual variance component that has a value close to zero.
Regression model
Initial analysis of the population pharmacokinetics of ranitidine was conducted without including any patient covariates in the model (BASE model). Individual Bayesian estimates obtained for each pharmacokinetic parameter were obtained from this BASE model and then plotted against the continuous covariates (weight and age) and the categorical covariates (disease state, concomitant drugs, route of administration and gender), to help identify potential relationships between the pharmacokinetic parameters and covariates. Following that, direct covariate testing within NONMEM was utilized to determine which pharmacokinetic parameters were significantly related to each candidate covariate and whether this relationship was linear or nonlinear. The main criterion for comparing models was change in the objective function value (OFV), a statistic which measures goodness of fit of the model and equals minus twice the log-likelihood of the data. However, goodness of fit was also investigated by examining the precision of parameter estimates (relative standard errors), the decrease in interindividual and residual variability and graphs of conditional weighted residuals and measured ranitidine concentrations plotted separately against predicted concentrations.
Significant covariates were added into the model simultaneously and tested for inclusion in the FINAL model using backward stepwise elimination. Starting with the least significant, covariates were then removed from the model individually and difference in OFV tested for significance at the 5% level of the stay criterion (an increase in OFV ≥3.841 with one degree of freedom). If the increase in OFV was <3.841, the covariate was then omitted from the model and the process repeated for the remaining covariates. At each stage of the process, covariates outside of the model (excluding the last to be removed) were then tested for inclusion into the model at a more stringent entry criterion of 1% significance (a decrease in OFV ≥6.635 with one degree of freedom) and were retained in the model if found to be significant. The process ended when there was no further significant change in OFV of the model.
Model evaluation
The population pharmacokinetic model was evaluated through nonparametric jack-knifing and bootstrap analyses. Bootstrap analysis (n = 1000 model fits with resampling) was performed twice, with or without gender as a covariate in the final model. Conditional weighted residuals and measured ranitidine concentrations were plotted separately against predicted concentrations to permit visual assessment of the deviations of model-predicted from observed concentrations. In addition, shrinkage estimates were calculated using the method described by Karlsson [20] and by Savic and Karlsson [21]. A visual predictive check was also generated for the final model and principal component analysis performed to investigate any possible subgroups within the patient population that were not identified during the model development. Principal component analysis was performed using SPSS® (version 17.0) for Windows (SPSS Inc., Chicago, IL, USA).
Results
Seventy-eight subjects with a total of 248 samples (median of two samples per patient, range 1–13) were included in the final analysis. The relative distribution of patient samples collected over time is shown in Figure 1. Thirteen subjects (21 samples) were excluded from the study because their ranitidine concentrations were all below the LOQ. Examination of the records of excluded patients revealed similar demographic and clinical characteristics to the remainder of the population. The demographic and clinical characteristics of the study participants are shown in Table 1.
Figure 1.
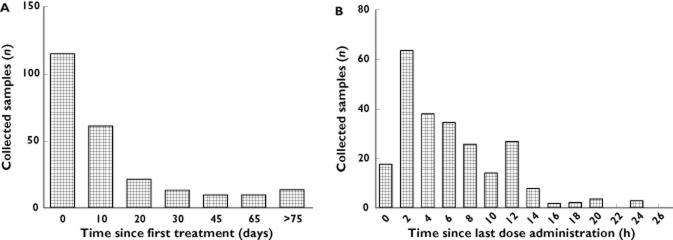
Histogram plots demonstrating the distribution of patient samples over the study duration (A) and over the time relative to last dose administration (B)
Table 1.
Demographic data of patients included in the study (n = 78)
| Characteristic | Value | ||
|---|---|---|---|
| Age (years), mean ± SD (range) | 4.57 ± 4.48 (range 15 days to 15.51 years) | ||
| Weight (kg), mean ± SD (range) | 16.27 ± 12.24 (range 1.3–47 kg) | ||
| Gender, n (male, female) | 37, 41 | ||
| Route of administration: | Intravenous | Oral | Both |
| n (%) | 16 (20.5%) | 12 (15.4%) | 50 (64.1%) |
| Dose (mg kg−1), mean ± SD | 1.18 ± 0.43 | 2.15 ± 1.32 | 2.10 ± 0.80 |
| Duration of treatment (days), mean ± SD | 15.48 ± 34.49 | 24.09 ± 42.15 | 41.62 ± 85.04 |
| Subjects with cardiac failure and/or surgery, n (%) | 17 (21.8%) | ||
| Cardiac failure | 14 (17.9%) | ||
| Cardiac surgery | 10 (12.8%) | ||
| Indication for cardiac surgery, n* | |||
| Patent ductus arteriosus | 2 | ||
| Atrial/ ventricular septal defect | 3 | ||
| Hypoplasia of left ventricle | 1 | ||
| Bilateral pleural effusion | 1 | ||
| Other | 3 | ||
| Subjects with renal failure, n (%) | 9 (11.5%) | ||
| Subjects receiving concomitant medications, n (%) | |||
| Atenolol | 8 (10.3%) | ||
| Carobel® (carob seed flour) | 9 (11.5%) | ||
| Cefotaxime | 9 (11.5%) | ||
| Digoxin | 9 (11.5%) | ||
| Fucidin® (sodium fusidate) | 8 (10.3%) | ||
| Furosemide | 17 (21.8%) | ||
| Gaviscon® (sodium alginate, sodium bicarbonate and calcium carbonate) | 11 (14.1%) | ||
| Primacor® (milrinone) | 8 (10.3%) | ||
| Morphine | 8 (10.3%) | ||
| Oxycarbazepine | 16 (20.5%) | ||
| Paracetamol | 20 (25.6%) | ||
| Potassium canreonate | 8 (10.3%) | ||
| Prednisolone | 11 (14.1%) | ||
| Spironolactone | 10 (12.8%) | ||
Continuous variables are presented as mean ± SD (range). Categorical variables are presented as number (percentage).
Patients who had undergone cardiac surgery received only intravenous therapy during the first week following surgery.
Pharmacokinetic modelling
The BASE model was best described by a one-compartment model with first-order absorption and elimination (implemented using NONMEM subroutines ADVAN2 and TRANS2). Pharmacokinetic parameters estimated from this model were CL, V, bioavailability (F1) and absorption rate constant (ka). Despite the fact that a number of subjects had less than four observations, the relatively larger number of patients recruited in the present study (compared with conventional pharmacokinetic studies) allowed four structural parameters to be estimated. Figure 1 suggested that the total numbers of observations relative to last dose administration were sufficiently abundant to allow the parameters to be estimated unambiguously. Both CL and V were allometrically scaled to an adult of 70 kg with power values of 0.75 and 1.0 for CL and V, respectively. Investigation of models where the power values were not fixed, but included as additional thetas (θs), did not result in any significant improvement in model fit. Inclusion of weight as an a priori covariate affecting both CL and V resulted in a significant improvement in goodness of fit and a 59.8 unit decrease in OFV. Population parameter estimates for CL and V from the BASE model were 32.4 l h−1 and 275 l, respectively. This model produced an OFV of −934.588.
Addition of several covariates resulted in a reduction of OFV ≥3.841 (P < 0.05). These covariates were age, gender, cardiac failure/surgery, concomitant administration of Carobel® (carob seed flour) or Gaviscon® (a combination of sodium alginate, sodium bicarbonate and calcium carbonate) on CL, and gender, renal dysfunction, concomitant administration of atenolol, Fucidin® (sodium fusidate), Primacor® (milrinone) or prednisolone on V. These significant covariates were then added to the BASE model and subjected to backward stepwise analysis using the rationale described above. None of the examined concomitant medications was significant enough to remain in the FINAL model. It is possible, however, that the small number of patients in each category of concomitant medication was not sufficient to test and quantify the true effect of these covariates. Backward elimination resulted in a FINAL model with gender as a significant covariate affecting CL and V, and cardiac failure/surgery affecting CL. The final OFV for this model was −960.61 (ΔOFV of −26.02, P value < 0.0005 compared with the BASE model).
Jack-knifing and bootstrapping were then performed on the model. With the exception of two subjects, all parameter estimates from the jack-knife analysis were within ±20% coefficient of variation (CV). Of the 1000 bootstrap runs, however, only 688 minimized successfully. Jack-knifing and bootstrap analyses also resulted in significant error regarding the covariate effect of gender on V (θGEND), with the standard error being significantly larger than 30% (70.8% for jack-knif and 63.6% for bootstrapping), and confidence intervals overlapping zero for both analyses. Therefore, this term was removed from the model. The effect of gender on CL was then found to be nonsignificant (ΔOFV = 0.10). Cardiac failure/surgery and weight were therefore the only significant covariates remaining in the model, with an OFV of −947.21. The structure of the FINAL model parameters was, therefore:
where WTi is the weight of the ith child and TVCL and TVV are the typical parameter estimates of CL and V, respectively. The parameter estimates for this model are shown in Table 2. The FINAL model resulted in a decrease in IIV for CL from 70.1 to 60.1% and for V from 86.3 to and 85.0%, when compared with the BASE model.
Table 2.
Ranitidine population parameter estimates from the BASE and FINAL models developed from the original data set of 78 patients, and mean parameter estimates from the FINAL model fitted to the bootstrap and jack-knifed samples
| Parameter | Original data set (n = 184) | 1000 Bootstrap samples | Jack-knifed samples | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| BASE model | FINAL model | Bootstrap | Jack-knife | |||||||
| Estimate | RSE% | Estimate | RSE% | Mean | RSE% | %Diff | Mean | RSE% | %Diff | |
| CL (l h−1) | ||||||||||
| θCL | 32.4 | 25.8 | 32.1 | 27.4 | 33.3 | 25.2 | 3.9% | 32.1 | 25.7 | 0.1% |
| V (l) | ||||||||||
| θV | 275 | 25.7 | 285 | 34.3 | 309.7 | 45.8 | 8.7% | 285.4 | 41.7 | 0.1% |
| ka (h−1) | ||||||||||
| θka | 0.429 | 23.5 | 1.31 | 26.1 | 1.305 | 52.6 | −0.4% | 1.309 | 36.2 | −0.1% |
| F1 | ||||||||||
| θF1 | 0.349 | 26.9 | 0.275 | 27.1 | 0.296 | 25.7 | 7.6% | 0.275 | 28.8 | 0.0% |
| θ(HEART,CL) | — | — | 0.463 | 23.5 | 0.485 | 23.6 | 4.8% | 0.463 | 25.4 | 0.0% |
| IIVCL (CV%) | ||||||||||
| ωCL | 70.1 | 28.5 | 60.1 | 33.0 | 57.8 | 19.6 | −3.8% | 60.0 | 20.4 | −0.2% |
| IIVV (CV%) | ||||||||||
| ωV | 86.3 | 58.1 | 85.0 | 44.5 | 83.8 | 31.5 | −1.4% | 85.0 | 24.6 | 0.0% |
| Residual (CV%) | ||||||||||
| σprop | 60.0 | 12.6 | 59.5 | 13.4 | 59.1 | 6.9 | −0.7% | 59.5 | 7.2 | 0.0% |
The BASE model includes weight as a covariate on clearance and volume, as follows:
Final model: 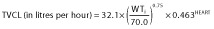 and
and 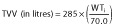
Definitions are as follows: IIVCL is the interindividual variability in clearance (CL); IIVV is the interindividual variability in the volume of distribution (V); σprop is the residual variability (proportional error model); RSE% is the percentage relative standard error of parameter estimates; and coefficient of variation (CV)% is the percentage coefficient of variation.
Percentage difference 
Model evaluation and validation
Plots of observed against population and individual predicted concentrations in the FINAL model (Figure 2) showed reasonable agreement around the line of identity, although with a slight downward bias, particularly at higher concentrations. The scatter plots of conditional weighted residuals vs. model-predicted ranitidine concentrations showed that they were randomly distributed, and weighted residuals lay within ±2 units of the null ordinate of perfect agreement (Figure 2). Estimates of η-shrinkage (of parameters for which IIV was identified, i.e. CL (ηCL,sh) and V (ηV,sh)) and ε-shrinkage for the final model (εsh) were acceptable (ηCL,sh = 0.18, ηV,sh = 0.29 and εsh = 0.10), with only V having notable shrinkage [21]. η- and ε-shrinkage values can be defined as the deviation of individual parameter estimates from their true values toward the population mean or the typical parameter estimates. A shrinkage magnitude of zero corresponds to the situation when the model is correct and individual data is sufficiently abundant to estimate the true individual parameters.
Figure 2.

Plots of measured vs. population predicted (A) and individual predicted (B) ranitidine concentrations from the FINAL model together with plots of conditional weighted residuals vs. population predicted ranitidine concentrations (C) and time relative to last dose administration (D)
The results of the visual predictive check performed on the FINAL population pharmacokinetic model are presented in Figure 3, stratified by route of administration, i.e. patients receiving intravenous vs. oral ranitidine. The visual predictive check plots are presented as concentration vs. time after administration of the dose. Individual predictions were used to represent concentrations reported as below the limit of quantification (n = 26). Results of the bootstrap analysis (after eliminating gender from the final model) are shown in Table 2. Of the 1000 bootstrap data sets, 969 minimized successfully, with 31 failing to minimize but not terminating abnormally. In addition, there was a close agreement in mean parameter estimates (FINAL model vs. bootstrap), with absolute differences of less than 9%. The final estimates from the jack-knife analysis were in general concordance with bootstrapping, with each data set being successfully minimized (see Table 2). All mean parameter estimates from jack-knifing were within ±1% of those from the FINAL model, demonstrating robustness of the model.
Figure 3.

Visual predictive check plots of the final model fitted to the full data set (A) and stratified by route of administration, intravenous (B) and oral (C). Continuous and dashed red lines indicate the median, fifth and 95th percentiles for the observed concentrations (filled circles). Shaded areas represent the upper, middle and lower confidence intervals for the 90% prediction intervals of simulated values (n = 1000 per each patient time point). A reference line representing the assay limit of quantification LOQ has been highlighted in all plots
Principal component analysis was then performed on the final model for the full data set. This involved using the final parameter estimates from the individual jack-knifed data sets, ascertaining which subjects had the most influential effect, and also determining whether there were any correlations between individuals or parameters that were not included in the final model. The use of principal component analysis in this manner means that a lack of a positive result is desirable. Eigenvalues greater than one were requested in the analysis; therefore, the first three components were extracted. The three components explained over 75% of the variance in parameter estimates, with less than 25% of the final variability in parameter estimates not being accounted for. The component loadings plot (Figure 4A), which is a visual representation of the three rotated components, did not reveal any significant correlation between any of the parameters that was not explained by the model. In addition, a scatter plot of each principal component against the others was obtained and examined for outliers (Figure 4B). Plots of the retained principal components were also used to identify influential subjects. The scatterplots did not reveal any groupings that would indicate any significant correlation between individuals. A review of demographics of outlying subjects failed to reveal any significant trends. Results of the principal component analysis therefore did not ascertain any underlying trends not identified by the earlier analysis, thus giving evidence for validity of the FINAL model.
Figure 4.

Principal component analysis performed on the FINAL model for the full data set. The component loadings plot of the three rotated components (A) and the scatterplots for the principal components (B) did not reveal significant correlation between the parameters, which was not explained by the model or any significant correlation between individuals
Discussion
Although ranitidine is not licensed for oral administration in children <3 years old or for parenteral administration in children <6 months old [22], both routes are commonly used for prophylaxis of stress ulcers and upper gastrointestinal bleeding in critically ill children and the treatment of gastro-oesophageal reflux disease in children [2]. When used off-label, dosage regimens are largely derived from data obtained in older children and adults, and pharmacokinetic-based dosage recommendations are lacking. The present study was undertaken, therefore, to explore ranitidine pharmacokinetics and identify covariates that explain the pharmacokinetic variability observed in critically ill children.
Ranitidine is rapidly absorbed following oral administration, with variable serum concentrations and a wide range of oral bioavailability between individuals [23, 24]. The pharmacokinetic parameters determined after intravenous and oral ranitidine administration, however, are remarkably similar when the two routes are compared at the level of an individual patient [25]. Plasma protein binding of ranitidine is approximately 15% [23] and, as it is hydrophilic [26, 27], it distributes primarily in total body water [28, 29]. Small amounts of the drug undergo metabolism in the liver [30]; however, renal excretion is the primary route of elimination of ranitidine and accounts for approximately 70% of an intravenous dose excreted unchanged in the urine [23].
The pharmacokinetic model developed in the present study revealed considerable IIV in the CL and V of ranitidine in critically ill children. This variability concurs with previously reported data obtained in critically ill adults and children in the paediatric intensive care unit [31, 32]. The estimated IIV (CV%) in the BASE model was 83.7 and 84.0% for CL and V, respectively (data not shown). However, when weight was allometrically incorporated into the model as a covariate affecting CL and V there was a change to the IIV (70.1 and 86.3% for CL and V, respectively) and a significant reduction in the OFV (59.8 units). Allometric size adjustment, with fixed exponents of 0.75 for CL and 1 for V, was used as a priori inclusion of weight because the method is well established, has a strong scientific and physiological basis [33–35] and has been adopted by many researchers during development of population pharmacokinetic models in children [35–37].
Final estimates obtained in the present study were a total CL of 32.10 l h−1 allometrically modelled for a 70 kg adult (1.32 l h−1 for an individual with a theoretical weight of 1 kg), V of 285 l (4.07 l for a 1 kg individual), ka of 1.31 h−1 and F1 of 27.5%. The final estimate of CL was comparable to values obtained from previous studies in critically ill children when adjusting for weight in an allometric model similar to that developed in the present study [32, 38]. The CL value reported by Orenstein et al. [39] (41.40 l h−1; 77.20 l h−1 when scaled to a weight of 70 kg) in children suffering from GOR, but otherwise healthy, was ∼2.4 times higher than that reported in the present study. However, this value was apparent clearance (CL/F), i.e. CL value divided by the bioavailable fraction of the drug (F), which accounts for the difference shown. Conversely, Wells et al. [40] reported a value of 0.88 l h−1 for 13 term neonates with an average weight of 3.49 kg (8.34 l h−1 when scaled allometrically to a weight of 70 kg), which is ∼3.8 times lower than the predicted estimate from the present study. This could be due to physiological immaturity of the renal and hepatic functions of neonates. Furthermore, neonates in that study were undergoing extracorporeal membrane oxygenation and would have been significantly more distressed than those in the present study, which may help explain the decreased clearance.
The covariate for renal dysfunction in the present study was not significant after backward stepwise elimination, and this resulted in marked differences to reported values from adult populations with renal failure. Garg et al. [41] reported a value of 9.14 l h−1 for 10 patients with a mean age of 57 years, and Koch et al. [42] reported a value of 22.21 l h−1 for 41 adults with renal failure with ages ranging from 20 to 69 years (when both values were scaled to a weight of 70 kg). The reason for lack of significant effect of renal dysfunction on ranitidine clearance in the present study, however, is unclear but could be due to haemofiltration or dialysis being performed to combat the disease state. This could be supported by the low serum creatinine levels recorded in these patients at the time of blood sampling; apart from one patient, serum creatinine levels did not exceed 100 μmol l−1 (median 34, range 16–98 μmol l−1). However, using the allometric model the final predicted estimates from the present study were, on average, 67.5% (range 34.2–98.5%) of those reported in healthy adults [43–48]. This could be due to the reduced health status of the critically ill paediatric population studied in the present study.
The final estimate of V was different from previously reported values in adults, being, on average, 1.86 times greater. The higher estimate of V in the present study compared with that observed in adults is to be expected because ranitidine distributes mainly in total body water, of which there is a higher percentage in children, and is not highly bound within plasma (∼15%) [26–29]. Furthermore, given that the estimated value of V in the present study is higher than the total body water expected in neonates, infants or children, the data suggest that there is additional binding or preferential accumulation within tissues in children, similar to that reported in adults [23]. The limited number of samples collected shortly (in the first 30 min) after ranitidine administration, however, did not enable accurate evaluation of the distribution phase; hence, a two-compartment model did not provide a better fit to the data. A one-compartment model was therefore chosen to describe the data in the present study.
Of the five main clinical conditions exhibited by patients in this study, namely renal dysfunction, cardiac failure/surgery, cancer, stomach and blood disorders (leucopenia, thrombocytopenia), only the cardiac failure/surgery covariate was found to be significant in the FINAL model, reducing total CL by a factor of 0.463. Given the relatively large value of this effect, further validation of the developed model by large prospective studies is warranted to eliminate the risk of covariate selection bias in data sets of small size similar to the present study (which can be associated with inflation of the estimated effect, especially given that covariates are unevenly distributed and variability is high) [49]. Once confirmed, the implications of altered ranitidine kinetics in paediatric patients with cardiac failure/surgery could be clinically relevant, affecting the doses necessary for safe and effective reduction of intragastric acidity and optimal control of symptoms. The decreased ranitidine CL found in the present study was in line with the prolonged elimination rate of other medications reported in similar studies of neonates and children following cardiac surgery [50].
Cardiac failure/surgery is known to alter pharmacokinetics of many drugs due to physiological upset [51]. For instance, postoperative renal dysfunction is one of the most severe complications of cardiac surgery, and is associated with increases in mortality, morbidity and subsequent length of stay in the intesive care unit [52]. The immature kidney of infants may be more susceptible to renal failure [53]. In one study, 20% of cases of acute renal failure in newborn infants were attributed to heart failure [54]. There is also a decrease in hepatic blood flow proportionate to the decrease in cardiac output [55] and an indirect relationship between cardiac index and hepatic blood flow [56]; therefore, both hepatic and renal blood flow decrease in proportion to the decrease in cardiac output [57], and this could account for the decreased CL of ranitidine associated with cardiac failure/surgery in our study. Ranitidine is one of the drugs most frequently requiring dosage adjustment in paediatric patients undergoing cardiac surgery and, although adverse effects from ranitidine are infrequent, the cost benefits of such an adjustment (through reducing unnecessary treatment) are significant [58]. In the present study, there was no evidence to suggest a difference in the dosing regimens selected for children with cardiac failure or those who had undergone heart surgery. Our results suggest that a 50% reduction in dose, coupled with careful monitoring, would be appropriate for patients with cardiac failure or heart surgery who require ranitidine.
Conclusion
This paper presents a study that investigated population pharmacokinetics of ranitidine in critically ill children receiving the drug either as multiple intravenous bolus doses, oral doses or a combination of the two. A one-compartment model best described the concentration profile, with four parameters incorporated, i.e. CL, V, ka and bioavailability. The model had an exponential structure for the interindividual errors imposed on CL and V. A proportional structure was used for the intra-individual error. The final parameter estimates for the population were 32.1 l h−1 for total CL and 285 l for V, both allometrically modelled for a 70 kg adult. The final estimates for ka and bioavailability were 1.31 h−1 and 27.5%, respectively. Apart from weight, the only other significant covariate was cardiac failure/surgery, which reduced CL by a factor of 0.463.
Acknowledgments
The authors wish to thank Action Medical Research and the Health and Social Care Research and Development office (N. Ireland) for financial support for the performance of this research.
Competing Interests
There are no competing interests to declare.
References
- 1.Crill CM, Hak EB. Upper gastrointestinal tract bleeding in critically ill pediatric patients. Pharmacotherapy. 1999;19:162–180. doi: 10.1592/phco.19.3.162.30914. [DOI] [PubMed] [Google Scholar]
- 2.Reveiz L, Guerrero-Lozano R, Camacho A, Yara L, Mosquera PA. Stress ulcer, gastritis, and gastrointestinal bleeding prophylaxis in critically ill pediatric patients: a systematic review. Pediatr Crit Care Med. 2010;11:124–132. doi: 10.1097/PCC.0b013e3181b80e70. [DOI] [PubMed] [Google Scholar]
- 3.Lopez-Herce J, Dorao P, Elola P, Delgado MA, Ruza F, Madero R. Frequency and prophylaxis of upper gastrointestinal hemorrhage in critically ill children: a prospective study comparing the efficacy of almagate, ranitidine, and sucralfate. Crit Care Med. 1992;20:1082–1089. doi: 10.1097/00003246-199208000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Chaïbou M, Tucci M, Dugas MA, Farrell CA, Proulx F, Lacroix J. Clinically significant upper gastrointestinal bleeding acquired in a pediatric intensive care unit: a prospective study. Pediatrics. 1998;102:933–938. doi: 10.1542/peds.102.4.933. [DOI] [PubMed] [Google Scholar]
- 5.Cochran EB, Phelps SJ, Tolley EA, Stidham GL. Prevalence of, and risk factors for, upper gastrointestinal tract bleeding in critically ill pediatric patients. Crit Care Med. 1992;20:1519–1523. doi: 10.1097/00003246-199211000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Nelson S, Chen E, Syniar GM, Christoffel KK. Prevalence of symptoms of gastroesophageal reflux during childhood: a pediatric practice-based survey. Arch Pediatr Adolesc Med. 2000;154:150–154. doi: 10.1001/archpedi.154.2.150. [DOI] [PubMed] [Google Scholar]
- 7.Rudolph CD, Mazur LJ, Liptak GS, Baker RD, Boyle JT, Colletti RB, Gerson WT, Werlin SL. Guidelines for evaluation and treatment of gastroesophageal reflux in infants and children. J Pediatr Gastroenterol Nutr. 2001;32:S1–31. doi: 10.1097/00005176-200100002-00001. [DOI] [PubMed] [Google Scholar]
- 8.Vandenplas Y, Hegar B. Diagnosis and treatment of gastro-oesophageal reflux disease in infants and children. J Gastroenterol Hepatol. 2000;15:593–603. doi: 10.1046/j.1440-1746.2000.02169.x. [DOI] [PubMed] [Google Scholar]
- 9.Engelhardt T, Webster NR. Pulmonary aspiration of gastric contents in anaesthesia. Br J Anaesth. 1999;83:453–460. doi: 10.1093/bja/83.3.453. [DOI] [PubMed] [Google Scholar]
- 10.Ku CM, Ong BC. Postoperative nausea and vomiting: a review of current literature. Singapore Med J. 2003;44:366–374. [PubMed] [Google Scholar]
- 11.Hoyt J. Aspiration pneumonitis: patient risk factors, prevention, and management. J Intensive Care Med. 1990;5:S2–9. [Google Scholar]
- 12.Iberti TJ. The hemodynamic effects of H2 antagonists in intensive care unit patients. J Intensive Care Med. 1990;5:S40–43. [Google Scholar]
- 13.Marik PE. Stress ulcer prophylaxis: a practical approach. J Intensive Care Med. 1999;14:1–8. [Google Scholar]
- 14.Gardner TB, Robertson DJ. Stress ulcer prophylaxis in non-critically ill patients: less may be more. Am J Gastroenterol. 2006;101:2206–2208. doi: 10.1111/j.1572-0241.2006.00847.x. [DOI] [PubMed] [Google Scholar]
- 15.Hing JP, Woolfrey SG, Greenslade D, Wright PMC. Analysis of toxicokinetic data using NONMEM: impact of quantification limit and replacement strategies for censored data. J Pharmacokinet Pharmacodyn. 2001;28:465–479. doi: 10.1023/a:1012247131190. [DOI] [PubMed] [Google Scholar]
- 16.Beal SL, Sheiner LB, Boeckmann AJ, NONMEM Users Guides . Ellicott City, MD: Icon Development Solutions; 1989. –2006. [Google Scholar]
- 17.Holford NH. Wings for NONMEM 2007. Available at http://wfn.sourceforge.net (last accessed 7 February 2012)
- 18.Wilkins JJ. NONMEMory: a run management tool for NONMEM. Comput Methods Programs Biomed. 2005;78:259–267. doi: 10.1016/j.cmpb.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 19.Jonsson EN, Karlsson MO. Xpose–an S-PLUS based population pharmacokinetic / pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58:51–64. doi: 10.1016/s0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 20.Karlsson MO. Model-building diagnostics. DIA Meeting, Philadelphia, PA, 5–6 December 2005.
- 21.Savic RM, Karlsson MO. Importance of shrinkage in empirical bayes estimates for diagnostics: problems and solutions. AAPS J. 2009;11:558–569. doi: 10.1208/s12248-009-9133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paediatric Formulary Committee. The BNF for Children 2010–2011. London: BMJ Group; 2011. [Google Scholar]
- 23.Roberts CJ. Clinical pharmacokinetics of ranitidine. Clin Pharmacokinet. 1984;9:211–221. doi: 10.2165/00003088-198409030-00003. [DOI] [PubMed] [Google Scholar]
- 24.Garg DC, Weidler DJ, Eshelman FN. Ranitidine bioavailability and kinetics in normal male subjects. Clin Pharmacol Ther. 1983;33:445–452. doi: 10.1038/clpt.1983.60. [DOI] [PubMed] [Google Scholar]
- 25.Blumer JL, Rothstein FC, Kaplan BS, Yamashita TS, Eshelman FN, Myers CM, Reed MD. Pharmacokinetic determination of ranitidine pharmacodynamics in pediatric ulcer disease. J Pediatr. 1985;107:301–306. doi: 10.1016/s0022-3476(85)80156-6. [DOI] [PubMed] [Google Scholar]
- 26.Kortejärvi H, Yliperttula M, Dressman JB, Junginger HE, Midha KK, Shah VP, Barends DM. Biowaiver monographs for immediate release solid oral dosage forms: ranitidine hydrochloride. J Pharm Sci. 2005;94:1617–1625. doi: 10.1002/jps.20392. [DOI] [PubMed] [Google Scholar]
- 27.GlaxoSmithKline Inc. ZANTAC® Product Monograph: Histamine H2-Receptor Antagonist. Mississauga, ON: GlaxoSmithKline Inc; 2009. Accessed at http://gsk.ca/english/docs-pdf/Zantac_PM_20090804_EN.pdf (last accessed 7 February 2012) [Google Scholar]
- 28.Brummel G, Lucking SE. Pharmacology: pharmacokinetics. In: Lucking SE, Maffei FA, Tamburro RF, Thomas NJ, editors. Pediatric Critical Care Study Guide: Text and Review. London: Springer; 2012. pp. 334–344. [Google Scholar]
- 29.Porter RS, Kaplan JL. The Merck Manual Online – Drug Distribution to Tissues. Readington, NJ: Merck Sharp & Dohme Corp; 2011. 2010–2011 ed. Accessed at http://www.merckmanuals.com/professional/clinical_pharmacology/pharmacokinetics/drug_distribution_to_tissues.html (last accessed 7 February 2012) [Google Scholar]
- 30.Brunton LL, Lazo JS, Parker KL, editors. Goodman & Gilman's The Pharmacological Basis of Therapeutics. 11th edn. New York: McGraw-Hill; 2006. [Google Scholar]
- 31.Ilett KF, Nation RL, Tjokrosetio R, Thompson WR, Oh TE, Cameron PD. Pharmacokinetics of ranitidine in critically ill patients. Br J Clin Pharmacol. 1986;21:279–288. doi: 10.1111/j.1365-2125.1986.tb05191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lugo RA, Harrison AM, Cash J, Sweeley J, Vernon DD. Pharmacokinetics and pharmacodynamics of ranitidine in critically ill children. Crit Care Med. 2001;29:759–764. doi: 10.1097/00003246-200104000-00014. [DOI] [PubMed] [Google Scholar]
- 33.Holford NH. A size standard for pharmacokinetics. Clin Pharmacokinet. 1996;30:329–332. doi: 10.2165/00003088-199630050-00001. [DOI] [PubMed] [Google Scholar]
- 34.Lugo RA, Harrison AM, Cash J, Sweeley J, Vernon DD. Population pharmacokinetic studies in pediatrics: issues in design and analysis. AAPS J. 2005;7:E475–487. doi: 10.1208/aapsj070248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson BJ, Woollard GA, Holford NH. A model for size and age changes in the pharmacokinetics of paracetamol in neonates, infants and children. Br J Clin Pharmacol. 2000;50:125–134. doi: 10.1046/j.1365-2125.2000.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rajagopalan P, Gastonguay MR. Population pharmacokinetics of ciprofloxacin in pediatric patients. J Clin Pharmacol. 2003;43:698–710. [PubMed] [Google Scholar]
- 37.Allegaert K, Anderson BJ, Cossey V, Holford NH. Limited predictability of amikacin clearance in extreme premature neonates at birth. Br J Clin Pharmacol. 2006;61:39–48. doi: 10.1111/j.1365-2125.2005.02530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wiest DB, O'Neal W, Reigart JR, Brundage RC, Gillette PC, Yost RL. Pharmacokinetics of ranitidine in critically ill infants. Dev Pharmacol Ther. 1989;12:7–12. [PubMed] [Google Scholar]
- 39.Orenstein SR, Blumer JL, Faessel HM, McGuire JA, Fung K, Li BUK, Lavine JE, Grunow JE, Treem WR, Ciociola AA. Ranitidine, 75 mg, over-the-counter dose: pharmacokinetic and pharmacodynamic effects in children with symptoms of gastro-esophageal reflux. Aliment Pharmacol Ther. 2002;16:899–907. doi: 10.1046/j.1365-2036.2002.01243.x. [DOI] [PubMed] [Google Scholar]
- 40.Wells TG, Heulitt MJ, Taylor BJ, Fasules JW, Kearns GL. Pharmacokinetics and pharmacodynamics of ranitidine in neonates treated with extracorporeal membrane oxygenation. J Clin Pharmacol. 1998;38:402–407. doi: 10.1002/j.1552-4604.1998.tb04443.x. [DOI] [PubMed] [Google Scholar]
- 41.Garg DC, Baltodano N, Jallad NS, Perez G, Oster JR, Eshelman FN, Weidler DJ. Pharmacokinetics of ranitidine In patients with renal failure. J Clin Pharmacol. 1986;26:286–291. doi: 10.1002/j.1552-4604.1986.tb03525.x. [DOI] [PubMed] [Google Scholar]
- 42.Koch KM, Liu M, Davis IM, Shaw S, Yin Y. Pharmacokinetics and pharmacodynamics of ranitidine in renal impairment. Eur J Clin Pharmacol. 1997;52:229–234. doi: 10.1007/s002280050279. [DOI] [PubMed] [Google Scholar]
- 43.Van Hecken AM, Tjandramaga TB, Mullie A, Verbesselt R, De Schepper PJ. Ranitidine-single dose pharmacokinetics and absolute bioavailability in man. Br J Clin Pharmacol. 1982;14:195–200. doi: 10.1111/j.1365-2125.1982.tb01961.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pérez JF, Juárez-Olguín H, Pérez CF, Pérez-Guillé G, Guillé-Pérez A, Camacho-Vieyra A, Toledo-López A, Carrasco-Portugal M, Lares-Asseff I. Effects of gender and phase of the menstrual cycle on the kinetics of ranitidine in healthy volunteers. Chronobiol Int. 2003;20:485–494. [PubMed] [Google Scholar]
- 45.Yin OQP, Tomlinson B, Chow AHL, Chow MSS. A modified two-portion absorption model to describe double-peak absorption profiles of ranitidine. Clin Pharmacokinet. 2003;42:179–192. doi: 10.2165/00003088-200342020-00005. [DOI] [PubMed] [Google Scholar]
- 46.Garg DC, Eshelman FN, Weidler DJ. Pharmacokinetics of ranitidine following oral administration with ascending doses and with multiple-fixed doses. J Clin Pharmacol. 1985;25:437–443. doi: 10.1002/j.1552-4604.1985.tb02873.x. [DOI] [PubMed] [Google Scholar]
- 47.Ammon S, Treiber G, Kees F, Klotz U. Influence of age on the steady state disposition of drugs commonly used for the eradication of Helicobactor pylori. Aliment Pharmacol Ther. 2000;14:759–766. doi: 10.1046/j.1365-2036.2000.00756.x. [DOI] [PubMed] [Google Scholar]
- 48.Olguín HJ, Flores J, Pérez G, Hernández G, Flores C, Guillé A, Camacho A, Toledo A, Carrasco M. Effect of food consistency on the pharmacokinetics of ranitidine in healthy volunteers. J Pharm Technol. 2002;18:178–181. [Google Scholar]
- 49.Ribbing J, Jonsson EN. Power, selection bias and predictive performance of the Population Pharmacokinetic Covariate Model. J Pharmacokinet Pharmacodyn. 2004;31:109–134. doi: 10.1023/b:jopa.0000034404.86036.72. [DOI] [PubMed] [Google Scholar]
- 50.Rigby-Jones AE, Nolan JA, Priston MJ, Wright PM, Sneyd JR, Wolf AR. Pharmacokinetics of propofol infusions in critically ill neonates, infants, and children in an intensive care unit. Anesthesiology. 2002;97:1393–1400. doi: 10.1097/00000542-200212000-00010. [DOI] [PubMed] [Google Scholar]
- 51.Shammas FV, Dickstein K. Clinical pharmacokinetics in heart failure. An updated review. Clin Pharmacokinet. 1988;15:94–113. doi: 10.2165/00003088-198815020-00002. [DOI] [PubMed] [Google Scholar]
- 52.Kuitunen A, Vento A, Suojaranta-Ylinen R, Pettilä V. Acute renal failure after cardiac surgery: evaluation of the RIFLE classification. Ann Thorac Surg. 2006;81:542–546. doi: 10.1016/j.athoracsur.2005.07.047. [DOI] [PubMed] [Google Scholar]
- 53.Rigden SPA, Barratt TM, Dillon MJ, De Leval M, Stark J. Acute renal failure complicating cardiopulmonary bypass surgery. Arch Dis Child. 1982;57:425–430. doi: 10.1136/adc.57.6.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gharehbaghi MM, Peirovifar A. Evaluating causes of acute renal failure in newborn infants. Pak J Med Sci. 2007;23:877–880. [Google Scholar]
- 55.Myers JD, Hickam JB. An estimation of the hepatic blood flow and splanchnic oxygen consumption in heart failure. J Clin Invest. 1948;27:620–627. doi: 10.1172/JCI102008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stenson RE, Constantino RT, Harrison DC. Interrelationships of hepatic blood flow, cardiac output, and blood levels of lidocaine in man. Circulation. 1971;43:205–211. doi: 10.1161/01.cir.43.2.205. [DOI] [PubMed] [Google Scholar]
- 57.Leithe ME, Margorien RD, Hermiller JB, Unverferth DV, Leier CV. Relationship between central hemodynamics and regional blood flow in normal subjects and in patients with congestive heart failure. Circulation. 1984;69:57–64. doi: 10.1161/01.cir.69.1.57. [DOI] [PubMed] [Google Scholar]
- 58.Moffett BS, Mott AR, Nelson DP, Gurwitch KD. Medication dosing and renal insufficiency in a pediatric cardiac intensive care unit: impact of pharmacist consultation. Pediatr Cardiol. 2008;29:744–748. doi: 10.1007/s00246-007-9170-3. [DOI] [PubMed] [Google Scholar]