

Abstract
Aims
To assess the safety, tolerability, pharmacokinetics (PK), pharmacodynamics (PD) and immunogenicity of CNTO 5825 following single-dose intravenous (i.v.) and subcutaneous (s.c.) administration in healthy and healthy atopic subjects.
Methods
Sixty-four subjects received a single dose of placebo or CNTO 5825 (0.1, 0.3, 1.0, 3.0, or 10 mg kg−1 i.v. in a dose-escalating manner, or 3.0 mg kg−1 s.c. in healthy subjects; and 10 mg kg−1 i.v. in healthy atopic subjects). Subjects were observed for 96 h postadministration and followed for 16 weeks. Safety and tolerability were monitored, and serum samples were collected to measure CNTO 5825 concentrations, antibodies to CNTO 5825 and PD biomarkers.
Results
Most adverse events were mild to moderate in severity and considered to be unrelated to CNTO 5825, with no dose-dependent trends seen. The two serious adverse events were considered to be unrelated to CNTO 5825. After i.v. administration, CNTO 5825 exhibited linear PK, with a terminal half-life of ∼22–32 days. After a single 3 mg kg−1 s.c. dose in healthy subjects, CNTO 5825 was absorbed into the systemic circulation with a median time to maximum serum concentration (tmax) of 5.45 days and absolute bioavailability of ∼75%. The PK profile of CNTO 5825 at 10 mg kg−1 was similar in both healthy and healthy atopic subjects. No antibodies to CNTO 5825 were detected through week 16. In the CNTO 5825-treated healthy atopic subjects, there was a significant reduction in serum IgE and C-C motif chemokine ligand 17 (P = 0.028 and 0.068 vs. placebo, respectively).
Conclusions
CNTO 5825 was well tolerated, had an acceptable safety profile, exhibited linear PK characteristics, and no detected antibodies to CNTO 5825.
Keywords: first-in-human study, interleukin-13, monoclonal antibody, safety
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Interleukin-13 (IL-13) is a cytokine associated with type II inflammatory responses. IL-13 is thought to have a pathogenic role in various diseases, such as asthma, atopic dermatitis and allergic rhinitis, and to play a role in exaggerated inflammation and tissue remodelling in these and potentially other diseases, such as chronic obstructive pulmonary disease, systemic sclerosis and idiopathic pulmonary fibrosis.
There are several treatments under development that target IL-13. Lebrikizumab is the most advanced anti-IL-13 experimental therapeutic targeting asthma, and a recent study showed that the primary end-point of the trial was met, with monthly subcutaneous lebrikizumab treatments showing a statistically significant increase in prebronchodilator forced expiratory volume in one second (FEV1) at 12 weeks in adult patients with asthma inadequately controlled with inhaled corticosteroids.
WHAT THIS STUDY ADDS
CNTO 5825, a human monoclonal antibody that inhibits the binding of human interleukin-13 (IL-13) to both IL-13 receptor α1 and IL-13 receptor α2, has been found to bind to human IL-13 with high affinity and specificity and appears to suppress the biological activity of IL-13.
A single subcutaneous (3 mg kg−1) or intravenous dose (0.1–10 mg kg−1) of CNTO 5825 is well tolerated in both healthy and healthy atopic subjects, exhibits linear pharmacokinetic characteristics with a long terminal half-life and no detectable antibodies to CNTO 5825, and produces a pharmacodynamic response consistent with its mechanism of action.
Introduction
Interleukin-13 (IL-13) is a cytokine strongly associated with type 2 helper T cell (Th2) immune responses. IL-13 is an approximately 12 kDa protein secreted from multiple cell types, including T cells, eosinophils, basophils, mast cells, smooth muscle cells, epithelial cells and macrophages. IL-13 is functionally related to interleukin-4 (IL-4), another Th2-associated cytokine; however, IL-13 is produced at much greater levels than IL-4 in vivo, and the general availability of both ligands and the particular receptor combinations expressed on responding cells is likely to dictate the overall importance of IL-13 vs. IL-4 in specific disease settings [1]. IL-13 shares components of its receptor signalling pathway with IL-4 [2] and initiates a signalling cascade with the engagement of IL-13 receptor α1 (IL-13Rα1), followed by subsequent coupling with the IL-4 receptor α (IL-4Rα), resulting in the phosphorylation and activation of signal transducer and activator of transcription 6 [3].
IL-13 signalling has been shown to mediate a variety of biological activities, including airway hyper-responsiveness, allergic inflammation, tissue eosinophilia, mastocytosis, immunoglobulin (Ig) E production, goblet cell hyperplasia, tissue remodelling and fibrosis. In addition, IL-13 has been shown to play a role in the growth of tumour cells [1], modulation of resistance to intracellular organisms (such as Leishmania species and Listeria monocytogenes) and gastrointestinal nematodes [1] and protection against induced autoimmune myocarditis [4].
Consistent with its biological activities, IL-13 is thought to play a role in the pathogenesis of several diseases associated with an inappropriate Th2 immune response, including asthma, allergic dermatitis and atopic rhinitis [1, 5]. Additionally, IL-13 is thought to be associated with the inflammation and tissue remodelling observed in patients with non-atopic asthma and chronic obstructive pulmonary disease and in patients with fibrotic diseases, including systemic sclerosis and idiopathic pulmonary fibrosis [6–8]. As such, inhibition of IL-13 may abrogate both the atopic and non-atopic manifestations of these diseases.
Evidence suggests that IL-13 has an important role in asthma; it is associated with human disease, and preclinical models have demonstrated that it induces many of the features associated with human asthma. IL-13 has a role in mediating airway hyper responsiveness, eosinophilic inflammation and mucus hypersecretion, the elements of asthma most closely linked to the manifestations of the disease [9].
In a clinical study in patients with poorly controlled asthma, monthly subcutaneous (s.c.) treatment with an anti-IL-13 under clinical development (lebrikizumab) was associated with a significant improvement in prebronchodilator forced expiratory volume in one second (FEV1) at week 12 and reductions of serum Th2 chemokines and IgE, supporting suppression of the IL-13–mediated biological effect [10]. This suggests that the blocking of IL-13 may be a good target for attenuating the progression of asthma and other Th2-related pathologies.
CNTO 5825 is a human anti-IL-13 monoclonal antibody (mAb) and is a potent and specific antagonist of IL-13 that inhibits the binding of IL-13 to both IL-13Rα1 and IL-13 receptor α2 (IL-13Rα2) cell surface receptors and consequently modulates IL-13–mediated signal transduction.
The objectives of this first-in-human study were to evaluate the safety, tolerability, pharmacokinetics (PK), immunogenicity and pharmacodynamics (PD) of both single intravenous (i.v.) and s.c. administrations of CNTO 5825 in healthy and healthy atopic subjects.
Methods
Ethical considerations
The ethics committee (University Hospital Antwerp, Edegem, Belgium) approved the study protocol and informed consent documents. The study was conducted in accordance with the Declaration of Helsinki and the regulations established in the USA for the Protection of Human Subjects (United States Code of Federal Regulations Title 21, Part 56). All subjects provided informed consent before participating in any study-related procedures.
Study population
Male and female subjects between the ages of 18 and 55 years were eligible. Subjects were required to have had no clinically relevant abnormalities as determined by medical history, physical examination, vital signs, serum chemistry, haematology, coagulation tests, serology, urine dipstick and 12-lead electrocardiogram. Prescription, herbal and over-the-counter medications were prohibited for 14 days prior to and throughout the duration of the study. Occasional use of paracetamol and continued use of multivitamins, both at recommended doses, was allowed. Subjects were required to be nonsmokers with no history of alcohol or drug abuse. Female subjects of childbearing potential were required to have a negative pregnancy test upon study entry. The healthy subjects in the atopic cohort had to have a positive Phadiatop allergy (IgE) test (Thermo Fisher Scientific, Uppsala, Sweden) at screening for inclusion in the study.
Study design and treatment
This study was conducted as a double-blind, placebo-controlled, single ascending dose study in healthy subjects. Sixty-four healthy adult subjects (37 males and 27 females) were enrolled in seven dose cohorts (cohort 1, 0.1 mg kg−1 i.v.; cohort 2, 0.3 mg kg−1 i.v.; cohort 3, 1.0 mg kg−1 i.v.; cohort 4, 3.0 mg kg−1 i.v.; cohort 5, 10 mg kg−1 i.v.; cohort 6, 3 mg kg−1 s.c.; and cohort 7, 10 mg kg−1 i.v. in healthy atopic subjects), and dose escalation occurred in a staggered parallel fashion. Cohort 7 consisted of only atopic subjects in order to explore the PD response in addition to safety/tolerability and PK in the highest safe/tolerable dose of CNTO 5825 in this population that is known to have elevated inflammatory markers. In cohorts 1–6, a total of eight subjects each were randomized 6:2 to receive a single-dose administration of either CNTO 5825 or placebo. Cohort 7 was composed of 16 subjects (12 assigned to CNTO 5825 and four to placebo).
The study-agent was administered as a 30 min i.v. infusion in cohorts 1–5 and 7 and a 3 mg kg−1 single s.c. injection in cohort 6. The decision to proceed to each higher i.v. dose cohort was based on a review of safety data collected for 6 days after study-agent administration from the previous dose cohort.
Safety assessments
Subjects were monitored for 16 weeks after administration of the study-agent. All treated subjects were observed in a hospital setting for 5 days postinfusion and underwent follow-up evaluations at 1, 2, 3, 4, 8, 12 and 16 weeks after administration of the study-agent. Safety assessments included monitoring for all adverse events (AEs) and examination of vital signs, physical examinations, electrocardiograms and laboratory parameters. All untoward events, including serious AEs (SAEs), occurring between the time of obtaining informed consent and the 16-week postadministration study follow-up were recorded, regardless of whether they were considered to be study related. The severity of an AE and its relationship to the study-agent were determined by the investigators, who were blinded to the treatment assignments. A safety-evaluable subject was defined as any subject who received a dose of CNTO 5825 or placebo.
Pharmacokinetic sampling and bioanalysis
For the measurement of CNTO 5825 concentrations in the serum, blood samples were obtained prior to study-agent administration, and at 1 and 2 (i.v. cohorts only), 8, 24, 48, 72 and 96 h after administration. Additional blood samples for the measurement of CNTO 5825 concentrations were obtained at 1, 2, 3, 4, 8, 12 and 16 weeks after study-agent administration. Serum CNTO 5825 concentrations were determined using a validated electrochemiluminescent immunoassay. Using this assay, the lowest quantifiable concentration in a sample was 0.078 μg ml−1 [lower limit of quantification (LLOQ) multiplied by minimum required dilution, 0.0078 μg ml−1 × 10].
Immunogenicity assessment
The development of antibodies to CNTO 5825 was determined from serum samples obtained prior to and at 2, 4, 8, 12 and 16 weeks after CNTO 5825 administration. Bioanalysis of antibodies to CNTO 5825 in human serum test samples was performed utilizing an electrochemiluminescent immunoassay.
Noncompartmental pharmacokinetic analysis
Noncompartmental analysis using WinNonlin (version 5.2.1; Pharsight Corporation, Mountain View, CA, USA) was performed to determine the PK parameters of CNTO 5825 following a single i.v. infusion or s.c. injection. The maximum serum concentration (Cmax) was obtained from inspection of the individual serum concentration–time data. The terminal elimination rate constant (λz) was determined by least-squares regression analysis of the log-linear portion of the terminal phase. The area under the serum concentration–time curve from time zero to infinity (AUC0–∞) was determined as AUC(0–tz) + Cz/λz, where Cz is the last measurable serum concentration at time tz. The terminal half-life (t1/2) was calculated as 0.693/λz. The total systemic clearance (CL) was determined by dividing the dose by the AUC0–∞. The volume of distribution during the terminal phase (Vz) was determined as CL/λz. The absolute bioavailability following a single s.c. administration was calculated using the AUC0–∞ following a single s.c. administration of 3 mg kg−1 divided by the AUC0–∞ following a single i.v. dose of 3 mg kg−1 (AUC0–∞,s.c./AUC0–∞,i.v.).
Pharmacodynamic assessments
Serum samples were obtained for the measurement of PD biomarkers at baseline (i.e. prior to study-agent administration and at weeks 1, 2, 3, 4, 8, 12 and 16 after study-agent administration). Serum IgE was measured by standard enzyme-linked immunosorbent assay (ELISA; Bethyl Laboratories, Inc., Montgomery, TX, USA; catalogue no. E88-108; LLOQ = 20 ng ml−1), C-C motif chemokine ligand (CCL) 5 by Meso Scale Discovery assay system (Human CCL5 (RANTES) Ultra-Sensitive Kit, catalogue no. K151BFC-4; Meso Scale Discovery, Gaithersburg, MD, USA; LLOQ = 0.2 pg ml−1, from manufacturer), and CCL11, CCL17 and C-X-C motif chemokine ligand 5(CXCL5) were measured together by Luminex system assay (LegendPlex Custom Panel; BioLegend, San Diego, CA, USA; LLOQ = 1.3 pg ml−1). The values for LLOQ were determined from the back-calculated concentration from the mean raw intensity value plus 10 standard deviations of six replicates of blank samples. All serum samples from the healthy atopic subjects in cohort 7 had concentrations above LLOQ for each of the analytes.
Statistical analysis
No formal statistical testing was performed on safety or PK data. A PK-evaluable subject was defined as any subject whose PK profiles had sufficient postdose blood samples that allowed accurate calculation of some or all of the specified PK parameters. Descriptive statistics were used to summarize data by dose cohort for PK-evaluable subjects.
Changes in PD biomarkers were assessed as both: (i) the integrated average change throughout the 16 week trial period (calculated as the AUC for the log2 [fold/baseline] across all time points measured from baseline through day 112, divided by 112 days; and (ii) the maximum decrease from baseline through day 112, expressed as the log2 [fold/baseline]. Statistical significance of changes in biomarker levels between the subjects who received CNTO 5825 and placebo in the atopic cohort 7 was evaluated post hoc using a one-tailed Student's unpaired t-test with Welch correction.
Results
Study population
Sixty-four healthy subjects were enrolled and comprised the safety-evaluable population. Subjects randomly assigned to cohorts 1–7 (0.1, 0.3, 1, 3 and 10 mg kg−1 i.v., 3 mg kg−1 s.c. and 10 mg kg−1 i.v. in atopic subjects, respectively) received the study-agent per protocol, with each cohort consisting of six CNTO 5825-treated subjects and two placebo-treated subjects, with the exception of cohort 7, which comprised 12 CNTO 5825-treated atopic subjects and four placebo-treated atopic subjects.
For the purpose of data analysis, placebo-treated subjects from each dose cohort were combined as the placebo-treatment group (n = 16). The distribution of age, weight, race and sex for each treatment group is shown in Table 1. Overall, the study population included 27 (42.2%) females and 37 (57.8%) males. The median age of the subjects was 46 years (range, 18–55 years), and the median bodyweight was 76.6 kg (range, 53.9–96.3 kg). Almost all subjects were white (n = 63; 98.4%). There were no noteworthy differences between cohorts in demographic characteristics.
Table 1.
Baseline demographic characteristics
| Pooled placebo subjects | Cohort 1, 0.1 mg kg−1 i.v. | Cohort 2, 0.3 mg kg−1 i.v. | Cohort 3, 1.0 mg kg−1 i.v. | Cohort 4, 3.0 mg kg−1 i.v. | Cohort 5, 10 mg kg−1 i.v. | Cohort 6, 3.0 mg kg−1 s.c. | Cohort 7, atopic subjects, 10 mg kg−1 i.v. | Total | |
|---|---|---|---|---|---|---|---|---|---|
| Subjects treated (n) | 16 | 6 | 6 | 6 | 6 | 6 | 6 | 12 | 64 |
| Age (years) | 41.8 ± 9.79 | 46.0 ± 3.41 | 44.5 ± 9.97 | 37.5 ± 10.27 | 44.3 ± 6.59 | 43.2 ± 6.27 | 45.0 ± 7.62 | 43.9 ± 9.41 | 43.1 ± 8.47 |
| Sex: female [n (%)] | 6 (37.5) | 2 (33.3) | 2 (33.3) | 3 (50.0) | 4 (66.7) | 2 (33.3) | 1 (16.7) | 7 (58.3) | 27 (42.2) |
| Race [n (%)] | |||||||||
| Mixed | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 1 (1.6) |
| White | 16 (100.0) | 6 (100.0) | 6 (100.0) | 6 (100.0) | 6 (100.0) | 5 (83.3) | 6 (100.0) | 12 (100.0) | 63 (98.4) |
| Weight (kg) | 76.0 ± 11.93 | 77.8 ± 10.54 | 72.0 ± 8.86 | 73.6 ± 9.90 | 70.3 ± 12.78 | 73.1 ± 9.40 | 79.0 ± 10.47 | 74.1 ± 14.03 | 74.7 ± 11.21 |
| Body mass index (kg m−2) | 25.3 ± 2.63 | 26.1 ± 1.81 | 24.3 ± 1.83 | 24.8 ± 3.33 | 24.5 ± 2.15 | 25.2 ± 2.70 | 25.8 ± 3.97 | 25.7 ± 2.67 | 25.3 ± 2.60 |
Data are reported as means ± SD unless otherwise noted.
Safety and tolerability
Adverse events
A summary of AEs that occurred in two or more CNTO 5825 treated-subjects is presented in Table 2. Thirty-two (66.7%) CNTO 5825-treated subjects and nine (56.3%) placebo-treated subjects had at least one AE during the study. There was no apparent dose response in the occurrence of AEs. The most frequently reported AE in subjects receiving CNTO 5825 was headache [eight (16.7%) vs. 2 (12.5%) for placebo]. Other AEs that were reported in two or more subjects and occurred more frequently in subjects that received CNTO 5825 compared with placebo included (in decreasing order of frequency): nasopharyngitis [six (12.5%) vs. none for placebo], back pain [three (6.3%) vs. none for placebo] and dizziness, epistaxis, erythema, hypersensitivity, influenza-like illness, limb injury, palpitations and vomiting [all occurring in two (4.2%) vs. none for placebo]. The majority of AEs were mild to moderate in severity, and most events were considered by the study investigator to be unrelated to CNTO 5825. No AEs were reported in the s.c. cohort. None of the AEs that occurred in this study led to any discontinuation of study-agent administration or subject termination from the study.
Table 2.
Summary of all adverse events that occurred in two or more CNTO 5825-treated subjects
| Pooled placebo subjects | Cohort 1, 0.1 mg kg−1 i.v. | Cohort 2, 0.3 mg kg−1 i.v. | Cohort 3, 1.0 mg kg−1 i.v. | Cohort 4, 3.0 mg kg−1 i.v. | Cohort 5, 10 mg kg−1 i.v. | Cohort 6, 3.0 mg kg−1 s.c. | Cohort 7, atopic subjects, 10 mg kg−1 i.v. | Combined | |
|---|---|---|---|---|---|---|---|---|---|
| Subjects treated (n) | 16 | 6 | 6 | 6 | 6 | 6 | 6 | 12 | 48 |
| Total number of subjects with adverse events [n (%)] | 9 (56.3) | 5 (83.3) | 2 (33.3) | 6 (100.0) | 4 (66.7) | 5 (83.3) | 0 (0.0) | 10 (83.3) | 32 (66.7) |
| Headache | 2 (12.5) | 3 (50.0) | 0 (0.0) | 1 (16.7) | 1 (16.7) | 1 (16.7) | 0 (0.0) | 2 (16.7) | 8 (16.7) |
| Nasopharyngitis | 0 (0.0) | 3 (50.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 2 (16.7) | 6 (12.5) |
| Back pain | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (33.3) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 3 (6.3) |
| Diarrhoea | 1 (6.3) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 1 (16.7) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 3 (6.3) |
| Oropharyngeal pain | 1 (6.3) | 1 (16.7) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (8.3) | 3 (6.3) |
| Dizziness | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (8.3) | 2 (4.2) |
| Vomiting | 0 (0.0) | 0 (0.0) | 1 (16.7) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (4.2) |
| Epistaxis | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 1 (8.3) | 2 (4.2) |
| Influenza-like illness | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (33.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (4.2) |
| Erythema | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (4.2) |
| Palpitations | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (4.2) |
| Hypersensitivity | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (16.7) | 2 (4.2) |
| Limb injury | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 2 (4.2) |
| Myalgia | 1 (6.3) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (8.3) | 2 (4.2) |
Two subjects had SAEs reported during the study. One SAE of cervical hernia, in a subject who received 0.1 mg kg−1 CNTO 5825, began on day 11 as a moderate headache, which worsened into neck pain and akinesia of the left arm, and was diagnosed as cervical hernia after a magnetic resonance imaging scan. The cervical hernia was considered unrelated to the study agent and was treated with surgery and resolved on day 94. A second subject, receiving 10 mg kg−1 CNTO 5825, experienced SAEs of dysmenorrhoea, dyspareunia, and persistent pain on day 63 and was hospitalized. The subject had a vaginal hysterectomy. Pathology results showed chronic cervicitis, unrelated to the study-agent, and the SAE was resolved on day 106. No deaths occurred during the study period.
Infections
Eight (16.7%) CNTO 5825-treated subjects and two (12.5%) placebo-treated subjects experienced infections or infestations. The infections or infestations in CNTO 5825-treated subjects included: nasopharyngitis [six (12.5%)], gastroenteritis [one (2.1%)] and pharyngitis [one (2.1%)]. Infections or infestations in the placebo-treated patients included a groin abscess [one (6.3%)] and oral herpes [one (6.3%)]. There were no subjects with serious infections in this study; all infections were considered by the investigators to be mild to moderate.
Allergic, infusion and injection-site reactions
CNTO 5825 administered as a 30 min i.v. infusion at dose levels ranging from 0.1 to 10 mg kg−1 was well tolerated. No anaphylaxis or severe allergic reactions were observed. Infusion reactions were defined as any AE that occurred during the infusion or within 60 min after the end of the infusion. According to this definition, there were two subjects who experienced an infusion reaction. A 52-year-old female who received placebo experienced a mild headache that started 19 min after the end of the infusion, which was considered by the investigator to be possibly related to study-agent administration. In addition, a 48-year-old female who received 1 mg kg−1 CNTO 5825 experienced vomiting 8 min after the start of the infusion, which was considered to be unrelated to the study-agent. No injection-site reactions were reported after s.c. administration of CNTO 5825.
Laboratory parameters, vital signs and electrocardiograms
There were no clinically significant changes observed in the safety laboratory, vital signs, physical examinations or 12-lead electrocardiogram measurements between CNTO 5825- and placebo-treated subjects or between baseline and post-treatment with CNTO 5825 for any dose group. There were no noteworthy differences between CNTO 5825 and placebo or between CNTO 5825 doses in post-baseline mean values at any time point.
Pharmacokinetic parameters from noncompartmental analysis
Mean serum CNTO 5825 concentration vs. time curves for subjects in each dose group are presented in Figure 1. Following a single i.v. infusion, CNTO 5825 serum concentrations exhibited a biphasic PK profile, with a relatively rapid distribution phase and a relatively slow terminal elimination phase. Mean terminal t1/2 values ranged from approximately 22 to 32 days and appeared to be independent of the dose administered.
Figure 1.
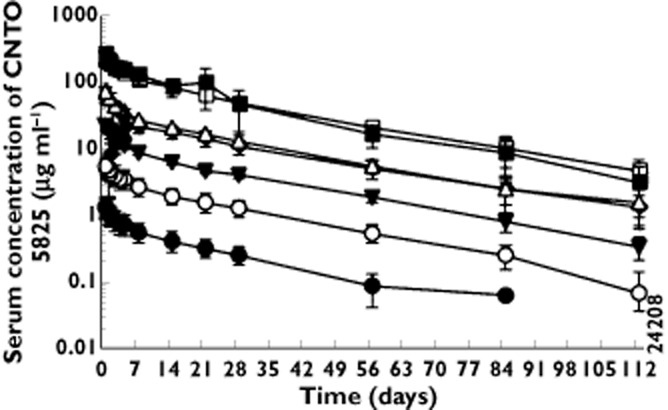
Mean (±SD) CNTO 5825 serum concentration–time profiles following a single intravenous (i.v.) infusion or subcutaneous (s.c.) injection of CNTO 5825 in healthy subjects.  , 0.1 mg kg–1 i.v.;
, 0.1 mg kg–1 i.v.;  , 0.3 mg kg–1 i.v.;
, 0.3 mg kg–1 i.v.;  , 1.0 mg kg–1 i.v.;
, 1.0 mg kg–1 i.v.;  , 3.0 mg kg–1 i.v.;
, 3.0 mg kg–1 i.v.;  , 10 mg kg–1 i.v.;
, 10 mg kg–1 i.v.;  , 10 mg kg–1 i.v., atopic subjects;
, 10 mg kg–1 i.v., atopic subjects;  , 3.0 mg kg–1 s.c.
, 3.0 mg kg–1 s.c.
A summary of the PK parameters for each i.v. dose cohort is shown in Table 3. Mean Cmax and AUC0–∞ values increased in an approximately dose-proportional manner after i.v. doses ranging from 0.1 to 10 mg kg−1. Systemic clearance and volume of distribution at terminal phase appeared to be independent of the dose administered, with mean values for CL ranging from 2.4 to 4.8 ml day−1 kg−1 and for Vz ranging from 74.5 to 155.7 ml kg−1 in all i.v. cohorts.
Table 3.
Summary of mean (±SD) pharmacokinetic parameters from noncompartmental analysis following a single intravenous or subcutaneous dose of CNTO 5825 in healthy subjects
| Cohort 1, 0.1 mg kg−1 i.v. | Cohort 2, 0.3 mg kg−1 i.v. | Cohort 3, 1.0 mg kg−1 i.v. | Cohort 4, 3.0 mg kg−1 i.v. | Cohort 5, 10 mg kg−1 i.v. | Cohort 6, 3.0 mg kg−1 s.c. | Cohort 7, atopic subjects, 10 mg kg−1 i.v. | |
|---|---|---|---|---|---|---|---|
| Subjects treated (n) | 6 | 6 | 6 | 6 | 6 | 6 | 12 |
| Cmax (μg ml−1) | 1.38 ± 0.266 | 5.94 ± 1.024 | 23.77 ± 4.282 | 79.42 ± 10.440 | 285.40 ± 76.911 | 22.99a ± 4.853 | 233.13 ± 16.814 |
| AUC0–∞ (μg day ml−1) | 21.44 ± 2.078 | 107.02 ± 13.553 | 364.13 ± 33.615 | 1212.91 ± 194.786 | 4510.91 ± 1276.174 | 905.55 ± 251.348 | 4300.23 ± 592.977 |
| CL (ml day−1 kg−1) | 4.78 ± 0.473 | 2.85 ± 0.356 | 2.82 ± 0.265 | 2.55 ± 0.414 | 2.39 ± 0.832 | 3.55b ± 1.043 | 2.37 ± 0.312 |
| Vz (ml kg−1) | 155.72 ± 65.358 | 95.73 ± 12.358 | 90.88 ± 12.550 | 113.02 ± 27.681 | 74.53 ± 14.791 | 142.58c ± 24.311 | 82.94 ± 15.302 |
| t1/2 (days) | 22.94 ± 10.555 | 23.55 ± 3.899 | 22.46 ± 3.228 | 31.92 ± 11.384 | 22.54 ± 4.503 | 29.40 ± 8.580 | 24.16 ± 2.089 |
Abbreviations and symbols are as follows:
median tmax = 5.45 days;
CL/F;
Vz/F; AUC0–∞, area under the serum concentration–time curve from time zero to infinity; CL, systemic clearance; Cmax, maximum serum concentration; F, absolute bioavailability; t1/2, terminal half-life; and Vz, volume of distribution during terminal phase.
After a single 3 mg kg−1 s.c. dose in healthy subjects, CNTO 5825 was slowly absorbed into the systemic circulation with median time (range) to maximum serum concentration of 5.45 (3.9–7.0) days. CNTO 5825 was eliminated from the systemic circulation with a mean terminal t1/2 of 29 days after s.c. administration (Table 3). The estimated absolute bioavailability following 3 mg kg−1 s.c. of CNTO 5825 was approximately 75%. The PK parameters from the healthy atopic subjects 10 mg kg−1 dose cohort (cohort 7) were similar to the values observed in the healthy subjects 10 mg kg−1 dose cohort (cohort 5).
Immunogenicity
None of the 48 subjects with appropriate samples after a single i.v. or s.c. dose was classified as positive for antibodies to CNTO 5825 16 weeks after study-agent administration.
Pharmacodynamics
In the atopic 10 mg kg−1 i.v. cohort, serum IgE levels were significantly reduced following dosing with CNTO 5825 compared with placebo (P = 0.028, AUC). The geometric mean of the maximum decrease in IgE levels was 23% (95% confidence interval 16–29%) for CNTO 5825 vs. 9% (95% confidence interval 4–14%) for placebo (P = 0.003, −15% difference). The average peak decrease in IgE levels occurred within 4 weeks of dosing and was generally sustained for the remainder of study duration to 16 weeks postdosing (Figure 2A). Importantly, baseline IgE levels were comparable in the placebo-treated and CNTO 5825-treated subjects in the atopic cohort, and significance was maintained when adjusting for baseline IgE levels in general linear models (GLM)/analysis of covariance (ancova) analysis (P = 0.020, AUC).
Figure 2.

Serum levels of IgE (A), CCL17 (B), CCL5 (C), CCL11 (D) and CXCL5 (E) in subjects in the 10 mg kg−1 i.v. atopic cohort following dosing of CNTO 5825 or placebo.  , placebo;
, placebo;  , CNTO 5825
, CNTO 5825
In the atopic cohort, CCL17 levels were decreased in the subjects treated with CNTO 5825 when compared with those treated with placebo (P = 0.068, AUC). The geometric mean of the maximum decrease in CCL17 levels was 42% for CNTO 5825 vs. 34% for placebo (P = 0.27). The average peak decrease in CCL17 levels occurred within 4 weeks of dosing and gradually returned to baseline levels through study end at 16 weeks postdosing (Figure 2B). Importantly, baseline CCL17 levels were comparable in the placebo- and CNTO 5825-treated subjects in the atopic cohort, and the trend was maintained when adjusting for baseline CCL17 levels in general linear models/ancova analysis (P = 0.052, AUC).
There were no significant changes in CCL5, CCL11 and CXCL5 serum concentrations in CNTO 5825-treated subjects when compared with placebo (Figure 2C–E).
Discussion
The goals of this first-in-human study were to determine the safety, tolerability, PK, PD and immunogenicity of CNTO 5825 following a single i.v. or s.c. dose in healthy and healthy atopic subjects.
The safety results from this study demonstrate that CNTO 5825 was safe and well tolerated by the 42 healthy subjects treated with a 30 min i.v. infusion of a single intravenous dose of CNTO 5825 at dose levels ranging from 0.1 to 10 mg kg−1 and the six subjects treated with a single s.c. dose of 3 mg kg−1. No subject experienced anaphylaxis or severe allergic reactions, one subject experienced a non-drug-related infusion reaction and no injection-site reactions were observed in the s.c. cohort. Among subjects receiving CNTO 5825, headache and nasopharyngitis were the most commonly reported AEs. There was a slightly higher percentage of subjects experiencing an infection in the combined CNTO 5825 groups compared with placebo. However, the number of subjects is small, and it is not possible to determine whether this difference is clinically meaningful. Generally, no trends in AEs were observed across the five i.v. dose groups in the dose escalation phase and the s.c. administration and atopic cohorts. Observed AEs were generally transient in nature, mild to moderate in severity, and considered by the study investigator to be unrelated to CNTO 5825. There were two SAEs observed in the CNTO 5825-treated subjects that were considered to be unrelated to CNTO 5825 and were resolved by the end of the study.
The Cmax and AUC0–∞ increased in an approximately dose-proportional manner. In addition, CL, Vz and t1/2 appeared to be independent of the dose administered. These observations suggest that CNTO 5825 exhibits linear PK over the i.v. dose range of 0.1–10 mg kg−1. The terminal elimination half-life was relatively long, ranging from approximately 22 to 32 days, and the values were similar after i.v. or s.c. administration. After s.c. administration of 3 mg kg−1, CNTO 5825 was slowly absorbed into systemic circulation (median tmax of 5.45 days), and the absolute bioavailability was relatively high, at approximately 75%. The PK parameters for CNTO 5825 in healthy atopic subjects were similar to values observed in healthy subjects, indicating that atopic status did not impact the PK of CNTO 5825.
None of the subjects tested positive for antibodies to CNTO 5825 after a single i.v. or s.c. dose, and there was no evidence of accelerated clearance of CNTO 5825 from the systemic circulation in any subject evaluated in this study.
After a single dose of CNTO 5825, significant PD effects of CNTO 5825 were observed for the 10 mg kg−1 i.v. atopic cohort. Serum total IgE was significantly decreased from baseline in the CNTO 5825 treatment group compared with the placebo treatment group, with a similar trend observed for CCL17. These results demonstrate that a single dose of CNTO 5825 in healthy atopic subjects impacts downstream immunological mediators directly related to the target and the disorder (i.e. atopy).
IgE is a critical protein in the disease mechanism of atopy and is directly and specifically regulated by IL-13 (although also similarly by IL-4). In the 10 mg kg−1 i.v. atopic cohort, treatment with CNTO 5285 significantly decreased serum total IgE levels in comparison to the placebo treatment group. The reduction of IgE levels was generally sustained, on average, from a peak inhibition around day 28 through day 112 (the end of the trial).
Specifically blocking IL-13, but not IL-4, resulted in a significant and sustained reduction in serum IgE levels, despite the similar capacity of both cytokines to regulate IgE expression. This redundancy of IgE regulation with IL-4 may limit the maximal inhibition that IL-13 antagonism can exert. As only a single dose of CNTO 5825 was administered, it is unknown if multiple doses given over a longer period of time could result in a greater reduction in IgE levels.
C-C motif chemokine ligand 17 is a chemokine that promotes Th2-type responses by stimulating chemoattraction and enhancing activation of Th2 cells [11]. IL-13 and IL-4 are the major regulators of CCL17 production by peripheral blood mononuclear cells [12, 13]. Overexpression of CCL17 has been reported particularly in atopic disorders [14] and asthma [15, 16]. Despite not being markedly elevated in atopic subjects in this study, the average change of CCL17 levels throughout the trial period showed a trend of being lower in CNTO 5825-treated subjects compared with the placebo-treated subjects.
Early clinical trials of anti-IL-13 protein therapies in asthma have shown promise. To date, based on published clinical data, no safety concerns seem to have been associated with any of these compounds [17]. Likewise, no safety signals have been detected with CNTO 5825 in this small single-dose phase 1 study. The observed long terminal elimination half-life, low systemic clearance and high absolute bioavailability of CNTO 5825 were consistent with the PK profile of similar anti-IL-13 mAbs, such as tralokinumab [18, 19], IMA-638 and IMA-026 [20]. Interestingly, a decrease in serum total IgE and CCL17 was observed in a multiple-dose study with lebrikizumab in patients with poorly controlled asthma, similarly observed in the healthy atopic subjects after a single dose of CNTO 5825 in this study [10].
In conclusion, this clinical study provides initial information on the safety, tolerability, PK, immunogenicity and PD of CNTO 5825 administered intravenously or subcutaneously in healthy and healthy atopic subjects. CNTO 5825 had an acceptable safety profile, exhibited linear PK characteristics over the dose range studied and produced a PD response consistent with its mechanism of action.
Acknowledgments
The authors would like to acknowledge Sofie Deleu, MD, and the study personnel at the Clinical Pharmacology Unit of Janssen Research & Development, LLC, Merksem, Belgium. The authors also would like to thank Joseph C. Marini, PhD, Ming Lu, PhD, and Honghui Zhou, PhD of Biologics Clinical Pharmacology at Janssen Research & Development, LLC, for their technical expertise and scientific input during data analysis and manuscript review; Daniel Horowitz, PhD, and Keying Ma, PhD, of Immunology Biomarker at Janssen Research & Development, LLC, for their technical contributions to the analysis of biomarker samples; and the Medical Affairs Publications Group of Janssen Services, LLC, for assistance in preparing the manuscript.
Competing Interests
All authors are or were employees of Janssen Research & Development, LLC, during the time the study was conducted and own(ed) shares of stock in Johnson & Johnson. All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: all authors (B.v.H., I.P.N., E.B.-T., M.J.L., A.P., H.M.D. and K.J.P.) had support from Janssen Research & Development, LLC, for the submitted work; all authors (B.v.H., I.P.N., E.B.-T., M.J.L., A.P., H.M.D. and K.J.P.) had employment with Janssen Research & Development, LLC, in the previous 3 years; and all authors (B.v.H., I.P.N., E.B.-T., M.J.L., A.P., H.M.D. and K.J.P.) disclose ownership of Johnson & Johnson stock, of which Janssen Research & Development, LLC, is a subsidiary. This study was funded by Janssen Research & Development, LLC, a subsidiary of Johnson & Johnson and the manufacturer of CNTO 5825. The clinicaltrials.gov identifier number is NCT01081691.
References
- 1.Wynn TA. IL-13 effector functions. Annu Rev Immunol. 2003;21:425–456. doi: 10.1146/annurev.immunol.21.120601.141142. [DOI] [PubMed] [Google Scholar]
- 2.LaPorte SL, Juo ZS, Vaclavikova J, Colf LA, Qi X, Heller NM, Keegan AD, Garcia KC. Molecular and structural basis of cytokine receptor pleiotropy in the interleukin-4/13 system. Cell. 2008;132:259–272. doi: 10.1016/j.cell.2007.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palmer-Crocker RL, Hughes CC, Pober JS. IL-4 and IL-13 activate the JAK2 tyrosine kinase and Stat6 in cultured human vascular endothelial cells through a common pathway that does not involve the gamma c chain. J Clin Invest. 1996;98:604–609. doi: 10.1172/JCI118829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cihakova D, Barin JG, Afanasyeva M, Kimura M, Fairweather D, Berg M, Talor MV, Baldeviano GC, Frisancho S, Gabrielson K, Bedja D, Rose NR. Interleukin-13 protects against experimental autoimmune myocarditis by regulating macrophage differentiation. Am J Pathol. 2008;172:1195–1208. doi: 10.2353/ajpath.2008.070207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wills-Karp M. Interleukin-13 in asthma pathogenesis. Immunol Rev. 2004;202:175–190. doi: 10.1111/j.0105-2896.2004.00215.x. [DOI] [PubMed] [Google Scholar]
- 6.Elias J. The relationship between asthma and COPD: lessons from transgenic mice. Chest. 2004;126:111S–116. doi: 10.1378/chest.126.2_suppl_1.111S. [DOI] [PubMed] [Google Scholar]
- 7.Riccieri V, Rinaldi T, Spadaro A, Scrivo R, Ceccarelli F, Franco MD, Taccari E, Valesini G. Interleukin-13 in systemic sclerosis: relationship to nailfold capillaroscopy abnormalities. Clin Rheumatol. 2003;22:102–106. doi: 10.1007/s10067-002-0684-z. [DOI] [PubMed] [Google Scholar]
- 8.Park SW, Ahn MH, Jang HK, Jang AS, Kim DJ, Koh ES, Park JS, Uh ST, Kim YH, Paik SH, Shin HK, Youm W, Park CS. Interleukin-13 and its receptors in idiopathic interstitial pneumonia: clinical implications for lung function. J Korean Med Sci. 2009;24:614–620. doi: 10.3346/jkms.2009.24.4.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wills-Karp M, Finkelman FD. Untangling the complex web of IL-4- and IL-13-mediated signaling pathways. Sci Signal. 2008;1:pe55. doi: 10.1126/scisignal.1.51.pe55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, Arron JR, Harris JM, Scheerens H, Wu LC, Su Z, Mosesova S, Eisner MD, Bohen SP, Matthews JG. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365:1088–1098. doi: 10.1056/NEJMoa1106469. [DOI] [PubMed] [Google Scholar]
- 11.Imai T, Baba M, Nishimura M, Kakizaki M, Takagi S, Yoshie O. The T cell-directed CC chemokine TARC is a highly specific biological ligand for CC chemokine receptor 4. J Biol Chem. 1997;272:15036–15042. doi: 10.1074/jbc.272.23.15036. [DOI] [PubMed] [Google Scholar]
- 12.Nomura T, Terada N, Kim WJ, Nakano K, Fukuda Y, Wakita A, Numata T, Konno A. Interleukin-13 induces thymus and activation-regulated chemokine (CCL17) in human peripheral blood mononuclear cells. Cytokine. 2002;20:49–55. doi: 10.1006/cyto.2002.1979. [DOI] [PubMed] [Google Scholar]
- 13.Syed F, Huang CC, Li K, Liu V, Shang T, Amegadzie BY, Griswold DE, Song XY, Li L. Identification of interleukin-13 related biomarkers using peripheral blood mononuclear cells. Biomarkers. 2007;12:414–423. doi: 10.1080/13547500701192652. [DOI] [PubMed] [Google Scholar]
- 14.Takeuchi H, Yamamoto Y, Kitano H, Enomoto T. Changes in thymus- and activation-regulated chemokine (TARC) associated with allergen immunotherapy in patients with perennial allergic rhinitis. J Investig Allergol Clin Immunol. 2005;15:172–176. [PubMed] [Google Scholar]
- 15.Leung TF, Wong CK, Chan IH, Ip WK, Lam CW, Wong GW. Plasma concentration of thymus and activation-regulated chemokine is elevated in childhood asthma. J Allergy Clin Immunol. 2002;110:404–409. doi: 10.1067/mai.2002.126378. [DOI] [PubMed] [Google Scholar]
- 16.Sekiya T, Yamada H, Yamaguchi M, Yamamoto K, Ishii A, Yoshie O, Sano Y, Morita A, Matsushima K, Hirai K. Increased levels of a TH2-type CC chemokine thymus and activation-regulated chemokine (TARC) in serum and induced sputum of asthmatics. Allergy. 2002;57:173–177. doi: 10.1034/j.1398-9995.2002.5720256.x. [DOI] [PubMed] [Google Scholar]
- 17.Oh CK, Geba GP, Molfino N. Investigational therapeutics targeting the IL-4/IL-13/STAT-6 pathway for the treatment of asthma. Eur Respir Rev. 2010;19:46–54. doi: 10.1183/09059180.00007609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh D, Kane B, Molfino NA, Faggioni R, Roskos L, Woodcock A. A phase 1 study evaluating the pharmacokinetics, safety and tolerability of repeat dosing with a human IL-13 antibody (CAT-354) in subjects with asthma. BMC Pulm Med. 2010;10:3. doi: 10.1186/1471-2466-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oh CK, Faggioni R, Jin F, Roskos LK, Wang B, Birrell C, Wilson R, Molfino NA. An open-label, single-dose bioavailability study of the pharmacokinetics of CAT-354 after subcutaneous and intravenous administration in healthy males. Br J Clin Pharmacol. 2010;69:645–655. doi: 10.1111/j.1365-2125.2010.03647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gauvreau GM, Boulet LP, Cockcroft DW, Fitzgerald JM, Carlsten C, Davis BE, Deschesnes F, Duong M, Durn BL, Howie KJ, Hui L, Kasaian MT, Killian KJ, Strinich TX, Watson RM, Y N, Zhou S, Raible D, O'Byrne PM. Effects of interleukin-13 blockade on allergen-induced airway responses in mild atopic asthma. Am J Respir Crit Care Med. 2011;183:1007–1014. doi: 10.1164/rccm.201008-1210OC. [DOI] [PubMed] [Google Scholar]