

Abstract
Aim
The objective of this work was to characterize the safety, tolerability and pharmacokinetics of ABT-288, a highly selective histamine H3 receptor antagonist, in healthy young adults and elderly subjects following single and multiple dosing in a phase 1 setting.
Methods
Single doses (0.1, 0.3, 1, 3, 10, 20 and 40 mg ABT-288) and multiple doses (0.5, 1.5, 3 and 6 mg ABT-288 once-daily for 14 days) were evaluated in young adults and multiple doses (0.5, 1.5, 3 and 5 mg ABT-288 once-daily for 12 days) were evaluated in elderly subjects using randomized, double-blind, placebo-controlled, dose-escalating study designs. The effect of food on ABT-288 pharmacokinetics (5 mg single dose) was evaluated using an open label, randomized, crossover design.
Results
ABT-288 safety, tolerability and pharmacokinetics were comparable in young and elderly subjects. Single doses up to 40 mg and multiple doses up to 3 mg once-daily were generally safe and well tolerated. The most frequently reported adverse events were hot flush, headache, abnormal dreams, insomnia, nausea and dizziness. ABT-288 exposure (AUC) was dose-proportional over the evaluated dose ranges. The mean elimination half-life ranged from 40 to 61 h across dose groups. Steady state was achieved by day 10 of once-daily dosing with 3.4- to 4.2-fold accumulation. Food did not have a clinically meaningful effect on ABT-288 exposure.
Conclusions
Based on the above results, 1 and 3 mg once-daily doses of ABT-288 were advanced to phase 2 evaluation in Alzheimer's patients.
Keywords: ABT-288, Alzheimer's, cognition, H3 receptors, pharmacokinetics, tolerability
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Histamine H3 receptor antagonists increase the release of the brain neurotransmitters that are known to modulate cognitive processes. H3 receptor antagonists have demonstrated pro-cognitive effects in preclinical studies. ABT-288 is a highly selective histamine H3 receptor antagonist designed for symptomatic treatment of cognitive disorders.
WHAT THIS STUDY ADDS
In the present article, we present the safety, tolerability and pharmacokinetics of ABT-288 in healthy young adults and in elderly human subjects. Results from these trials, along with preclinical data, were the basis for advancing ABT-288 to proof-of-concept phase 2 evaluation in Alzheimer's disease and the presented studies guided dose selection for phase 2 testing. In general, results presented in the current article provide the most comprehensive characterization of the safety and tolerability profiles of pharmacological H3 receptor antagonism in young adults and elderly human subjects.
Introduction
Alzheimer's disease (AD) affects 24 million people worldwide and its prevalence is projected to double over the next 20 years [1]. AD is a progressive neurodegenerative disorder manifested by cognitive deterioration, progressive impairment of activities of daily living, and a variety of neuropsychiatric and behavioural disturbances. Although the cause of AD has not been clearly established, profound loss of cortical cholinergic neurotransmission is a nearly invariant feature of advanced AD [2], and this loss correlates well with level of cognitive decline.
Approved symptomatic therapies for AD, including acetylcholinesterase inhibitors and the NMDA receptor antagonist memantine, have modest efficacy and their therapeutic benefit is short-lived [3, 4]. It is also unclear whether long term use of these therapies is associated with reduction in institutionalization or progression of disability [3]. Furthermore, these drugs have common and dose-limiting side effects 4. Given these limitations, additional classes of medications with different mechanisms of action and improved efficacy and safety profiles are highly desirable for the treatment of AD.
Histamine is an important biogenic amine that is involved in a number of physiological responses. The effects of histamine occur through activation of four G-protein coupled receptor subtypes, H1, H2, H3 and H4 that differ in their pharmacology, signal transduction characteristics and molecular biology [5–8].
Histamine H3 auto- and hetero-receptors are highly expressed in brain regions, such as the prefrontal cortex, hippocampus and hypothalamus, that play an important role in attention, learning and memory [9–11]. The presynaptic H3 receptor, when activated, decreases the release of histamine, acetylcholine and norepinephrine from cholinergic neurons. Inhibition of the H3 receptor with a pharmacologically active antagonist increases the release of these procognitive neurotransmitters and thus the availability to the post-synaptic receptors [12, 13]. Accordingly, in multiple non-human experimental paradigms, selective H3 receptor antagonists have shown the potential to improve wakefulness, attention and performance in different cognitive domains [14, 15]. It was thus hypothesized that H3 receptor antagonists might provide a new pharmacological approach to improve cognitive impairment associated with AD [16–18].
ABT-288, or 2-[4′-((3aR,6aR)-5-methyl-hexahydro-pyrrolo[3,4-b]pyrrol-1-yl)-biphenyl-4-yl]-2H-pyridazin-3-one, is a competitive H3 receptor antagonist with high affinity for human and rat H3 receptors (Ki = 1.9 and 8.2 nm, respectively) and greater than 5000-fold H3 receptor selectivity vs. other histaminergic receptors [19]. In preclinical evaluations, ABT-288 enhanced the release of histamine, acetylcholine and dopamine in vivo, and exhibited efficacy in rodent behavioural tests across multiple cognitive domains affected in disorders of cognition such as AD. ABT-288 demonstrated favourable pharmacokinetic properties and safety profiles in preclinical species [19]. As such, ABT-288 was a good candidate to evaluate in humans.
In the present article, we present the safety, tolerability and pharmacokinetics of ABT-288, a highly selective H3 receptor antagonist, in healthy young adults and in elderly human subjects. Results from these trials, along with preclinical data [19], were the basis for advancing ABT-288 to proof-of-concept phase 2 evaluation in AD and the presented studies guided dose selection for phase 2 testing. In general, results presented in the current article provide the most comprehensive characterization of the safety and tolerability profiles of pharmacological H3 receptor antagonism in young adults and elderly human subjects.
Methods
Study designs
Study 1 (evaluation in healthy young adults):
This was a single centre, double-blind, randomized, placebo-controlled, three-in-one, phase 1 study of ABT-288 in healthy young adults. The objectives of this study were to assess the safety, tolerability and pharmacokinetics of escalating single and multiple oral doses of ABT-288 in healthy young adult subjects and to evaluate the effect of food on the pharmacokinetics of a single oral dose of ABT-288. The study consisted of three parts: single dose escalation (part 1), effect of food on the pharmacokinetics of ABT-288 (part 2) and multiple dose escalation (part 3). The study design is illustrated in Table 1.
Table 1.
Designs and dose regimens of ABT-288 healthy young adults and elderly studies
| Study | Treatment group | N | Treatment | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Period 1 | Period 2 | Period 3 | Period 4 | |||||||
| Young adults Part 1 (single dose escalation) | 1 | 3 | 0.1 mg | 1 mg | Placebo | 40 mg | ||||
| 3 | 0.1 mg | Placebo | 10 mg | 40 mg | ||||||
| 3 | Placebo | 1 mg | 10 mg | Placebo | ||||||
| 2 | 3 | 0.3 mg | 3 mg | Placebo | NE | |||||
| 3 | 0.3 mg | Placebo | 20 mg | NE | ||||||
| 3 | Placebo | 3 mg | 20 mg | NE | ||||||
| Young adults Part 2 (food effect) | 3 | 6 | 5 mg fasting | 5 mg non-fasting | ||||||
| 6 | 5 mg non-fasting | 5 mg fasting | ||||||||
| Young adults Part 3 (multiple dose escalation) | 4 | 9 | 0.5 mg once-daily for 14 days | |||||||
| 3 | Placebo | |||||||||
| 5 | 9 | 3 mg once-daily for 14 days | ||||||||
| 3 | Placebo | |||||||||
| 6 | 9 | 6 mg once-daily for 14 days* | ||||||||
| 3 | Placebo | |||||||||
| 7 | 9 | 1.5 mg once-daily for 14 days | ||||||||
| 3 | Placebo | |||||||||
| Elderly subjects (multiple dose escalation) | 1 | 6 | 0.5 mg once-daily for 12 days | |||||||
| 6 | 1.5 mg once-daily for 12 days | |||||||||
| 4 | Placebo | |||||||||
| 2 | 6 | 3 mg once-daily for 12 days | ||||||||
| 2 | Placebo | |||||||||
| 3† | 6 | 5 mg once-daily for 12 days | ||||||||
| 2 | Placebo | |||||||||
Dosing with 6 mg once-daily was stopped on day 6 due to intolerance;
only five subjects enrolled in this dose group and they all received ABT-288; NE, not evaluated. Non-fasting: with a high fat/high calorie breakfast.
Single-dose escalation (part 1):
The single dose escalation was a first-in-man evaluation of ABT-288 and followed a randomized, double-blind, placebo-controlled, four-period, incomplete crossover design. Seven single dose regimens (0.1, 0.3, 1, 3, 10, 20 and 40 mg) were evaluated. The starting dose was selected based on the projected human pharmacokinetics and the anticipated pharmacologically active exposures in humans to ensure lack of pharmacological activity at the starting dose and therefore ensure safety of the subjects. The 0.1 mg dose represented a 580-fold safety margin from the human equivalent dose (HED) of the no-observed adverse event level (NOAEL) in monkeys (most sensitive species in toxicology studies). Eighteen healthy adult subjects in general good health participated in this part of the study. Subjects were equally divided into two groups (group 1 and group 2) such that each group contained nine subjects. Within each group, the subjects were randomly assigned in equal numbers (three subjects each) to one of three sequences of regimen administration as shown in Table 1. A minimum 14 day washout was required between consecutive doses for each subject. Each dose of ABT-288 or placebo was taken orally with water (240 ml) after a minimum 10 h fast.
Effect of food on pharmacokinetics of ABT-288 (part 2):
The effect of food on the pharmacokinetics of ABT-288 was evaluated using an open label, two-period, fasting and non-fasting (high fat/high calorie breakfast), randomized, complete crossover design. Twelve healthy adult subjects participated in this part of the study. For the fasting regimen, ABT-288 was taken orally with water (240 ml) following a minimum 10 h fast and approximately 4 h before lunch. For the non-fasting regimen, ABT-288 was taken orally with water (240 ml) approximately 30 min after the start of a high fat/high calorie breakfast. The breakfast provided approximately 1000 kcal, with approximately 50% of the calories from fat. The dose for the food effect evaluation (5 mg) was selected such that it was slightly higher than the predicted therapeutic doses of ABT-288. The washout interval between the doses of ABT-288 in the two periods of part 2 was 11 days.
Multiple dose escalation (part 3):
The multiple dose escalation was conducted using a randomized, double-blind, placebo-controlled, sequential group design. Four oral doses of ABT-288 were planned to be tested with each dose administered once-daily for 14 days (0.5, 3, 6 and 12 mg or matching placebo). However, dosing in the 6 mg once-daily dose group (group 6) was stopped after day 6 due to poor tolerability. Consequently, the subsequent dose group (group 7) received a lower dose (1.5 mg once-daily) for 14 days. Forty-eight healthy adult subjects participated in this part of the study (12 per dose group with nine subjects receiving ABT-288 and three subjects receiving placebo) as summarized in Table 1. Doses on the intensive pharmacokinetic sampling days were administered orally with water (240 ml) under fasting conditions following a minimum 10 h fast. Doses on the remaining days were administered under non-fasting conditions.
Study 1 was conducted at Covance Clinical Research Unit (CRU), Inc. (Madison, WI, USA). The study protocol was reviewed and approved by the Covance CRU East Institutional Review Board.
Study 2 (evaluation in elderly subjects):
This was a single centre, double-blind, randomized, placebo-controlled, dose-escalating, multiple dose study of ABT-288 in elderly subjects. The study was conducted in three parts as illustrated in Table 1. In part 1, 16 subjects were randomly assigned to one of three 12 day regimens: ABT-288 0.5 mg once-daily, ABT-288 1.5 mg once-daily or placebo once-daily in a 3:3:2 ratio. In each of parts 2 and 3, eight subjects were to be randomly assigned to a 12 day regimen of either ABT-288 or matching placebo once-daily in a 3:1 ratio. The ABT-288 dose in part 2 was 3 mg and eight subjects were enrolled and randomized as planned. The ABT-288 dose in part 3 was 5 mg. Only five subjects were enrolled and all received ABT-288. Each dose of study drug was taken orally with approximately 240 ml of water, approximately 30 min after the start of a standardized breakfast.
Study 2 was conducted at West Coast Clinical Trials, LLC (Cypress, CA, USA). The study protocol was reviewed and approved by the Compass Institutional Review Board. The two studies described in this article were conducted in accordance with all applicable regulatory and Good Clinical Practice guidelines and following the ethical principles originating in the Declaration of Helsinki.
In both studies, the subjects, investigator, subinvestigators and other site personnel were blinded, with the exception of a pharmacist who prepared the study drug doses. The sponsor as the third party was unblinded (with the exception of the Abbott study monitor who was blinded) to allow for an expedited review of the safety, tolerability and pharmacokinetic data and to make informed decisions regarding possible modification (lowering) of planned doses for subsequent groups if needed.
Three hard-gelatin capsule strengths (0.1, 1 and 5 mg) of ABT-288 were utilized for dosing in the two studies. For each dose group, the placebo capsules matched ABT-288 capsules in terms of number, appearance and size.
Subjects
Subjects signed a written informed consent form prior to enrolment in the studies. Subjects were considered eligible for enrolment if they were men or women between 18 and 45 years (study 1), or 65 years of age or older (study 2), were in general good health based on medical history, physical examination, brief neurological examination, vital signs, 12-lead electrocardiogram (ECG) evaluation and clinical laboratory profile and their body mass index (BMI) was within 18 to 30 kg m−2 (study 1) or 18 to 35 kg m−2 (study 2). Females in study 1 were post-menopausal for at least 2 years or surgically sterile, and were not pregnant or breast-feeding. Males in both studies were surgically sterile, sexually inactive or used a barrier method (i.e. condom) of birth control starting on participation of the study and continuing until 30 days after the last study drug administration. Subjects were excluded if they had a history of major medical illness (study 1) or had clinically significant illness within 1 month prior to the study (study 2), had abnormal results in ECG or laboratory tests (including HIV and hepatitis tests), had consumed alcohol within 48 h from the start of the study, were tobacco users within 6 months preceding study drug administration, had taken (or were required to take regularly) prescription or over-the-counter medications within 2 weeks of dosing (study 1) or used any psychoactive medications within the 2 week period prior to study drug administration, with the exception of selected antidepressants and/or low dose anxiolytics/hypnotics (study 2), had a history of allergies to any medication, had a recent (6 month) history of drug or alcohol abuse, used known inhibitors or inducers of drug metabolizing enzymes within 1 month prior to study drug administration, had consumed grapefruit or grapefruit products within the 72 h period prior to study drug administration, had donated or lost 550 ml or more blood volume (including plasmapheresis) or received a transfusion of any blood product within 8 weeks prior to study drug administration, had received any investigational drug within 6 weeks prior to study drug administration, had an estimated creatinine clearance (CLcr) less than 40 ml min−1 (study 2) or was considered by the investigator, for any reason, to be an unsuitable candidate for receiving ABT-288.
Safety monitoring
Safety was evaluated based on assessments of adverse events, physical examinations, brief neurological examination, laboratory tests (haematology, clinical chemistry and urinalysis), vital signs, orthostatic vital signs and 12-lead ECGs. All observed or volunteered adverse events were recorded after administration of each dose with regard to their time of onset, severity, duration and possible relationship to the study drug.
Blood and urine sampling
Blood samples for measurement of plasma ABT-288 concentrations were collected by venipuncture into 6 ml evacuated sodium heparin containing collection tubes. For parts 1 and 2 of study 1, blood samples were collected within 10 min prior to dosing (0 h) and at 0.5, 1, 1.5, 2, 3, 4, 6, 9,12, 16, 24, 36, 48, 60, 72, 96, 120 and 144 h after dosing in each study period. For part 3 of study 1, blood samples were collected (i) prior to dosing (0 h) and at 0.5, 1, 1.5, 2, 3, 4, 6, 9, 12, 16 and 24 h after dosing on days 1 and 7; (ii) prior to dosing (0 h) on days 5, 6, 12 and 13; and (iii) prior to dosing (0 h) and at 0.5, 1, 1.5, 2, 3, 4, 6, 9, 12, 16, 24, 36, 48, 60, 72, 96, 120, 144, 168, 192, 216 and 240 h after dosing on day 14. For study 2, blood samples were collected (i) prior to dosing (0 h) and at 0.5, 1, 1.5, 2, 3, 4, 6, 9, 12, 16 and 24 h after dosing on day 1; (ii) prior to dosing (0 h) on days 4, 7, 10 and 11; and (iii) prior to dosing (0 h) and at 0.5, 1, 1.5, 2, 3, 4, 6, 9, 12, 16, 24, 36, 48, 96, 168 and 240 h after dosing on day 12. The blood samples were centrifuged within 30 min of collection using a refrigerated centrifuge to separate the plasma. The plasma samples were frozen within 1 h after collection and maintained at −20°C or colder.
Urine samples for measurement of ABT-288 concentrations were collected in part 3 of study 1 and in study 2. Urine was collected in containers without preservatives over the following intervals: 0 to 6, 6 to 12, 12 to 18 and 18 to 24 h after dosing on days 7 and 14 (study 1) and day 12 (study 2). All urine collected over each interval for a subject was thoroughly mixed and the volume was measured and recorded. One 3 ml aliquot from each collection interval was then separated for analysis. The urine samples were frozen within 1 h after collection and maintained at approximately −70°C or colder.
Analytical methods
Plasma concentrations of ABT-288 were determined using a validated liquid chromatography method with tandem mass spectrometric detection at Abbott Laboratories (Abbott Park, IL, USA). ABT-288 was extracted from human plasma using protein precipitation with acetonitrile on a semi-automated liquid handler. Stable isotope-labeled ABT-288 (ABT-288 13CD3) was used as an internal standard. Chromatographic separation was achieved using a Synergi Max-RP column (4 μm, 2.0 × 150 mm; Phenomenex, Torrace, CA, USA) and Alltima HP C18 EPS guard column (2.1 × 7.5 mm, 5 μm; Alltech Corporation, Deerfield, IL, USA). The mobile phase consisted of 0.001% trifluoroacetic acid, 0.01% formic acid, 1 mm ammonium formate in 25/75 (v/v) acetonitrile/water. The flow rate was 0.3 ml min−1 and the chromatography ran for 3.3 min. ABT-288 and its internal standard were monitored by an API 4000 tandem mass spectrometer on positive ionization mode with Analyst software (Applied Biosystems, Foster City, CA, USA). The following transitions were monitored: m/z 373 to 342 for ABT-288 and m/z 377 to 114 for its internal standard. The validated plasma concentration range was from 0.0164 to 82.8 ng ml−1 (r2 ≥ 0.997) with inter-run variability (% coefficient of variation [%CV]) ≤ 6.4% and mean bias between −5.4% and 1.6%. Urine concentrations of ABT-288 were determined using the same chromatographic method described above for plasma samples after a 10 times dilution of urine samples with the mobile phase. Validated urine concentration range was from 0.273 to 152 ng ml−1 (r2≥ 0.997) with inter-run variability (%CV) ≤ 3.3% and mean bias between 4.2% and 6.6%.
Pharmacokinetic analysis
The plasma pharmacokinetic parameters of ABT-288 were estimated using a standard non-compartmental method with WinNonlin-Professional™ software (Version 5.2; Pharsight Corporation, Mountain View, CA, USA). The estimated pharmacokinetic parameters included the maximum observed plasma concentration (Cmax) and time to Cmax (tmax), the apparent terminal phase elimination rate constant (λz) and the terminal phase elimination half-life (t1/2). For single dose evaluations (parts 1 and 2 of study 1), the area under the plasma concentration–time curve (AUC) from time 0 to time of the last measurable concentration (AUC(0,tlast)) and the AUC from time 0 to infinity (AUC(0,∞)) were calculated. For multiple dose evaluations, the AUC from time 0 to 24 h (AUC(0,24h)) and the minimum observed plasma concentration (Cmin) on days 7 and 14 (study 1, part 3) and day 12 (study 2) were calculated. The apparent oral clearance (CL/F) was calculated from the dose and AUC(0,∞) (single dose evaluation) or steady-state AUC(0,24 h) (multiple dose evaluations). The accumulation ratio (Rac) was calculated as the ratio of ABT-288 AUC(0,24 h) on last day of dosing to day 1.
The percentage of ABT-288 dose recovered in urine (fe %) at steady-state was calculated as the amount of ABT-288 recovered in the urine (Au) over the time interval 0 to 24 h, divided by the administered dose and multiplied by 100. Renal clearance (CLR) of ABT-288 was calculated as Au/AUC(0,24h).
Statistical analyses
Statistical analyses were performed using SAS software (Version 8.2; SAS Institute Inc., Cary, NC USA).
To assess dose proportionality following single doses of ABT-288 in part 1 of study 1, an analysis of variance (anova) was performed on the difference in the dose-normalized Cmax, dose-normalized AUC, tmax and λz between the highest and lowest doses administered to a subject, but the data of a subject who was not administered the highest dose within his/her sequence group were excluded. Subjects were classified by group, but the test to compare highest and lowest doses was performed on the average of the mean differences for the two groups. For each parameter, an analysis using a mixed effects model was also performed using data from all doses. The model included effects for dose level and subjects were viewed as a random sample. A test for trend with dose was performed on a contrast in the dose level effects chosen to provide good power for an approximately linear function of dose. The logarithmic transformation was employed for Cmax and AUC.
To assess the effect of food on the pharmacokinetics of ABT-288 in part 2 of study 1, an anova was performed for tmax, and the logarithms of Cmax and AUC. Point estimates and 90% confidence intervals (CIs) for the bioavailability of the non-fasting regimen relative to the fasting regimen were obtained from the analysis of Cmax and AUC. The CIs were used to perform a two one-sided test procedure for bioequivalence assessment.
To assess dose proportionality following multiple dosing, analysis of covariance (ancova) was performed on day 14 (part 3 of study 1) and day 12 (study 2) for the following pharmacokinetic parameters: tmax, λz (determined from the concentrations after the last dose of a regimen), dose-normalized Cmin and logarithms of dose-normalized Cmax and AUC. Within the framework of the ancova, the hypothesis of no difference between the lowest and the highest doses was tested. In addition, for the four dose levels of study 2, a test was performed on a contrast in the dose level effects chosen to provide good power for an approximately linear function of dose.
To determine whether steady-state was reached in the multiple dose study in young adults, two analyses were conducted. First, a test was performed on the change from study day 7 to study day 14 in ABT-288 Cmin and log-transformed Cmax and AUC in the framework of a one way anova, with subjects classified by dose level. Second, since the comparison between day 7 and day 14 led to the conclusion of difference in exposure between these 2 days, a repeated-measures analysis, with dose level, study day and the interaction of dose level and study day as factors, was performed on the dose-normalized pre-dose concentrations of days 12, 13 and 14. The hypothesis of no difference between day 14 and each of days 12 and 13 was tested at a significance level of 0.05. For the study in elderly subjects, a similar analysis was conducted on the pre-dose concentrations of days 10, 11 and 12.
Sample size
The probability that a given adverse event would not be observed in different numbers of subjects administered an assigned ABT-288 dose was calculated for various true population incidence rates. Based on these calculations, for a true incidence rate of 20%, the probability of detecting an adverse event was 74% with six subjects and 87% with nine subjects. For doses other than the highest dose administered in an evaluation, assuming a given adverse event was not observed with the dose under consideration and the next higher dose, one could conservatively consider this outcome as though the lower of the two doses was administered in both groups. Under such a scenario, the probability for detecting an adverse event of true incidence rate of 20% was 93% with 12 subjects. As such, the number of subjects utilized in different parts of the present studies (six or nine subjects to receive ABT-288) was deemed satisfactory for a phase 1 setting.
Results
Demographics and subject disposition
A total of 78 young adult male and female subjects were enrolled in study 1. In part 2 of study 1 (food effect), one subject withdrew consent prior to dosing in period 2. This subject was excluded from the pharmacokinetic statistical analyses. During part 3, one subject in the 6 mg once-daily dose was prematurely discontinued from the study after 4 days of dosing due to adverse events. Dosing was stopped early on study day 6 for all subjects in the 6 mg once-daily dose group due to an increased incidence of adverse events.
A total of 29 elderly male and female subjects were enrolled in study 2. Two subjects in the 5 mg once-daily dose group discontinued from the study (on days 3 and 9) due to adverse events. Although eight subjects were planned to be enrolled in the 5 mg dose group of study 2, only five subjects enrolled in this dose group due to slow enrolment. Since the blinded randomization code of this dose group had the first serial six subject numbers assigned to ABT-288 and the last two serial subject numbers assigned to placebo, all five enrolled subjects received ABT-288.
A summary of the demographics of the participants in the two studies is presented in Table 2. Two female subjects enrolled in study 1, one in group 1 (part 1) and one in group 4 (part 3, received 0.5 mg ABT-288 once-daily). Fourteen female subjects enrolled in study 2 and they were distributed across treatments as follows: placebo (4), 0.5 mg (3), 1.5 mg (4), 3 mg (2) and 5 mg (1).
Table 2.
Demographic characteristics of study participants
| Study 1: Healthy young adults | Study 2: Elderly subjects | |||
|---|---|---|---|---|
| Demographic | Part 1 (n = 18) | Part 2 (n = 12) | Part 3 (n = 48)* | (n = 29)* |
| Age (years) | 32 ± 7 | 29 ± 6 | 28 ± 7 | 71 ± 5 |
| Weight (kg) | 78.2 ± 8.2 | 81.9 ± 9.7 | 77.5 ± 10.2 | 71.2 ± 11.7 |
| Height (cm) | 176 ± 8 | 178 ± 6 | 177 ± 8 | 165 ± 9 |
| Sex | 17 males (94%), 1 female (6%) | 12 males (100%) | 47 males (98%), 1 female (2%) | 15 males (52%), 14 females (48%) |
| Race | 15 White (83%), 3 Black (17%) | 11 White (92%), 1 White/Other (8%) | 38 White (79%), 6 Black (13%), 2 Asian (4%), 1 Other (2%), 1 Mixed (2%) | 22 White (76%), 4 Black (14%), 3 Asian (10%) |
Demographics are presented as mean ± SD for age, weight and height and as n (%) for sex and race.
Demographic characteristics were comparable for subjects assigned to study drug or placebo.
Safety and tolerability
Healthy young adults (study 1):
Single doses of ABT-288 up to 40 mg and multiple doses of ABT-288 up to 3 mg once-daily for 14 days were generally safe and well tolerated by the subjects. The incidence of adverse events appeared to increase with increasing dose (parts 1 and 3). The most frequently reported adverse events are presented in Table 3 for the single dose escalation and in Table 4 for the multiple dose escalation. The minimally intolerated single dose was not established and, therefore, the maximum tolerated single dose was not defined. However, the frequency and intensity of adverse events suggested it was not practical to continue dose escalation beyond 40 mg. The 3 mg once-daily dose was the maximum tolerated multiple dose and 6 mg once-daily was the minimally intolerable multiple dose.
Table 3.
Adverse events reported by at least 3 subjects administered ABT-288 in the single dose escalation
| Adverse event | Single doses in healthy young adults (study 1 part 1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo n = 18 | 0.1 mg n = 6 | 0.3 mg n = 6 | 1 mg n = 6 | 3 mg n = 6 | 10 mg n = 6 | 20 mg n = 6 | 40 mg n = 6 | Overall ABT-288 n = 18 | |
| Any adverse event | 12 (66.7) | 3 (50.0) | 1 (16.7) | 2 (33.3) | 3 (50.0) | 5 (83.3) | 6 (100.0) | 5 (83.3) | 14 (77.8) |
| Hot flush | 0 | 0 | 0 | 0 | 0 | 2 (33.3) | 3 (50.0) | 5 (83.3) | 9 (50.0) |
| Nausea | 0 | 0 | 0 | 0 | 0 | 3 (50.0) | 2 (33.3) | 4 (66.7) | 8 (44.4) |
| Dizziness | 1 (5.6) | 0 | 0 | 0 | 1 (16.7) | 2 (33.3) | 1 (16.7) | 2 (33.3) | 6 (33.3) |
| Headache | 3 (16.7) | 1 (16.7) | 0 | 1 (16.7) | 0 | 1 (16.7) | 2 (33.3) | 0 | 5 (27.8) |
| Dysgeusia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 (50.0) | 3 (16.7) |
| Anxiety | 1 (5.6) | 0 | 0 | 0 | 0 | 0 | 0 | 3 (50.0) | 3 (16.7) |
Number and (percentage) of subjects reporting an adverse event judged by the investigator as possibly or probably related to study drug are presented.
Table 4.
Adverse events reported by at least three subjects administered ABT-288 in either of the two multiple dose evaluations
| Adverse events | Multiple doses in healthy young adults (study 1 part 3) | Multiple doses in elderly subjects (study 2) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo n = 12 | 0.5 mg n = 9 | 1.5 mg n = 9 | 3 mg n = 9 | 6 mg n = 9 | Overall ABT-288 n = 36 | Placebo n = 6 | 0.5 mg n = 6 | 1.5 mg n = 6 | 3 mg n = 6 | 5 mg n = 5 | Overall ABT-288 n = 23 | |
| Any adverse event | 6 (50.0) | 7 (77.8) | 6 (66.7) | 7 (77.8) | 8 (88.9) | 28 (77.8) | 6 (100) | 6 (100) | 5 (83.3) | 4 (66.7) | 5 (100) | 20 (87) |
| Headache | 1 (8.3) | 2 (22.2) | 2 (22.2) | 2 (22.2) | 4 (44.4) | 10 (27.8) | 1 (16.7) | 3 (50.0) | 3 (50.0) | 1 (16.7) | 3 (60.0) | 10 (43.5) |
| Dizziness | 1 (8.3) | 1 (11.1) | 0 | 1 (11.1) | 3 (33.3) | 5 (13.9) | 0 | 3 (50.0) | 0 | 1 (16.7) | 0 | 4 (17.4) |
| Dysgeusia | 0 | 0 | 0 | 1 (11.1) | 2 (22.2) | 3 (8.3) | 0 | 0 | 0 | 0 | 1 (20) | 1 (4.3) |
| Abnormal dreams | 1 (8.3) | 0 | 1 (11.1) | 1 (11.1) | 5 (55.6) | 7 (19.4) | 3 (50.0) | 2 (33.3) | 1 (16.7) | 0 | 2 (40.0) | 5 (21.7) |
| Sleep disorder | 0 | 1 (11.1) | 1 (11.1) | 0 | 3 (33.3) | 5 (13.9) | 0 | 0 | 0 | 0 | 1 (20) | 1 (4.3) |
| Insomnia | 0 | 0 | 1 (11.1) | 1 (11.1) | 1 (11.1) | 3 (8.3) | 0 | 1 (16.7) | 0 | 1 (16.7) | 0 | 2 (8.7) |
| Initial insomnia | 0 | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 1 (16.7) | 3 (60.0) | 4 (17.4) |
| Nightmare | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 2 (40.0) | 3 (13.0) |
| Nausea | 0 | 2 (22.2) | 0 | 1 (11.1) | 3 (33.3) | 6 (16.7) | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 2 (8.7) |
| Energy increased | 0 | 3 (33.3) | 0 | 0 | 1 (11.1) | 4 (11.1) | 0 | 0 | 0 | 0 | 0 | 0 |
| Palpitations | 1 (8.3) | 0 | 0 | 2 (22.2) | 2 (22.2) | 4 (11.1) | 0 | 0 | 0 | 0 | 0 | 0 |
Number and (percentage) of subjects reporting an adverse event judged by the investigator as possibly or probably related to study drug are presented.
In part 1 (escalating single doses), 14 (14/18, 77.8%) subjects who received ABT-288 and 12 (12/18, 66.7%) subjects who received placebo reported at least one treatment-emergent adverse event. The incidence of adverse events was higher in the higher dose groups (10 to 40 mg) and increased as the dose increased. The adverse events reported by at least three subjects treated with ABT-288 in order of decreasing frequency were hot flush, nausea, dizziness, headache, dysgeusia and anxiety (Table 3).
In part 2 (food effect), five (5/11, 45.5%) subjects in the fasting regimen and two (2/12, 16.7%) subjects in the non-fasting regimen reported at least one treatment-emergent adverse event. The adverse events assessed by the investigator as possibly or probably related to treatment with ABT-288 were palpitations, nightmare and hyperhidrosis. These adverse events were reported by one subject each.
In part 3 (multiple doses), seven (7/9, 77.8%), six (6/9, 66.7%), seven (7/9, 77.8%) and eight (8/9, 88.9%) subjects who received multiple doses of 0.5, 1.5, 3 or 6 mg ABT-288 once-daily, respectively, reported at least one treatment-emergent adverse event. Six (6/12, 50.0%) subjects who received placebo reported at least one treatment-emergent adverse event. The adverse events reported by at least three subjects administered ABT-288 in order of decreasing frequency were headache, abnormal dreams, nausea, dizziness, sleep disorder, increased energy, palpitations, insomnia and dysgeusia (Table 4).
All adverse events were classified as mild or moderate in severity with the exception of a severe adverse event of hypophosphataemia (<0.9 mg dl−1) in the 40 mg single dose group that occurred 1 day after dosing and resolved without treatment.
No deaths or other serious adverse events occurred during the study. During part 3 of the study, one subject in the 6 mg once-daily dose group was prematurely discontinued from the study after 4 days of dosing due to adverse events. Dosing was stopped early on study day 6 for all subjects in the 6 mg once-daily dose group due to general intolerance and an increase in the frequency and severity of adverse events such as headache, abnormal dreams, nausea, sleep disorder, palpitations and disorientation.
No other clinically significant abnormalities were detected in other safety analyses including vital signs, ECG variables and laboratory measurements.
Elderly subjects (study 2):
Overall, ABT-288 was generally safe and well tolerated in elderly subjects receiving multiple doses up to 3 mg once-daily, which was determined to be the maximum tolerated dose. The 5 mg once-daily dose was considered to be the minimally intolerated dose due to the rate of moderate/severe adverse events and withdrawals due to adverse events.
Twenty subjects (20/23, 87%) of those who received ABT-288 and six subjects (6/6, 100%) who received placebo reported at least one treatment-emergent adverse event. Adverse event frequency was similar across dose groups: 0.5 mg (100%), 1.5 mg (83.3%), 3 mg (66.7%), 5 mg (100%) and placebo (100%). The most common treatment-emergent adverse events reported by at least three subjects administered ABT-288 in order of decreasing frequency were headache, abnormal dreams, initial insomnia, dizziness and nightmares (Table 4).
Although the study protocol allowed concomitant use of anti-depressant and/or hypnotic drugs, only one subject enrolled in the 5 mg dose group was using zolpidem at bedtime for treatment of insomnia and continued zolpidem use during the study. This subject completed the study and did not report insomnia as an adverse event.
All adverse events were classified as mild or moderate in severity with the exception of one severe adverse event of worsening of asthma in the 5 mg once-daily dose group in a subject with medical history of asthma. Two subjects in the 5 mg once-daily dose group discontinued from the study, one subject discontinued on day 9 due to initial insomnia and the second subject discontinued on day 3 due to initial insomnia and headache. No deaths or other serious adverse events occurred during the study.
Results of other safety analyses including individual subject changes, changes over time and individually clinically significant values for vital signs, ECG variables, physical examinations and laboratory measurements were unremarkable for each treatment group.
No difference between males and females in ABT-288 tolerability was distinguishable based on the limited number of females enrolled.
Pharmacokinetics
Single dose escalation in healthy young adults:
The mean plasma concentration vs. time profiles of ABT-288 after administration of escalating single oral doses to healthy young adults are presented in Figure 1. The single dose pharmacokinetic parameters of ABT-288 are summarized in Table 5. Plasma concentrations of ABT-288 reached peak concentrations 2 to 6 h after oral dosing (Table 5). Afterwards, ABT-288 concentrations declined bi-exponentially with time (Figure 1), with harmonic mean terminal elimination half-lives of 41 to 61 h. ABT-288 oral clearance ranged from 37 to 44 l h−1 (Table 5).
Figure 1.
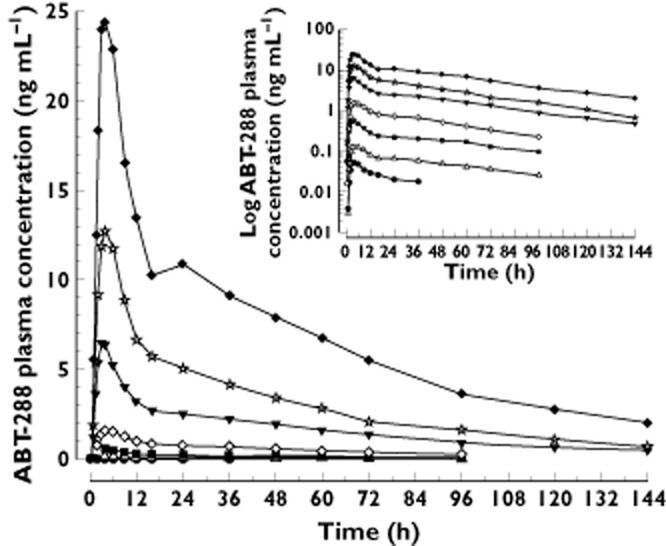
Mean plasma concentrations (linear and log scales) vs. time profiles of ABT-288 after administration of escalating single oral doses to healthy young adults. •, 0.1 mg; ▵; 0.3 mg; ▪, 1 mg; ◊, 3 mg; ▾, 10 mg; ☆, 20 mg; ♦, 40 mg
Table 5.
Pharmacokinetic parameters of ABT-288 (mean ± SD) after administration of escalating single oral doses to healthy young adults
| Parameter | 0.1 mg (n = 6)* | 0.3 mg (n = 6) | 1 mg (n = 6) | 3 mg (n = 6) | 10 mg (n = 6) | 20 mg (n = 6) | 40 mg (n = 6) |
|---|---|---|---|---|---|---|---|
| Cmax (ng ml−1) | 0.06 ± 0.01 | 0.14 ± 0.05 | 0.58 ± 0.10 | 1.62 ± 0.46 | 6.80 ± 1.52 | 13.2 ± 2.19 | 26.5 ± 4.25 |
| tmax (h)† | 4 (3–6) | 5 (4–6) | 4 (2–4) | 4 (4–6) | 3 (2–4) | 4 (3–6) | 4 (2–6) |
| t1/2 (h)‡ | ND | 56 ± 11 | 61 ± 15 | 43 ± 10 | 47 ± 5 | 41 ± 9 | 49 ± 11 |
| AUC(0,tlast) (ng h ml−1) | 1.1 ± 0.5 | 5.1 ± 1.4 | 19 ± 2.9 | 55 ± 10 | 230 ± 62 | 440 ± 79 | 970 ± 220 |
| AUC(0,∞) (ng h ml−1) | ND | 7.3 ± 1.6 | 28 ± 7.2 | 70 ± 9.1 | 270 ± 74 | 480 ± 96 | 1130 ± 340 |
| CL/F (l h−1) | ND | 43 ± 7.8 | 37 ± 9.1 | 44 ± 6.3 | 40 ± 9.6 | 43 ± 7.6 | 38 ± 11 |
The concentrations of ABT-288 at the 0.1 mg level were close to or below the LLOQ. Consequently robust estimates of t1/2, and AUC(0,∞) could not be obtained.
Median and range (minimum-maximum) are presented.
Harmonic mean ± pseudo-standard deviation is presented. ND, Not determined.
The dose-normalized Cmax and AUC(0,∞) values vs. dose relationships following single-dosing of ABT-288 are presented in Figures 2A. For the highest to lowest (within a subject) dose comparison, the dose-normalized Cmax and AUC(0,∞) values of the highest doses were statistically significantly higher (P < 0.05) than those of the lowest doses. The elimination rate constant (λz) values for the highest doses were statistically significantly higher than those for lower doses (P < 0.05). Results from the trend analysis indicated that there were statistically significant trends (P < 0.05) for an increase in ABT-288 dose-normalized Cmax in the 0.1 to 40 mg dose range. There were no statistically significant trends (P > 0.05) for changes in dose-normalized AUC(0,∞) or λz over the dose range for which those parameters could be calculated (0.3 to 40 mg). Overall, ABT-288 appeared to exhibit a slightly more than dose-proportional increase in Cmax (particularly over the 0.1 to 3 mg dose range, Figure 2A) and dose-proportional increase in AUC in the wide single dose range evaluated.
Figure 2.
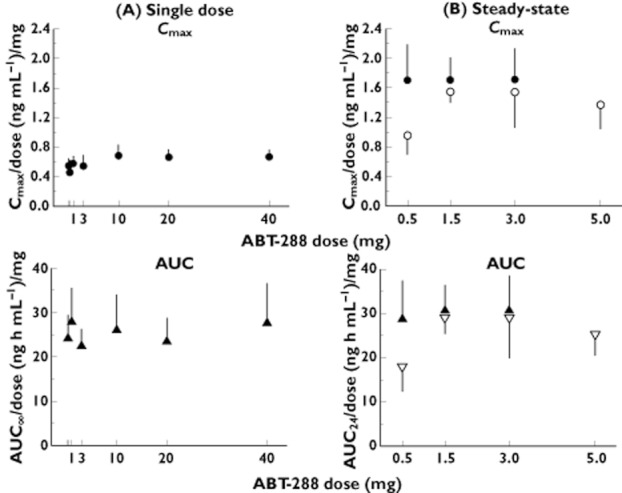
Dose-normalized ABT-288 Cmax and AUC values (mean and SD) vs. dose plots following single dosing (A) or at steady-state (B). Cmax •, young adults; ○, elderly subjects; AUC ▴, young adults; ▿, elderly subjects
Effect of food on the pharmacokinetics of ABT-288 in healthy young adults:
The mean plasma concentration vs. time profiles of ABT-288 after administration of a single oral dose (5 mg) under fasting and non-fasting (high fat/high calorie breakfast) conditions are presented in Figure 3. ABT-288 tmax was statistically significantly delayed (1.7 h delay, P < 0.05) for the non-fasting regimen compared with the fasting regimen. However, there were no statistically significant differences (P > 0.05) between the non-fasting and fasting regimens in ABT-288 Cmax, AUC(0,tlast) or AUC(0,∞). The 90% CIs for evaluating bioavailability and the corresponding point estimates of the non-fasting regimen relative to the fasting regimen are presented in Table 6. Food had a minimal effect on the central values of ABT-288 Cmax, AUC(0,tlast) and AUC(0,∞) (<5% change). The 90% CIs for assessing the bioavailability of ABT-288 (Cmax, AUC(0,tlast) and AUC(0,∞)) under non-fasting conditions relative to fasting conditions were within the 0.8 to 1.25 range indicating bioequivalence.
Figure 3.
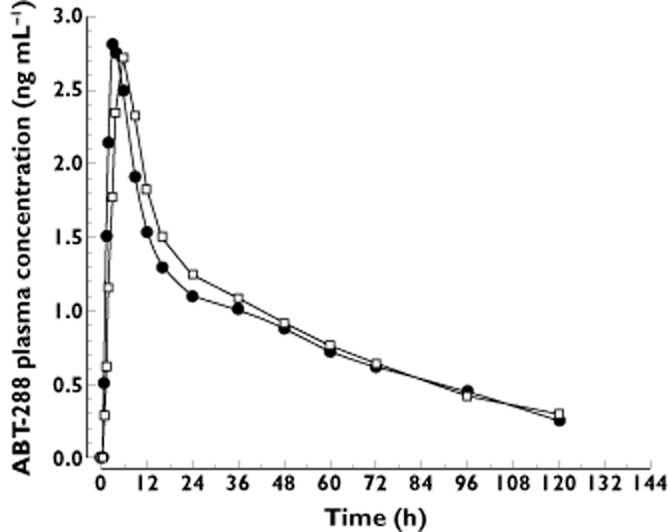
Mean plasma concentrations vs. time profiles of ABT-288 after administration of a single oral dose (5 mg) of ABT-288 under fasting and non-fasting (high fat/high calorie breakfast) conditions to healthy subjects. •, 5 mg, fasting; □, 5 mg, non-fasting
Table 6.
Bioavailability (and 90% CIs) of the non-fasting regimen of ABT-288 relative to the fasting regimen (5 mg single dose)
| Parameter | Central value* | Relative bioavailability | ||
|---|---|---|---|---|
| Point estimate† | 90% CIs | |||
| Non-fasting | Fasting | |||
| Cmax (ng ml−1) | 2.87 | 2.92 | 0.985 | 0.871, 1.114 |
| AUC(0,tlast) (ng h ml−1) | 105 | 100 | 1.042 | 0.995, 1.091 |
| AUC(0,∞) (ng h ml−1) | 127 | 124 | 1.022 | 0.960, 1.088 |
Antilogarithm of the least squares means for logarithms.
Antilogarithm of the difference (test − reference) of the least squares means for logarithms.
Multiple dose escalation in healthy young adults:
The mean plasma concentration vs. time profiles of ABT-288 after administration of escalating multiple once-daily oral doses to healthy young adults are presented in Figure 4. Dosing in the 6 mg dose group was discontinued after day 6 due to poor tolerability and therefore no steady-state profile or parameters are reported for this dose group. The steady-state (day 14) pharmacokinetic parameters of ABT-288 are summarized in Table 7. The median steady-state accumulation ratio (day 14 : day 1 AUC(0,24 h) ratio) of ABT-288 ranged from 3.4 to 3.7. The percentage of ABT-288 dose eliminated unchanged in urine was characterized for the 1.5 mg dose and was low (8.6 ± 3.5%) with a renal clearance of 2.8 ± 1.0 l h−1.
Figure 4.
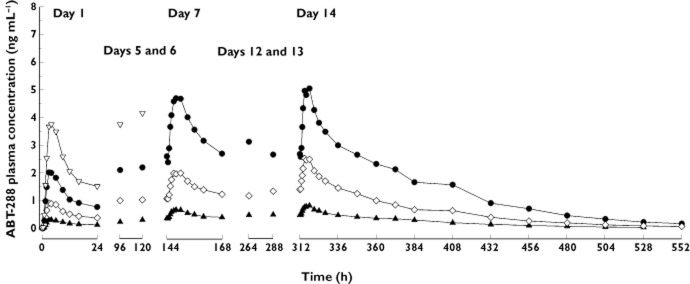
Mean plasma concentrations vs. time profiles of ABT-288 after administration of escalating once-daily oral doses of ABT-288 to healthy young adults. ▴, 0.5 mg once-daily; ◊, 1.5 mg once-daily; •, 3 mg once-daily; ▿, 6 mg once-daily
Table 7.
Pharmacokinetic parameters of ABT-288 (mean ± SD) after administration of escalating multiple once-daily doses of ABT-288 to healthy young adults and elderly subjects
| Parameter | Healthy young adults | Elderly subjects | |||||
|---|---|---|---|---|---|---|---|
| 0.5 mg (n = 9) | 1.5 mg (n = 9) | 3 mg (n = 9) | 0.5 mg (n = 6) | 1.5 mg (n = 6) | 3 mg (n = 6) | 5 mg (n = 3) | |
| Day 14 | Day 12 | ||||||
| Cmax (ng ml−1) | 0.85 ± 0.24 | 2.56 ± 0.451 | 5.14 ± 1.25 | 0.48 ± 0.13 | 2.32 ± 0.23 | 4.62 ± 1.43 | 6.86 ± 1.55 |
| tmax (h) | 6 (2–6) | 3 (3–6) | 6 (2–6) | 3 (1.5–9) | 3.5 (3–6) | 4 (2–9) | 4 (3–12) |
| Cmin (ng ml−1) | 0.44 ± 0.15 | 1.33 ± 0.23 | 2.48 ± 0.67 | 0.27 ± 0.08 | 1.37 ± 0.17 | 2.71 ± 1.11 | 3.47 ± 0.40 |
| t1/2* (h) | 40 ± 10 | 45 ± 7.5 | 49 ± 11 | 50 ± 20 | 52 ± 8.3 | 53 ± 17 | 53 ± 13 |
| AUC(0,24 h) (ng h ml−1) | 15 ± 4.0 | 46 ± 7.6 | 92 ± 24 | 8.5 ± 2.3 | 43 ± 4.9 | 87 ± 27 | 120 ± 20 |
| CL/F (l h−1) | 36 ± 8.5 | 34 ± 6.4 | 35 ± 13 | 64 ± 26 | 35 ± 4.1 | 38 ± 15 | 42 ± 6.7 |
| Rac† | 3.6 (2.6–4.1) | 3.7 (2.7–4.5) | 3.4 (2.6–4.1) | 3.7 (2.7–7.3) | 4.2 (3.2–4.9) | 4.1 (2.4–4.7) | 4.2 (3.8–4.6) |
Harmonic mean ± pseudo-standard deviation; half-life from the plasma concentrations during the terminal phase after administration of the last dose.
Observed accumulation ratio (calculated as the ratio of AUC(0,24 h) on day 14 or 12 to AUC(0,24 h) on day 1), median and range are presented. ND, Not determined.
The dose-normalized ABT-288 Cmax and AUC(0,24 h) vs. dose relationships are presented in Figure 2B. Statistical comparison between the highest and lowest ABT-288 dose levels (3 mg vs. 0.5 mg) revealed no statistically significant differences (P > 0.05) for any of the pharmacokinetic parameters tested indicating that exposures of ABT-288 were dose proportional over the 0.5 to 3 mg once-daily dose range.
Comparison between day 14 and day 7 pharmacokinetic parameters indicated that ABT-288 exposure was statistically significantly higher (7 to 24% higher across doses, P < 0.05) on study day 14 than on study day 7. Therefore, steady-state was not completely reached on day 7. Statistical comparison between pre-dose concentrations for all multiple dose regimens tested on day 14 with those on days 12 and 13 indicated study day 14 was not statistically significantly different (P > 0.05) from study day 12 or study day 13. Therefore, steady-state is achieved for ABT-288 by day 12 of once-daily dosing.
Multiple dose escalation in elderly subjects:
The steady-state (day 12) pharmacokinetic parameters of ABT-288 in elderly subjects are summarized in Table 7 in comparison with the pharmacokinetic parameters in healthy young adults. The median accumulation ratio of ABT-288 on day 12 ranged from 3.7 to 4.2 across doses. The mean percentages of ABT-288 dose excreted in urine on study day 12 were low (4% and 11%) with mean renal clearance values of 1.5 to 4.4 l h−1 across dose groups.
The dose-normalized ABT-288 Cmax and AUC(0,24h) vs. dose relationships are presented in Figure 2B in comparison with healthy young adults. Statistical comparison between the highest and lowest ABT-288 doses (5 mg vs. 0.5 mg) revealed no statistically significant differences (P > 0.05) for any of the pharmacokinetic parameters, except for ABT-288 Cmax, where the 5 mg dose had a statistically significantly higher dose-normalized Cmax than the 0.5 mg dose (P < 0.05). There were no statistically significant differences (P > 0.05) between the 5 mg dose and either of the 1.5 or 3 mg doses in ABT-288 dose-normalized Cmax.
The overall mean of the pre-dose concentrations for all multiple dose regimens tested on study day 12 was not significantly different (P > 0.05) from the overall mean on study day 10 or study day 11, indicating that steady-state was achieved for ABT-288 by day 10 of once-daily dosing.
Sex was not a significant covariate (P > 0.1) for ABT-288 exposure.
Discussion
The first study presented in this manuscript was a three-in-one first-in-man study of the selective histamine H3 receptor antagonist, ABT-288, in healthy young adults. The study characterized the safety, tolerability and pharmacokinetics of escalating single and multiple oral doses of ABT-288 and evaluated the effect of food on the pharmacokinetics of a single oral dose of ABT-288. This study demonstrated that ABT-288 has a favourable pharmacokinetic profile in humans, characterized most noticeably by a long elimination half-life (∼50 h), convenient for once-daily dosing with conventional immediate release formulations, approximately dose and time linear pharmacokinetics, which simplifies dose selection and dose adjustments if needed, and lack of any significant food effect, thereby ABT-288 can be administered regardless of meals. With regard to the safety and tolerability, the study demonstrated that the single dose tolerability of ABT-288 is not predictive of the multiple dose tolerability even after accounting for pharmacokinetic accumulation (3- to 4-fold accumulation at steady-state consistent with the long half-life). Single doses of ABT-288 up to 40 mg were tolerated, whereas multiple doses up to 3 mg once-daily (steady-state exposures equivalent to 9 to 12 mg single doses) were only tolerated in young adults. The 6 mg once-daily dose was terminated in young adults due to increased incidence of adverse events and overall poor tolerability. The safety profile of ABT-288 appears to be consistent with other H3 receptor antagonists with typical sleep-related adverse events (insomnia, sleep disturbances, abnormal dreams) and other likely histamine-mediated adverse events (headache, nausea, sweating/hot flushes) [20–23]. Titration to improve the tolerability of higher doses was considered. However, it was not expected to be beneficial for ABT-288 given its long elimination half-life which results in slow accumulation with repeated dosing (steady-state is not reached by day 7 of dosing).
Given that the target patient population (AD patients) is an elderly population, it was important to determine whether the pharmacokinetics, safety or tolerability of ABT-288 were different between young adults and elderly subjects before proceeding to phase 2 evaluations. Study 2 presented in this article was designed to answer these questions. Since study 1 demonstrated that steady-state was reached for ABT-288 by day 12 of dosing, study 2 utilized a 12 day dosing duration. Additionally, dosing in study 2 was conducted under non-fasting conditions on all days since study 1 demonstrated that food did not have a clinically meaningful effect on ABT-288 exposure. ABT-288 5 mg once-daily was selected as the highest dose in study 2 to determine whether a dose slightly less than the 6 mg evaluated in study 1 would have improved tolerability.
ABT-288 was generally safe at the evaluated dose range (up to 5 mg once-daily) in elderly subjects and the highest tolerated dose was 3 mg once-daily, similar to young adults. Additionally, the adverse events profile was comparable between young adults and elderly subjects. ABT-288 had comparable pharmacokinetics in young adults and elderly subjects at the 1.5 and 3 mg once-daily doses (Figure 2B). However, ABT-288 exposure at the 0.5 mg once-daily dose in elderly subjects was approximately 60% of that in young adults. The reason for the discrepancy in exposure at the 0.5 mg dose only is not clear and could be an artifact of the small sample size and between-subject variability.
ABT-288 plasma concentrations of 0.1 to 2 ng ml−1 were efficacious in the Five-Trial Inhibitory Avoidance model in SHR pups (note that ABT-288 has moderate and comparable plasma protein binding across species) which corresponded to H3 receptor occupancy of ∼10 to 40%, suggesting that low to moderate receptor occupancy may be associated with cognitive improvement [19]. Additional behavioural models and data reported for other H3 receptor antagonists suggested that target receptor occupancy of ∼60% is needed for efficacy. Electroencephalogram studies in rats suggested that ABT-288 exposures (and therefore H3 receptor occupancy) required for wakefulness are higher than those required for the precognitive effects in behavioural models [19]. Positron emission tomography studies, sleep evaluations and quantitative electroencephalography using two H3 receptor antagonists, MK-0249 and MK-3134, suggested that the alerting effects are appreciable with H3 receptor antagonism in humans at receptor occupancy of 67% and higher 24. The incidence and severity of insomnia observed at the intolerable doses of ABT-288 in the present studies (6 mg once-daily in young subjects and 5 mg once-daily in elderly subjects) suggest that these doses already have high H3 receptor occupancy and that the precognitive effects of ABT-288 in humans, if replicated from preclinical behavioural models, should be observed at lower doses.
A receptor occupancy study in humans was not conducted for ABT-288. However, based on the in vitro affinity for human H3 receptors and assuming that the free concentrations of ABT-288 at the synapses are comparable with the free plasma concentrations (ABT-288 is highly permeable compound), it is predicted that 6 mg once-daily of ABT-288 achieves receptor occupancy of ∼75% or higher. Similarly, 1 and 3 mg once-daily dose of ABT-288 are predicted to result in (peak to trough) H3 receptor occupancy of 40 to 25% and 70 to 50%, respectively. Similar calculation was successful in predicting the ABT-288 H3 receptor occupancy in rats using the rat in vitro Ki value as verified experimentally. A positron emission tomography study is needed for accurate characterization of the human H3 receptor occupancy by the doses evaluated in the present studies.
Given the limits of tolerability in humans, as characterized in the present studies, and the preclinical efficacious exposures, two dose levels (1 mg and 3 mg once-daily) of ABT-288 were selected for evaluation in the proof-of-concept phase 2 study in AD. The 3 mg once-daily dose was selected because it was the highest tolerated dose which maximizes the chance of observing efficacy, if efficacy indeed is associated with moderate to high receptor occupancy. The 1 mg once-daily dose was selected in order to examine the possibility of achieving efficacy with low levels of receptor occupancy as suggested by the SHR pup Five-Trial Inhibitory Avoidance model. The two doses are well separated in terms of exposure as demonstrated by the moderate pharmacokinetic variability (coefficient of variation in ABT-288 exposure within 20 to 30%) observed in the present studies.
In conclusion, in the present studies, ABT-288 was safe and well tolerated over single doses up to 40 mg and multiple doses up to 3 mg once-daily for 14 days in young adults and 12 days in elderly subjects. The most frequently reported adverse events were hot-flush, headache, abnormal dreams, insomnia, nausea and dizziness. ABT-288 exposure (AUC) was dose-proportional over the evaluated dose range, particularly at doses of 1 mg and above. ABT-288 mean elimination half-life ranged from 40 to 61 h. Food did not have a clinically meaningful effect on ABT-288 exposure. Steady-state was achieved for ABT-288 by study day 10 of once-daily dosing. The safety, tolerability and pharmacokinetic profiles of ABT-288 justified further evaluation in humans for symptomatic treatment of AD. Detailed reports of this evaluation are warranted.
Acknowledgments
The authors thank Lillian Lee of Abbott Clinical Pharmacology for providing assistance with development of the manuscript.
Competing Interests
The studies presented in this manuscript were funded by Abbott Laboratories. All authors are employees and share holders of Abbott Laboratories and the authors declare no other relationships or activities that could appear to have influenced the submitted work.
References
- 1.Qiu C, De Ronchi D, Fratiglioni L. The epidemiology of the dementias: an update. Curr Opin Psychiatry. 2007;20:380–385. doi: 10.1097/YCO.0b013e32816ebc7b. [DOI] [PubMed] [Google Scholar]
- 2.Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet. 1976;2:1403. doi: 10.1016/s0140-6736(76)91936-x. [DOI] [PubMed] [Google Scholar]
- 3.Courtney C, Farrell D, Gray R, Hills R, Lynch L, Sellwood E, Edwards S, Hardyman W, Raftery J, Crome P, Lendon C, Shaw H, Bentham P. Long-term donepezil treatment in 565 patients with Alzheimer's disease (AD2000): randomised double-blind trial. Lancet. 2004;363:2105–2115. doi: 10.1016/S0140-6736(04)16499-4. [DOI] [PubMed] [Google Scholar]
- 4.National Institute for Health and Clinical Excellence (NICE) Donepezil, galantamine, rivastigmine and memantine for the treatment of Alzheimer's disease. In: NICE Technology Appraisal Guidance 217 (issue date March 2011). Available at http://www.nice.org.uk/guidance/TA217 (last accessed 30 October 2012)
- 5.Leurs R, Smit MJ, Timmerman H. Molecular pharmacological aspects of histamine receptors. Pharmacol Ther. 1995;66:413–463. doi: 10.1016/0163-7258(95)00006-3. [DOI] [PubMed] [Google Scholar]
- 6.Hough LB. Genomics meets histamine receptors: new subtypes, new receptors. Mol Pharmacol. 2001;59:415–419. [PubMed] [Google Scholar]
- 7.Bakker RA, Timmerman H, Leurs R. Histamine receptors: specific ligands, receptor biochemistry, and signal transduction. Clin Allergy Immunol. 2002;17:27–64. [PubMed] [Google Scholar]
- 8.Brown RE, Stevens DR, Haas HL. The physiology of brain histamine. Prog Neurobiol. 2001;63:637–672. doi: 10.1016/s0301-0082(00)00039-3. [DOI] [PubMed] [Google Scholar]
- 9.Martinez-Mir MI, Pollard H, Moreau J, Arrang JM, Ruat M, Traiffort E, Schwartz JC, Palacios JM. Three histamine receptors (H1, H2 and H3) visualized in the brain of human and non-human primates. Brain Res. 1990;526:322–327. doi: 10.1016/0006-8993(90)91240-h. [DOI] [PubMed] [Google Scholar]
- 10.Drutel G, Peitsaro N, Karlstedt K, Wieland K, Smit MJ, Timmerman H, Panula P, Leurs R. Identification of rat H3 receptor isoforms with different brain expression and signaling properties. Mol Pharmacol. 2001;59:1–8. [PubMed] [Google Scholar]
- 11.Lovenberg TW, Roland BL, Wilson SJ, Jiang X, Pyati J, Huvar A, Jackson MR, Erlander MG. Cloning and functional expression of the human histamine H3 receptor. Mol Pharmacol. 1999;55:1101–1107. [PubMed] [Google Scholar]
- 12.Arrang JM, Garbarg M, Schwartz JC. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature. 1983;302:832–837. doi: 10.1038/302832a0. [DOI] [PubMed] [Google Scholar]
- 13.Blandina P, Bacciottini L, Giovannini MG, Mannaioni PF. H3 receptor modulation of the release of neurotransmitters in vivo. In: Rob L, Henk T, editors. Pharmacochemistry Library. Amsterdam: Elsevier; 1998. pp. 27–40. [Google Scholar]
- 14.Hancock AA, Fox GB. Perspectives on cognitive domains, H3 receptor ligands and neurological disease. Expert Opin Investig Drugs. 2004;13:1237–1248. doi: 10.1517/13543784.13.10.1237. [DOI] [PubMed] [Google Scholar]
- 15.Witkin JM, Nelson DL. Selective histamine H3 receptor antagonists for treatment of cognitive deficiencies and other disorders of the central nervous system. Pharmacol Ther. 2004;103:1–20. doi: 10.1016/j.pharmthera.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 16.Esbenshade TA, Browman KE, Bitner RS, Strakhova M, Cowart MD, Brioni JD. The histamine H3 receptor: an attractive target for the treatment of cognitive disorders. Br J Pharmacol. 2008;154:1166–1181. doi: 10.1038/bjp.2008.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brioni JD, Esbenshade TA, Garrison TR, Bitner SR, Cowart MD. Discovery of histamine H3 antagonists for the treatment of cognitive disorders and Alzheimer's disease. J Pharmacol Exp Ther. 2011;336:38–46. doi: 10.1124/jpet.110.166876. [DOI] [PubMed] [Google Scholar]
- 18.Bitner RS, Markosyan S, Nikkel AL, Brioni JD. In-vivo histamine H3 receptor antagonism activates cellular signaling suggestive of symptomatic and disease modifying efficacy in Alzheimer's disease. Neuropharmacology. 2011;60:460–466. doi: 10.1016/j.neuropharm.2010.10.026. [DOI] [PubMed] [Google Scholar]
- 19.Esbenshade TA, Browman KE, Miller TR, Krueger KM, Komater-Roderwald V, Zhang M, Fox GB, Rueter L, Robb HM, Radek RJ, Drescher KU, Fey TA, Bitner RS, Marsh K, Polakowski JS, Zhao C, Cowart MD, Hancock AA, Sullivan JP, Brioni JD. Pharmacological properties and pro-cognitive effects of ABT-288, a potent and selective histamine H3 receptor antagonist. J Pharmacol Exp Ther. 2012;343:233–245. doi: 10.1124/jpet.112.194126. [DOI] [PubMed] [Google Scholar]
- 20.Lin JS, Dauvilliers Y, Arnulf I, Bastuji H, Anaclet C, Parmentier R, Kocher L, Yanagisawa M, Lehert P, Ligneau X, Perrin D, Robert P, Roux M, Lecomte JM, Schwartz JC. An inverse agonist of the histamine H(3) receptor improves wakefulness in narcolepsy: studies in orexin-/- mice and patients. Neurobiol Dis. 2008;30:74–83. doi: 10.1016/j.nbd.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 21.Schwartz JC. The histamine H3 receptor: from discovery to clinical trials with pitolisant. Br J Pharmacol. 2011;163:713–721. doi: 10.1111/j.1476-5381.2011.01286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inocente CD, Arnulf I, Bastuji H, Thibault-Stoll A, Raoux A, Reimão R, Lin JS, Franco P. Pitolisant, an inverse agonist of the histamine H3 receptor: an alternative stimulant for narcolepsy-cataplexy in teenagers with refractory sleepiness. Clin Neuropharmacol. 2012;35:55–60. doi: 10.1097/WNF.0b013e318246879d. [DOI] [PubMed] [Google Scholar]
- 23.Weisler RH, Pandina GJ, Daly EJ, Cooper K, Gassmann-Mayer C. Randomized clinical study of a histamine H3 receptor antagonist for the treatment of adults with attention-deficit hyperactivity disorder. CNS Drugs. 2012;26:421–434. doi: 10.2165/11631990-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 24.Iannone R, Renger J, Potter W, Dijk D, Boyle J, Palcza J, Zhao B, Bergman A, Van Laere K, Bormans G, Sanabria S, Calder N, De Lepeleire I, Van Hoydonck P, Marsilio S, Cerchio K, Declercq R, Fox-Bosetti S, Verma A, Van Bortel L, Achten E, Ma J, Hargreaves R, Koblan K, Chodakewitz J, Gottesdiener K, Murphy G. The relationship between brain receptor occupancy (RO) and alerting effects in humans support MK-0249 and MK-3134 as inverse agonists at the histamine subtype-3 pre-synaptic receptor (H3R). American College Neuropsychopharmacology 48th Annual Meeting (acnp), Hollywood, FL, 2009 Dec 6–10; 198 (abstract)