

Abstract
Neutrophils are essential for successful host eradication of bacterial pathogens and for survival to polymicrobial sepsis. During inflammation, the bone marrow provides a large reserve of neutrophils that are released into the peripheral circulation where they traverse to sites of infection. Although neutrophils are essential for survival, few studies have investigated the mechanisms responsible for neutrophil mobilization from the bone marrow during polymicrobial sepsis. Using a cecal ligation and puncture model of polymicrobial sepsis, we demonstrate that neutrophil mobilization from the bone marrow is not dependent on TLR4, MyD88, TRIF, IFNARα/β or CXCR2 pathway signaling during sepsis. In contrast, we observe that bone marrow CXCL12 mRNA abundance and specific CXCL12 levels are sharply reduced, while splenic CXCR4 mRNA and cell surface expression are increased during sepsis. Blocking CXCL12 activity significantly reduced the blood neutrophilia by inhibiting bone marrow release of granulocytes during sepsis. CXCL12 inhibition, however, had no impact on the expansion of bone marrow neutrophil precursors and hematopoietic progenitors. Bone marrow neutrophil retention by CXCL12 blockade prevented blood neutrophilia, inhibited peritoneal neutrophil accumulation, allowed significant peritoneal bacterial invasion and elevated polymicrobial sepsis mortality. We conclude that changes in the pattern of CXCL12 signaling during sepsis are essential for neutrophil bone marrow mobilization and host survival while having little impact on bone marrow granulopoiesis.
Keywords: myeloid cells, mouse, cecal ligation and puncture, CXCR2
Introduction
Sepsis is the leading cause of death in the critically ill with 750,000 cases and 210,000 deaths annually (1, 2). Sepsis mortality has been attributed to derangements in the innate immune system (3). Neutrophils are a fundamental component of innate immunity and essential for bacterial eradication and polymicrobial sepsis survival in humans and animals (4-6). Excessive neutrophil activity during inflammatory states can induce unwanted tissue damage and organ dysfunction (7). Hence, neutrophil production, release, and clearance are tightly regulated. Under normal circumstances, 1-2% of the total bone marrow neutrophil population is released into the circulation (8). During episodes of infection, the bone marrow provides a large neutrophil reserve (9).
Although cytokines, chemokines, leukotrienes, proteases, integrins, and bacterial products (10) have been implicated in bone marrow neutrophil release, attention has centered on CXC chemokine and toll-like receptor (TLRs) signaling during neutrophil release. Evidence suggests that CXCR4 and its ligand, stromal cell derived factor-1 (SDF-1/CXCL12), and CXCR2 and its ligands macrophage inflammatory protein-2 (MIP-2/CXCL2) and KC/GRO-1α (CXCL1), initiate neutrophil bone marrow release under normal conditions. In healthy mice, interactions between CXCR4 and CXCL12 are essential for maintaining bone marrow neutrophil populations (11, 12). CXCR4 blockade disrupts the CXCR4/CXCL12 axis and increases neutrophil release from the bone marrow (13-15). Exogenous administration of CXCL1, CXCL2, and CXCL12 increase neutrophil mobilization from the bone marrow, while CXCR2 antagonism inhibits neutrophil bone marrow release(7-10, 16).
TLR signaling impacts neutrophil release directly or via chemokine signaling. TLRs recognize pathogen associated molecular regions on invading pathogens(17, 18) and initiate a multitude of inflammatory processes. MyD88-/- mice, with TLR signaling deficits, fail to increase CXCL1 and CXCL2 blood concentrations during polymicrobial sepsis (19), and Pseudomonas pneumonia (20). This data suggest that CXCL1 and CXCL2 elevations involved in neutrophil bone marrow release depend on intact TLR signaling pathways. Although a growing body of evidence associates bone marrow neutrophil mobilization with TLR, CXCR4/CXCL12, CXCR2/CXCL1 and CXCL2 signaling during homeostasis, the impact of these mediators on bone marrow neutrophil mobilization during polymicrobial sepsis has received little attention.
Using a cecal ligation and puncture model of polymicrobial sepsis, we investigated the role of TLR, CXCL12, and CXCR2 signaling in neutrophil bone marrow mobilization. The results indicate that bone marrow neutrophil mobilization is not dependent on TLR4, MyD88, TRIF, IFNARα/β or CXCR2 signaling pathways. Sepsis alters the tissue expression patterns of CXCL12, inhibiting mRNA abundance and chemokine levels in the bone marrow compared to blood, plasma and spleen levels. CXCL12 blockade prevented bone marrow neutrophil mobilization, yet had little impact on bone marrow neutrophil and hematopoietic progenitor expansion. The data suggest that in polymicrobia sepsis, CXCL12 inhibition resulted in peritoneal cavity neutropenia, unopposed bacterial invasion, and a 40% elevation in polymicrobial sepsis mortality. These findings suggest that CXCL12 signaling patterns are altered in sepsis, are essential for bone marrow neutrophil mobilization, and are necessary for host survival during polymicrobial sepsis.
Materials and Methods
Mice
All experiments were approved by the Institutional Animal Care and Use Committee at the University of Florida or Merck Research Laboratories (formerly DNAX). Specific pathogen-free C57BL/6, C3H/HeJ (TLR4 receptor mutation) and their control (C3H/HeOuJ) mice, were purchased from The Jackson Laboratory (Bar Harbor, ME). All mice were maintained at the University Of Florida College Of Medicine and were studied between 8-12 weeks of age. MyD88-/- mice and TRIF-/- mice on a B6x129(F1) background were a kind gift of Dr. Shizuo Akira to Merck Research Laboratories, and were maintained at Merck Research Laboratories, Palo Alto, CA. IFN-αβR/A129 mice on the 129S6/SvEv background (H-2b) and wild-type Sv129 mice were purchased from B & K Universal (Hull, East Yorkshire, UK).
Inhibitors
When indicated, mice were injected with either 500 μL/day i.p anti-CXCL12 or 500 μL/day i.p anti-CXCR2 polyclonal goat antisera beginning 12 hours prior to the initiation of sepsis. The anti-CXCL12 and anti-CXCR2 antibody preparations were gifts from Dr. Robert Strieter at the University of Virginia, Charlottesville, VA. As a control, heat-inactivated polyclonal goat serum (Sigma) was used (500 μL/day i.p). All inhibitors were injected 12 hrs before the induction of sepsis.
Cecal Ligation and Puncture
For induction of polymicrobial sepsis, mice underwent sham laparotomy or cecal ligation and puncture induced by ligation of the cecum and a double enterotomy created with either a 27 or 23 gauge needle(21, 22). With the smaller enterotomy (27 gauge needle), mortality was approximately 10-15%, while mortality with the larger enterotomy (23 gauge needle) was 20-30%. In both cases, death occurred predominantly within the first three days; thereafter, surviving mice developed abscesses surrounding the devitalized cecum as previously described (23-26). Survival analyses were performed with the larger enterotomy (23 gauge needle) in the CLP model with shams serving as surgery controls without infection in each experiment. Mice with CLP were pretreated with either vehicle control, heat inactivated goat serum with irrelevant IgG (500 μL/day i.p), or with anti-CXCL12 antisera (n=20 mice/group) (500 μL/day i.p anti-CXCL12) beginning 12 hours prior to the onset of sepsis and continuing daily for 8 days. The survival analyses were repeated twice.
Flow Cytometry
Spleens, bone marrow cells, and whole blood were analyzed by flow cytometry as previously described (21, 22, 25, 27) in sham and CLP mice created with the smaller enterotomy (27 gauge needle). Antibodies included anti-GR-1 (RB6-8C5) conjugated to PerCp5.5, anti-CD11b (Integrin aM, chain Mac-1a chain (M1/70)) conjugated to Pacific Blue, anti-F4/80 Antigen (Pan Macrophage Marker (BM8)) conjugated to APC, anti-CD31(MEC 13.3) conjugated to PE, Fc-Block (CD16/CD32 Fc g III/II Receptor (2.4G2)), Lineage Cocktail conjugated to biotin [CD3e(145-2C11), CD11b(M1/70), CD45R/B220(RA3-6B2), Ly6G and Ly6C(RB6-8C5), TER-119(TER-119)], Sca-1 conjugated to PE or PECy7 (D7), c-Kit conjugated to either FITC or APC (2B8). CD34 conjugated to either Alexa Fluor 647 or FITC (RAM34), FcγR conjugated to Pac Blue (CD16/32 clone 93), and Sytox Blue. F4/80, CD11b, CD34, and FcγR specific antibodies were purchased from eBioscience and all other antibodies were purchased from BD Pharmingen. Spleens, whole blood, and bone marrow were harvested at 12 hrs after either CLP or sham surgery and single cell suspensions were created by passing the cells through 70 μm pore sized cell strainers (Falcon). Erythrocytes were then lysed using ammonium chloride lysis buffer and washed two times using PBS without calcium, phenol red, or magnesium. Samples were acquired and analyzed using an LSRII flow cytometer (BD Biosciences). A minimum of 5 × 104 live, non-debris cells (Sytoxnegative) were collected for analysis.
Cytokine Production
At days 0, 1, 3, and 5 after sham or CLP procedure, whole blood was harvested by cardiac puncture and immediately centrifuged at 14,000 rpms for 10 minutes. The plasma supernatant was then carefully isolated and stored at -80°C until the time of analysis. The plasma samples were analyzed for cytokines using Luminex™ technology with reagents obtained from Upstate Cell Signaling Solutions (Beadlyte™ Mouse Multi-Cytokine Detection System) (Temecula, CA).
Real time RT-PCR
RNA was extracted using TRIzol™ (Invitrogen, Carlsbad, CA) and treated with DNase (1 unit/μg; Invitrogen) at room temperature for 15 minutes. The RNA was reverse-transcribed using Superscript II First Strand cDNA Synthesis (Invitrogen). Real-time PCR was performed with an Opticon 2 continuous fluorescence detector (MJ Research, Waltham, MA) using SYBR Green core reagents and AmpliTaq™ gold DNA polymerase from Perkin-Elmer Applied Biosystems (Foster City, CA). Gene expression was normalized to 18S rRNA (Ambion). Primers were designed using the Primer3 Output program (Whitehead Institute for Biomedical Research, Cambridge, MA) and were as follows: CXCR4 (NM_009911): Forward: CACGGCTGTAGAGCGAGTGT, Reverse: TGCCGACTATGCCAGTCAAG; CXCL12 (NM_001012477): Forward: AGCCCAAAGGACTTTCCAGT, Reverse: ACAAGGCATCTGTCGAGGAG; CXCR2: Forward: GGGAACTCCTTGGTGATGCT, Reverse: AGGTAGCGGTCCATGCTGAT; KC (NM_008176): Forward: ACTGCACCCAAACCGAAGTC, Reverse: TGTCAGAAGCCAGCGTTCAC; CXCL2 (NM_009149): Forward: ACTCTCAAGGGCGGTCAAAA, Reverse: GGCACATCAGGTACGATCCA. Optimal reaction concentrations were determined over 4 logs of linear amplification. Forty-five cycles of PCR were performed in duplicate for each primer. The fold change in gene transcript quantity compared with 18S rRNA was measured using the comparative (2-ΔΔCt) method, where Ct equals the cycle threshold.
Statistics
Continuous variables were first tested for normality and equality of variances. Differences among groups in flow cytometric analyses were evaluated by analysis of variance for multiple groups and Student’s t-test for two groups. Post-hoc comparisons were performed using Student-Newman Keuls multiple range tests. Statistical analysis for survival was performed using a Fisher’s Exact test of significance. In all cases, significance was designated at the 95% confidence level using a two-tailed test.
Results
Acute polymicrobial sepsis induces bone marrow granulocyte mobilization
During acute infection and sepsis, neutrophils are rapidly mobilized from the bone marrow into the peripheral circulation where they traverse to sites of inflammation. Using a modestly lethal cecal ligation and puncture (CLP) (LD10%) model of polymicrobial sepsis, we investigated several of the mediators that may be responsible for mobilizing neutrophils from the bone marrow into the peripheral circulation. At serial time points from 3 to 96 hours after the induction of sepsis, a dramatic difference was observed in the relative percentage and absolute number of total neutrophils in the femurs and blood between sham and CLP treated animals (Figure 1, Panels A & B). The neutrophil bone marrow decline and blood elevation was most apparent at 12 hours after CLP with a 50% reduction in the percentage of total bone marrow GR-1+CD11b+ cells and a concomitant three-fold increase in the percentage of total blood neutrophils, compared with sham and healthy naive control animals (Figure 1, Panels C & D).
Figure 1. Polymicrobial sepsis induces bone marrow neutrophil efflux into the circulation.
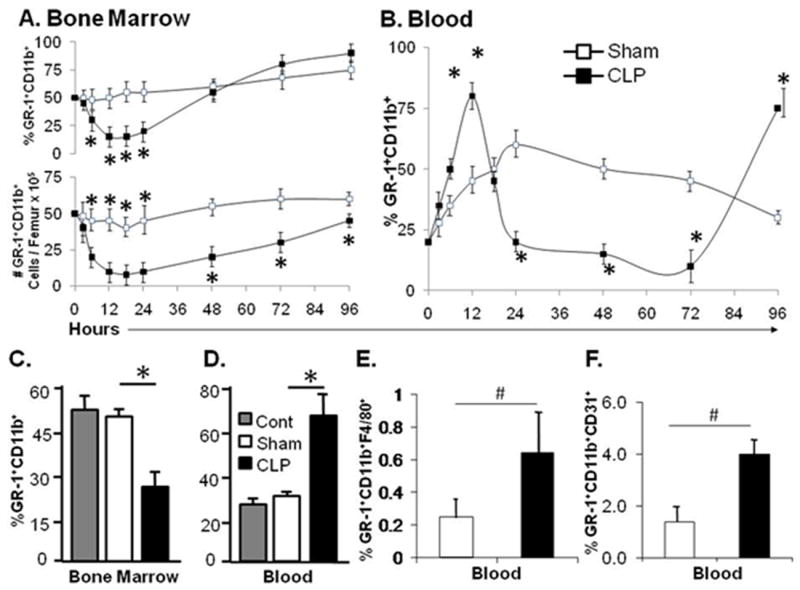
Panel A and B. Percentage and cell number of GR-1+CD11b+ neutrophil fluctuations in the bone marrow and blood 96 hours after polymicrobial sepsis. Panels C and D. Percentage of total GR-1+CD11b+ neutrophils in the blood and bone marrow 12 hours after sham or cecal ligation and puncture procedure (CLP). Panel E. Percentage of the total GR-1+CD11b+ population that is F4/80+ in the blood after either sham or CLP induced sepsis. Panel F. Percentage of the total GR-1+CD11b+ population that stain positive for CD31 in the blood after either sham or CLP induced sepsis. Values for panels A-E represent the mean and standard error of 5 animals per group from three independent experiments. B and C * p<0.01 by analysis of variance between Cont (healthy animal), Sham, and CLP treated mice. D and E # p<0.01, by Student’s t-test between Sham and CLP.
The bone marrow myeloid compartment consists of a large, heterogenous, population of cells that possess both mature and immature monocyte, macrophage, dendritic cell, erythroid, and neutrophil phenotypes (28). During episodes of inflammatory stress and acute infection, the vast majority of cells released from the bone marrow consist of mature neutrophils with lymphocytes and monocytes comprising the minority of cells (data not shown) (9, 10, 28). Immune phenotyping with the cell surface markers F4/80, a marker of monocytes and macrophages, and CD31, a marker of immature cells, demonstrates that less than one percent of the total blood GR-1+CD11b+ population expressed F4/80, and less than five percent are CD31 positive 12 hours after sepsis initiation (Figure 1 Panels, E & F). As neutrophils mature Ly6G and CD11b cell surface expression becomes more dense. Cell surface analysis revealed that the blood neutrophils at 12 hrs after sepsis induction have a high cell surface density of both Ly6G and CD11b compared to the remaining immature bone marrow neutrophils (data not shown). The immune phenotype of the circulating GR-1+CD11+ population is consistent with that of more mature granulocytes.
Bone marrow neutrophil release is independent of CXCR2 signaling during polymicrobial sepsis
Increases in the expression of chemokines CXCL1 and CXCL2 are used as indicators of acute inflammation and have been associated with peripheral recruitment of neutrophils to sites of local infection(29). In order to understand and characterize the comprehensive impact of polymicrobial sepsis on bone marrow neutrophil release, we first characterized the expression of CXCL1, CXCL2, and CXCR2 mRNA in total bone marrow, blood, and spleen cells 12 hours after CLP. The data demonstrate a dramatic elevation in both the total bone marrow and splenocyte mRNA abundance of CXCL1 at 12 hours. Plasma CXCL1 concentrations peak at 24 hours after the initiation of sepsis (Figure 2, panels A, B and C). Contrary to CXCL1, CXCL2 mRNA abundances actually decline in the bone marrow after sepsis, while mRNA abundances increase in the spleen. Likewise, CXCR2 expression changes in a similar trend compared to CXCL2 expression, with reductions in the mRNA abundance in the bone marrow and elevations in the spleen (Figure 2, panel D). The data suggests that elevations in peripheral CXCL1 and CXCL2 mRNA and chemokine levels coincide temporally with blood neutrophilia and may be involved in bone marrow neutrophil release during polymicrobial sepsis.
Figure 2. Neutrophil egression from the bone marrow does not depend on CXCR2 signaling during sepsis.
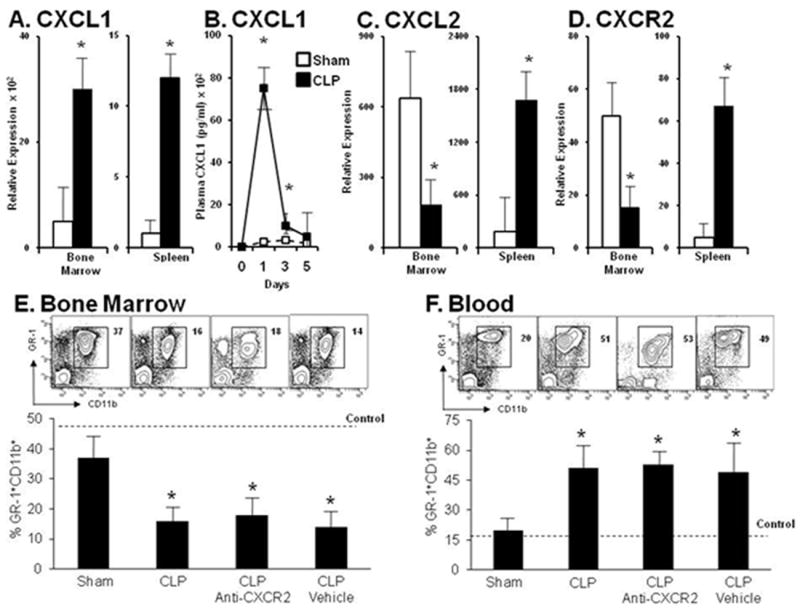
Panel A. Relative transcription level of CXCL1 in the bone marrow and spleen at 12 hours after the induction of sepsis. Panel B. Blood plasma elevation of CXCL1 levels extends through day 5 compared with sham levels. Panel C. Relative transcriptional level of CXCL2 in the bone marrow and spleen in sham and CLP treated mice12 hours after sepsis. Panels D. Relative transcriptional level of CXCR2 in the bone marrow and spleen of sham and CLP treated mice 12 hours after sepsis. Panel E. Depicts representative contour plots and bar graphs of GR-1+CD11b+ levels in the bone marrow of mice receiving CXCR2 blockade or vehicle control in sham and CLP treated animals. Panel F. Depicts representative contour plots and bar graphs of GR-1+CD11b+ levels in the blood of mice receiving CXCR2 blockade or vehicle control in sham and CLP treated animals. Values for panels A-D represent the mean and standard error of 5-7 animals per group from two independent experiments. Values for panels E and F represent the mean and standard error of 5 animals per group from three independent experiments. A, C, D, E and F * p<0.01 by Student’s t-test between sham and CLP groups. B *p<0.05 by analysis of variance and a post hoc Dunn’s test of significance at each time point compared to time point 0.
Based on our prior results, we sought to determine the impact of CXCR2 blockade on neutrophil bone marrow release during polymicrobial sepsis. Incorporating an anti-CXCR2 antibody which blocks ligand binding to CXCR2, we investigated the implications of CXCR2 blockade on bone marrow neutrophil release during polymicrobial infection. Mice were pretreated by i.p. injection with anti-CXCR2 antibodies or vehicle control 12 hours before the onset of CLP. Mouse bone marrow, blood, and spleen cells were harvested 12 hours after the induction of sepsis and analyzed for the total percentages of GR-1+CD11b+ cells. Sepsis produces a 50% reduction in the bone marrow granulocyte population at 12 hours (Figure 2, panel E). However, CXCR2 blockade has no significant effect on either the reduction in the numbers or percentages of GR-1+CD11b+ cells in the bone marrow. Concomitantly, sepsis produces a more than two-fold increase in the percentages of total blood granulocytes in the septic mice compared to mice undergoing sham procedure (Figure 2, panel F), and CXCR2 blockade has no effect on this response.
Bone marrow neutrophil release is also not dependent on toll-like receptor signaling
Interruption of TLR signaling during acute infection has been associated with significant reductions in peripheral CXCL1 and CXCL2 levels(19, 20) which may inhibit neutrophil bone marrow mobilization and peripheral recruitment. Even though our experiments demonstrate that neutrophil bone marrow mobilization is not dependent on CXCR2 signaling, other reports indicate that peripheral neutrophil recruitment requires intact TLR signaling (30). Because TLR signaling occurs through MyD88 and TRIF dependent pathways, and may also involve type I interferon (IFN) signaling, the CLP model of polymicrobial sepsis was performed in MyD88−/−, TRIF−/−, IFN-α/ßR−/− and C3H/HeJ (TLR4 mutant) mice. As shown in Figure 3, panels A and B, mice devoid of MyD88 signaling demonstrate no deficit in neutrophil mobilization from the bone marrow into the peripheral circulation. We next induced polymicrobial sepsis in the TRIF−/−, IFN-α/ßR−/− and C3H/HeJ mice and find no attenuation in neutrophil reduction in the bone marrow (Figure 3, panels C, D and E) and elevation in the blood (data not shown).
Figure 3. Effects of transgenic mice on bone marrow neutrophil egression during polymicrobial sepsis.

Panels A and B. Changes in the percentage of total neutrophils (GR-1+CD11b+ cells) in the bone marrow and blood of MyD88-/- and wild type control C57BL/6 or B6.129 mice 12 hours after either sham or sepsis induction. MyD88-/-animals demonstrated similar bone marrow neutrophil mobilization into the circulation compare to wild type control mice. Panels C, D and E represent the bone marrow release of neutrophils 12 hours after either sepsis or sham procedure in IFNα/βR-/-, TRIF-/-, or C3H/HeJ (TLR4 mutant) transgenic mice respectively. There was no attenuation in the bone marrow neutrophil reduction in any of these transgenic animals. Values for panels A-E represent the mean and standard error of 5-7 animals per group from three independent experiments. A-E * p<0.01 by Student’s t-test between Sham and CLP groups.
CXCL12 expression changes in the bone marrow and spleen during acute infection
Considering the previous results, we hypothesized that neutrophil bone marrow egression may be dependent on CXCL12 signaling during polymicrobial sepsis. To understand the impact of CLP-induced sepsis on the fluctuation of CXCL12, we first characterized the mRNA abundance of CXCL12 and CXCR4 in total bone marrow and spleen cells at 12 hours after sepsis induction. The results demonstrate a reciprocal change between the bone marrow and spleen CXCL12 expression after sepsis, with the CXCL12 mRNA abundance and protein levels in total bone marrow cells significantly reduced while mRNA abundance and protein levels in the spleen remain constant or are modestly elevated (Figure 4 panel A, B). The significant reduction in bone marrow CXCL12 mRNA and protein expression during sepsis is not surprising given the dramatic mobilization of neutrophils from bone marrow to the peripheral circulation. The decrease in total bone marrow CXCL12 transcripts and protein levels agrees with the work of Semerad et al., who demonstrated that sterile inflammation also induces a decrease in the bone marrow expression of CXCL12 (12). Considering the splenic accumulation of immature myeloid cells that occurs after sepsis, the fact that splenic CXCL12 mRNA and protein levels are modestly elevated in septic animals compared with sham controls is not surprising.
Figure 4. CXCL12 Blockade inhibits neutrophil egression from the bone marrow into the systemic circulation.
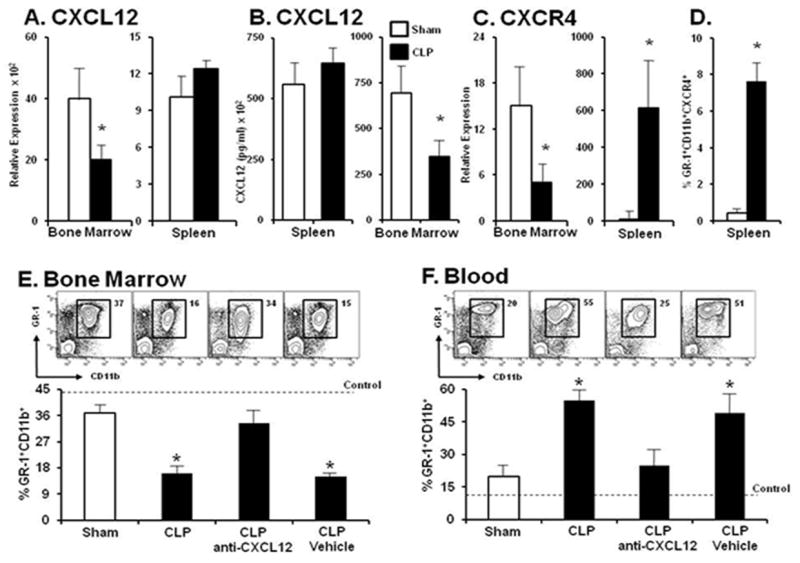
Panel A. Relative transcription level of CXCL12 in total bone marrow and spleen cells harvested at 12 hours after the induction of either sham procedure or CLP sepsis. Panel B. Splenic CXCL12 chemokine level in total splenocytes and bone marrow aspirates at 12 hours after sham or sepsis. Panel C, Relative expression of the chemokine receptor CXCR4 in total bone marrow and spleen cells between sham and CLP treated mice 12 hours after sepsis. Panel D. Percentage of GR-1+CD11b+ cells that express CXCR4 in the spleen at 12 hours after sham procedure or CLP induction. Panel E. Fluctuations in the percentage of total GR-1+CD11b+ neutrophils in the bone marrow in the presence or absence of CXCL12 inhibition 12 hours after sham procedure or CLP sepsis. CXCL12 blockade inhibited neutrophil release from the bone marrow. Panel F. Neutrophil elevation in the blood was inhibited by CXCL12 blockade. Values for panels A-C represent the mean and standard error of 5-7 animals per group from three independent experiments. A-C * p<0.01 by Student’s t-test between Sham and CLP groups. Values for panels D and E represent the mean and standard error of 5-7 animals per group from 4 independent experiments. A and B * p<0.01 by Student’s t-test between Sham and CLP groups. Dashed line represents neutrophils observed in healthy naive wild type C57BL/6 mice.
Similar to the reciprocal changes in CXCL12 mRNA abundance in the bone marrow and spleen are the changes in CXCR4 expression which are dramatically reduced in the bone marrow and elevated in the spleen during sepsis. The increased expression of CXCR4 in the spleen was confirmed at the protein level by cell surface expression analysis (data not shown). Although the total number of splenic GR-1+CD11b+ granulocytes is reduced early after sepsis(21, 22), a greater number of the remaining splenic granulocytes express over six-fold more CXCR4 on their cells surface compared to sham controls (Figure 4, panels C and D).
Using an anti-CXCL12 antisera, we investigated the impact of CXCL12 blockade on bone marrow neutrophil release during polymicrobial infection. In healthy mice, such treatments would be expected to result in a bone marrow neutropenia and marked blood neutrophilia (13-15) by disrupting the CXCR4/CXCL12 interaction culminating in bone marrow neutrophil mobilization (11). G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4 (11). CXCR4 desensitization is associated with tissue localization of hemopoietic progenitor cells (31); however their effects in sepsis are unknown. Mice were pretreated by i.p. injection with either anti-CXCL12 or control antisera twelve hours before the onset of sepsis. Mouse bone marrow, blood, and spleen were harvested 12 hours after CLP and analyzed for the total percentages of GR-1+CD11b+ cells. Expectedly, sepsis results in a greater than 50% reduction in the bone marrow granulocyte population compared to the sham group. In contrast, mice that received CXCL12 blockade exhibit little reduction in the total percentages of bone marrow GR-1+CD11b+ cells, with granulocyte levels equivalent to sham animals (Figure 4, panel E). Concomitantly, sepsis produces a more than twofold increase in the percentages of total blood granulocytes, while treatment with anti-CXCL12 completely prevents the elevation in blood granulocyte levels (Figure 4, panel F).
CXCL12 blockade attenuates bone marrow lymphocyte reduction during acute polymicrobial infection
Given the fact that CXCR4/CXCL12 signaling has not only been implicated in hematopoietic progenitor cell release from the bone marrow (11), but also hematopoietic stem and progenitor cells proliferation and differentiation (32, 33), we sought to determine whether CXCL12 blockade imposed any upstream effects on neutrophil precursors in the bone marrow during CLP-induced sepsis. Twelve hours after the initiation of polymicrobial sepsis, total bone marrow cells were harvested and the major myeloid lineage precursor populations were evaluated in the presence or absence of CXCL12 blockade. Since granulocyte production begins with lineagenegativesca-1positivec-kitpositive hematopoietic stem cells (LSKs), which generate lineagenegativesca-1negativec-kitpositiveCD34positiveFcλRlow common myeloid progenitor cells (CMPs), which further differentiate into lineagenegativesca-1negativec-kitpositiveCD34positiveFcλRhigh granulocyte-monocyte progenitors (GMPs) that ultimately develop into immature neutrophils (34), we chose to evaluated the bone marrow LSK, CMP, and GMP populations after the induction of sepsis in the presence or absence of CXCL12 blockade (Figure 5, panels A, B, & C). The results demonstrate that CXCL12 blockade has no significant effect on the bone marrow LSK, CMP, and GMP populations, suggesting that CXCL12 does not regulate neutrophil or neutrophil precursor development during acute polymicrobial infection.
Figure 5. CXCL12 blockade attenuates B-lymphocyte efflux from the bone marrow during polymicrobial sepsis.
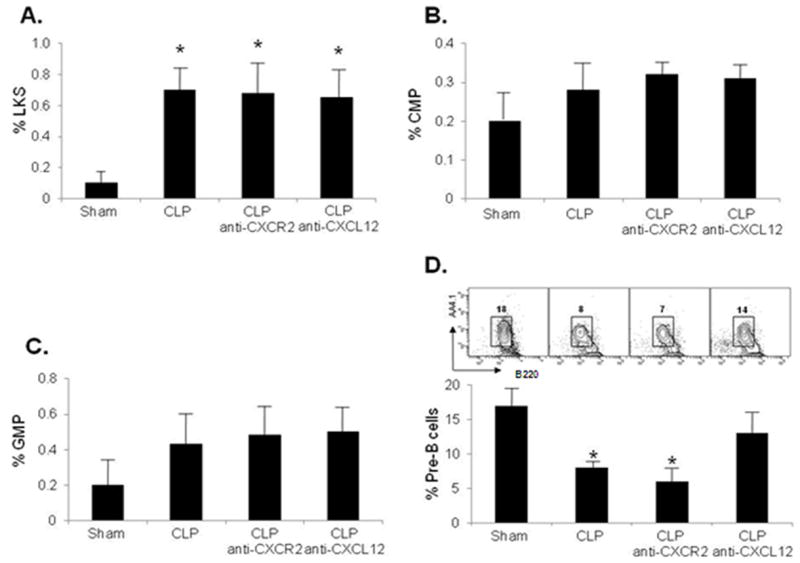
Panels A, B and C. Either CXCL12 or CXCR2 blockade have no impact on the bone marrow levels of hematopoietic stem cell (Panel A), common myeloid progenitor cell (Panel B), or granulocyte-monocyte progenitor cell (Panel C) levels in the bone marrow during polymicrobial sepsis. Panel D. Immature B lymphocyte CD19+AA4.1highB220intermediate bone marrow efflux is attenuated by CXCL12 blockade and not CXCR2 inhibition during polymicrobial sepsis. Changes in the B lymphocyte bone marrow levels are evident from the representative contour plots in each group. Values for panels A through D represent the mean and standard error of 5-7 animals per group from 3 independent experiments. A-D *p<0.05 analysis of variance and a post hoc Dunn’s test of significance for each group compared to sham animals.
The CXCR4/CXCL12 signaling axis also regulates B lymphopoiesis and myelopoiesis by confining these precursors within the supportive fetal liver and bone marrow microenvironments for further maturation (15). In response to non-infectious stimuli, bone marrow B-lymphocytes and mature granulocytes are mobilized to the peripheral circulation through a CXCL12 associated mechanism(34). Since our prior data demonstrates that CXCL12 inhibition during polymicrobial sepsis attenuated the release and ultimate reduction in bone marrow neutrophils (Figure 4), we surmised that CXCL12 blockade should alleviate the bone marrow B lymphocyte reduction during sepsis. CXCL12 blockade during sepsis did indeed attenuate the inflammation-associated mobilization of CD19+AA4.1highB220intermediate immature B lymphocytes compared with sepsis alone (Figure 5, panel D). These results underscore the codependence of granulocyte and B lymphocyte bone marrow egression on CXCL12 signaling during polymicrobial sepsis.
CXCL12 blockade reduces bone marrow neutrophil mobilization and peritoneal accumulation, leading to bacterial expansion during polymicrobial sepsis
Our studies thus far suggested that CXCL12 signaling is vital for bone marrow mobilization of neutrophils during the early response to sepsis. We next determined whether CXCL12 blockade affected neutrophil mobilization during later phases of sepsis. Using an anti-CXCL12 antibody we investigated the impact of CXCL12 blockade on bone marrow neutrophil release during polymicrobial sepsis over a 96 hour period. Mice were pretreated with either anti-CXCL12 or control antisera twelve hours before the onset of sepsis. Mouse bone marrow, blood, and peritoneal washings were harvested at 3 to 96 hours after sham surgery or CLP and the relative percentages and absolute numbers of GR-1+CD11b+ cells were determined. There was a significant reduction in the bone marrow neutrophil population in the CLP group compared to the sham group and while CXCL12 blockade prevented this reduction (Figure 6, Panels A & B). Moreover, while the sham and CLP-treated animals exhibited dramatic elevations in blood neutrophil numbers, the CLP mice pretreated with CXCL12 blocking antibody displayed only a modest elevation in blood neutrophil numbers (Figure 6, Panel C).
Figure 6. CXCL12 blockade inhibits peritoneal neutrophil accumulation and allows bacterial proliferation following sepsis.
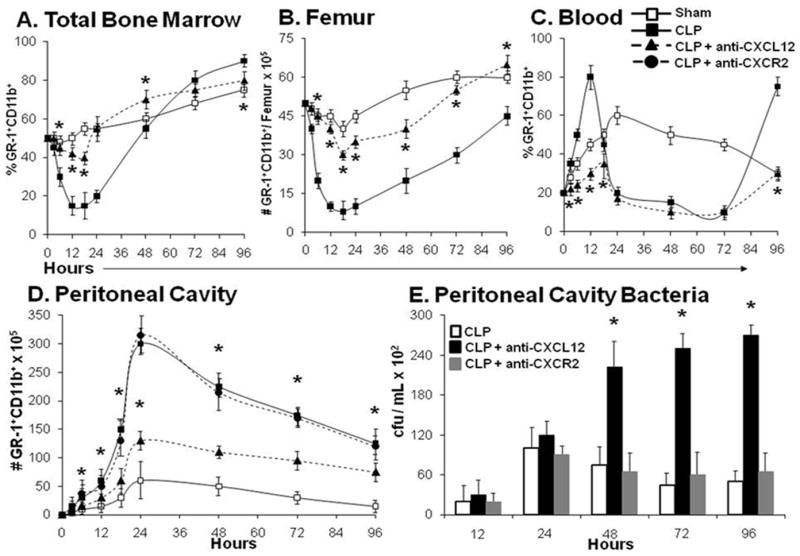
Anti-CXCL12 or anti-CXCR2 were administered 12 hours before the onset of sepsis and once daily for the next 4 days. Panels A-D, Demonstrate the effect of CXCL12 blockade over a 96 hour period following sham or CLP sepsis on the relative percentage and absolute number of bone marrow, blood and peritoneal cavity neutrophils. Panel E. CXCL12 blockade enhanced intra-peritoneal bacterial proliferation following CLP sepsis. Values for panels A through E represent the mean and standard error of 5-7 animals per group from 3 independent experiments. A-D p<0.05 analysis of variance and a post hoc Dunn’s test of significance for each group compared to sham animals. *p<0.01 CLP-anti-CXCL12 compared with CLP alone. Panel E. *p<0.01 CLP-anti-CXCL12 compared with CLP alone using a Student’s t test.
Since neutrophils are essential for host survival during sepsis, we examined whether CXCL12 blockade also affects neutrophil recruitment to the peritoneum. Peritoneal lavage from mice pretreated with i.p. injection of anti-CXCL12 twelve hours prior to the onset of CLP revealed significant reductions in the absolute number of peritoneal neutrophils beginning at six and extending through 96 hours when compared to CLP treatment alone (Figure 6D). The reduction in blood and peritoneal neutrophil numbers in anti-CXCL12 treated septic mice coincides with significantly diminished bacterial clearance in these animals (Figure 6E). This data suggest that CXCL12 inhibition prohibits bone marrow neutrophil mobilization, which results in blood and peritoneal neutropenia and unopposed bacterial invasion.
CXCL12 blockade reduces survival during polymicrobial sepsis
Neutrophils are a fundamental component of the innate immune system and essential for successful host eradication of bacterial pathogens. During episodes of sepsis or bacterial challenge, the bone marrow provides a large reserve of neutrophils that are released into the peripheral circulation and home to sites of infection(9). Numerous reports have demonstrated that survival to polymicrobial sepsis is dependent on neutrophil-mediated microbe eradication at local sites of infection (4, 5). In order to determine the significance of CXCL12 blockade on neutrophil bone marrow egression and polymicrobial sepsis survival, we examined survival of animals receiving CXCL12 inhibition for seven days with mice receiving vehicle control alone after the induction of (LD30) polymicrobial sepsis. The absolute survival in the mice receiving CXCL12 blockade is reduced by 40%, such that 70% of the CXCL12 anti-sera treated mice die (Figure 7).
Figure 7. CXCL12 blockade reduces survival to polymicrobial sepsis.
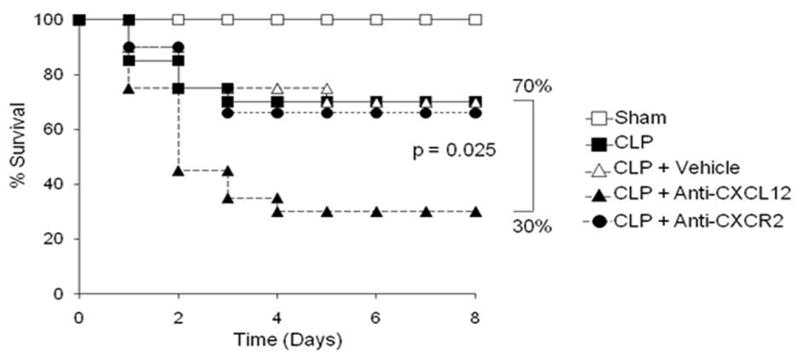
Anti-CXCL12 or anti-CXCR2 were administered 12 hours before the onset of sepsis and once daily for the next 7 days. CXCL12 blockade produced a significant reduction (40%) in survival following the initiation of polymicrobial sepsis. The survival analysis was repeated two times with n=20 mice/group in each experiment. Statistical analysis was done using a Fisher’s Exact test of significance p=0.025.
Discussion
Survival to polymicrobial sepsis requires the mobilization of functional neutrophils to local sites of infection (4, 5). However, few studies have investigated the mechanisms involved in bone marrow mobilization of neutrophils in response to a polymicrobial sepsis challenge. Although previous studies have identified several mediators capable of bone marrow neutrophil mobilization (19, 20), few studies have focused on the mechanisms of neutrophil release from the bone marrow during a microbial challenge. Although colony stimulating factors (CSFs) can induce BM neutrophil mobilization, in part through down-regulating CXCR4 (35-37), deletion of G-CSF does not affect neutrophil mobilization in response to Candida albicans infection (35). During episodes of acute streptococcal infection, release of neutrophils into the blood is accelerated; however, the specific mechanisms mediating that release were not investigated (35, 38-40).
In this study we used a global approach to identify signaling pathways involved in polymicrobial sepsis induced neutrophil mobilization. The results indicate that bone marrow neutrophil mobilization into the circulation is not dependent on TLR4, MyD88, TRIF, IFNARα/β or CXCR2 signaling pathways. In contrast, CXCL12 blockade prevented the release of neutrophils from the bone marrow during sepsis, resulting in a failure to increase blood and peritoneal neutrophil counts. CXCL12 blockade during sepsis was accompanied by a 5 fold increase in peritoneal bacteria colony forming units when compared with CLP treatment alone. This failure to clear bacteria led to a 40% increase in sepsis mortality in anti-CXCL12 treated mice. These findings are in striking contrast to the role that CXCR4/CXCL12 plays in neutrophil release from bone marrow in the healthy animal. In this case, local bone marrow signaling through CXCR4/CXCL12 appears to be important in retention of neutrophils and immature B-cells in the bone marrow. Blocking the CXCR4/CXCL12 axis causes neutrophilia in the otherwise healthy animal (13-15). However, unlike the healthy animal, there are marked alterations in the expression of CXCR4 and CXCL12 in polymicrobial sepsis which can explain the differential effects of blockade. Although CXCR4/CXCL12 signaling plays an essential role in bone marrow retention of neutrophils in healthy naive animals, changes in the expression of CXCL12 in peripheral tissues may establish gradients that promote neutrophil emigration from the bone marrow during sepsis. During steady state exogenous CXCL12 administration has a modest effect on bone marrow neutrophil mobilization, but during malarial infection, CXCL12 levels are dramatically increased in the periphery (41, 42). Moreover exogenous CXCL12 administration during malarial infection results in a reduction in malarial loads(42). Although this evidence suggests that CXCR4/CXCL12 signaling may be necessary for host defense to infection, the role of peripheral CXCL12 elevation on neutrophil mobilization is still unclear. Our data indicate that polymicrobial sepsis decreases CXCR4 and CXCL12 expression in the bone marrow, but increases the expression of CXCR4 and CXCL12 in the spleen. This data supports the hypothesis that a CXCL12 gradient may exist between the bone marrow and the periphery, and may be pivotal for neutrophil release during infection. Similarly, this may explain the difference in the effects of CXCR4 blockade in healthy animals associated with neutrophil release from the bone marrow, whereas CXCL12 blockade during sepsis or inflammatory conditions impedes bone marrow neutrophil release by prohibiting peripheral CXCL12 gradients as mentioned above and as observed in pathologic conditions like acute mylogenous leukemia (43). Although CXCL12 blockade also inhibited immature B-cell efflux from the bone marrow as would be expected based on the data of Ueda and coauthors (34), it is not unreasonable to mention that the anti-CXCL12 effect may stem from an anti-apoptotic property that serves to inhibit B-cell apoptosis that occurs during sepsis (44).
Much to our surprise, disruption of either TLR signaling or CXCR2 pathways has no significant effect on the neutrophil response to polymicrobial sepsis. Although increases in CXCL1 and CXCL2 levels have been the hallmark of inflammation in murine models of polymicrobial sepsis, their relevance on neutrophil bone marrow mobilization has been rarely observed. This notion is supported by recent findings by Link and coauthors who suggest that CXCR2 signaling is a second chemokine axis that interacts antagonistically with CXCR4 during neutrophil release from the bone marrow (16). Holzmann and colleagues demonstrated that the production of CXCL1 and CXCL2 during polymicrobial sepsis is dependent on intact TLR signaling through the MyD88 pathway (19). Further work by Ayala demonstrated that antibody inhibition of CXCL2, but not CXCL1, resulted in reduced neutrophil accumulation in the lung following polymicrobial sepsis (29). Our initial findings that CXCL1, CXCL2 and CXCR2 levels increase in response to infection, provided early enthusiasm that neutrophil emigration from the bone marrow may depend on those chemokine elevations during polymicrobial sepsis. Furthermore, coupled with the evidence provided by Holzmann implicating TLR signaling in general, and MyD88 signaling in particular, as paramount for CXCL1 and CXCL2 elevations during sepsis, we were optimistic that neutrophil bone marrow release would ultimately be inhibited by disrupting TLR signaling through either the MyD88, TRIF, or IFNAR pathways. However, neutrophil release from the bone marrow was not inhibited by disruption of TLR or CXCR2 pathways.
In conclusion, we provide evidence that neutrophil efflux from the bone marrow during acute infection depends on CXCL12/CXCR4 signaling, and not on inflammatory TLR or CXCR2 signaling. We further identify CXCL12 as an important survival factor in polymicrobial sepsis necessary for both neutrophil mobilization from the bone marrow and neutrophil recruitment to peripheral sites of infection. Without CXCL12, there is a failure in pathogen clearance, resulting in increased sepsis mortality.
Acknowledgments
This work was supported by grants R37 GM-40586 and R01 GM-62041 (LLM), awarded by the National Institute of General Medical Sciences. U.S.P.H.S. MJD, KAO and AGC were all supported by a training grant in burn and trauma research (T32 GM-08431); RDW was supported by a training grant in cancer research (T32 CA 106493) awarded by the National Cancer Institute.
Bibliography
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 3.Hotchkiss RS, I, Karl E. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 4.Belaaouaj A, McCarthy R, Baumann M, Gao Z, Ley TJ, Abraham SN, Shapiro SD. Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat Med. 1998;4:615. doi: 10.1038/nm0598-615. [DOI] [PubMed] [Google Scholar]
- 5.Czermak BJ, Sarma V, Pierson CL, Warner RL, Huber-Lang M, Bless NM, Schmal H, Friedl HP, Ward PA. Protective effects of C5a blockade in sepsis. Nat Med. 1999;5:788. doi: 10.1038/10512. [DOI] [PubMed] [Google Scholar]
- 6.Dunn DL. Antibody immunotherapy of gram-negative bacterial sepsis in an immunosuppressed animal model. Transplantation. 1988;45:424. doi: 10.1097/00007890-198802000-00036. [DOI] [PubMed] [Google Scholar]
- 7.Kollef MH, Schuster DP. The acute respiratory distress syndrome. N Engl J Med. 1995;332:27. doi: 10.1056/NEJM199501053320106. [DOI] [PubMed] [Google Scholar]
- 8.Semerad CL, Liu F, Gregory AD, Stumpf K, Link DC. G-CSF is an essential regulator of neutrophil trafficking from the bone marrow to the blood. Immunity. 2002;17:413. doi: 10.1016/s1074-7613(02)00424-7. [DOI] [PubMed] [Google Scholar]
- 9.Metcalf D. Control of granulocytes and macrophages: molecular, cellular, and clinical aspects. Science. 1991;254:529. doi: 10.1126/science.1948028. [DOI] [PubMed] [Google Scholar]
- 10.Christopher MJ, Link DC. Regulation of neutrophil homeostasis. Curr Opin Hematol. 2007;14:3. doi: 10.1097/00062752-200701000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Petit I, Szyper-Kravitz M, Nagler A, Lahav M, Peled A, Habler L, Ponomaryov T, Taichman RS, Arenzana-Seisdedos F, Fujii N, Sandbank J, Zipori D, Lapidot T. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002;3:687. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- 12.Semerad CL, Christopher MJ, Liu F, Short B, Simmons PJ, Winkler I, Levesque JP, Chappel J, Ross FP, Link DC. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood. 2005;106:3020. doi: 10.1182/blood-2004-01-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, Liles WC, Li X, Graham-Evans B, Campbell TB, Calandra G, Bridger G, Dale DC, Srour EF. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005;201:1307. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liles WC, Broxmeyer HE, Rodger E, Wood B, Hubel K, Cooper S, Hangoc G, Bridger GJ, Henson GW, Calandra G, Dale DC. Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood. 2003;102:2728. doi: 10.1182/blood-2003-02-0663. [DOI] [PubMed] [Google Scholar]
- 15.Ma Q, Jones D, Springer TA. The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity. 1999;10:463. doi: 10.1016/s1074-7613(00)80046-1. [DOI] [PubMed] [Google Scholar]
- 16.Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. 2010;120:2423. doi: 10.1172/JCI41649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430:257. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- 18.Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001;2:947. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- 19.Weighardt H, Kaiser-Moore S, Vabulas RM, Kirschning CJ, Wagner H, Holzmann B. Cutting edge: myeloid differentiation factor 88 deficiency improves resistance against sepsis caused by polymicrobial infection. J Immunol. 2002;169:2823. doi: 10.4049/jimmunol.169.6.2823. [DOI] [PubMed] [Google Scholar]
- 20.Ramphal R, Balloy V, Huerre M, Si-Tahar M, Chignard M. TLRs 2 and 4 are not involved in hypersusceptibility to acute Pseudomonas aeruginosa lung infections. J Immunol. 2005;175:3927. doi: 10.4049/jimmunol.175.6.3927. [DOI] [PubMed] [Google Scholar]
- 21.Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, O’Malley K A, Wynn JL, Antonenko S, Al-Quran SZ, Swan R, Chung CS, Atkinson MA, Ramphal R, Gabrilovich DI, Reeves WH, Ayala A, Phillips J, Laface D, Heyworth PG, Clare-Salzler M, Moldawer LL. MyD88-dependent expansion of an immature GR-1+CD11b+ population induces T cell suppression and Th2 polarization in sepsis. J Exp Med. 2007;204:1463. doi: 10.1084/jem.20062602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delano MJ, Thayer T, Gabrilovich S, Kelly-Scumpia KM, Winfield RD, Scumpia PO, Cuenca AG, Warner E, Wallet SM, Wallet MA, O’Malley KA, Ramphal R, Clare-Salzer M, Efron PA, Mathews CE, Moldawer LL. Sepsis induces early alterations in innate immunity that impact mortality to secondary infection. J Immunol. 2011;186:195. doi: 10.4049/jimmunol.1002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moldawer LL, Efron PA. Cecal ligation and puncture. Curr Protoc Immunol. 2010;Chapter 19(Unit 19 13) doi: 10.1002/0471142735.im1913s91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scumpia PO, Delano MJ, Kelly-Scumpia KM, Weinstein JS, Wynn JL, Winfield RD, Xia C, Chung CS, Ayala A, Atkinson MA, Reeves WH, Clare-Salzler MJ, Moldawer LL. Treatment with GITR agonistic antibody corrects adaptive immune dysfunction in sepsis. Blood. 2007;110:3673. doi: 10.1182/blood-2007-04-087171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scumpia PO, Delano MJ, Kelly KM, O’Malley KA, Efron PA, McAuliffe PF, Brusko T, Ungaro R, Barker T, Wynn JL, Atkinson MA, Reeves WH, Salzler MJ, Moldawer LL. Increased natural CD4+CD25+ regulatory T cells and their suppressor activity do not contribute to mortality in murine polymicrobial sepsis. J Immunol. 2006;177:7943. doi: 10.4049/jimmunol.177.11.7943. [DOI] [PubMed] [Google Scholar]
- 26.Oberholzer A, Oberholzer C, Bahjat KS, Ungaro R, Tannahill CL, Murday M, Bahjat FR, Abouhamze Z, Tsai V, LaFace D, Hutchins B, Moldawer LL, Clare-Salzler MJ. Increased survival in sepsis by in vivo adenovirus-induced expression of IL-10 in dendritic cells. J Immunol. 2002;168:3412. doi: 10.4049/jimmunol.168.7.3412. [DOI] [PubMed] [Google Scholar]
- 27.Wynn JL, Scumpia PO, Winfield RD, Delano MJ, Kelly-Scumpia K, Barker T, Ungaro R, Levy O, Moldawer LL. Defective innate immunity predisposes murine neonates to poor sepsis outcome but is reversed by TLR agonists. Blood. 2008;112:1750. doi: 10.1182/blood-2008-01-130500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cyster JG. Lymphoid organ development and cell migration. Immunol Rev. 2003;195:5. doi: 10.1034/j.1600-065x.2003.00075.x. [DOI] [PubMed] [Google Scholar]
- 29.Lomas JL, Chung CS, Grutkoski PS, LeBlanc BW, Lavigne L, Reichner J, Gregory SH, Doughty LA, Cioffi WG, Ayala A. Differential effects of macrophage inflammatory chemokine-2 and keratinocyte-derived chemokine on hemorrhage-induced neutrophil priming for lung inflammation: assessment by adoptive cells transfer in mice. Shock. 2003;19:358. doi: 10.1097/00024382-200304000-00011. [DOI] [PubMed] [Google Scholar]
- 30.Miller LS, O’Connell RM, Gutierrez MA, Pietras EM, Shahangian A, Gross CE, Thirumala A, Cheung AL, Cheng G, Modlin RL. MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. Immunity. 2006;24:79. doi: 10.1016/j.immuni.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 31.Shen H, Cheng T, Olszak I, Garcia-Zepeda E, Lu Z, Herrmann S, Fallon R, Luster AD, Scadden DT. CXCR-4 desensitization is associated with tissue localization of hemopoietic progenitor cells. J Immunol. 2001;166:5027. doi: 10.4049/jimmunol.166.8.5027. [DOI] [PubMed] [Google Scholar]
- 32.Nie Y, Han YC, Zou YR. CXCR4 is required for the quiescence of primitive hematopoietic cells. J Exp Med. 2008;205:777. doi: 10.1084/jem.20072513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stellos K, Langer H, Daub K, Schoenberger T, Gauss A, Geisler T, Bigalke B, Mueller I, Schumm M, Schaefer I, Seizer P, Kraemer BF, Siegel-Axel D, May AE, Lindemann S, Gawaz M. Platelet-derived stromal cell-derived factor-1 regulates adhesion and promotes differentiation of human CD34+ cells to endothelial progenitor cells. Circulation. 2008;117:206. doi: 10.1161/CIRCULATIONAHA.107.714691. [DOI] [PubMed] [Google Scholar]
- 34.Ueda Y, Yang K, Foster SJ, Kondo M, Kelsoe G. Inflammation controls B lymphopoiesis by regulating chemokine CXCL12 expression. J Exp Med. 2004;199:47. doi: 10.1084/jem.20031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Basu S, Hodgson G, Zhang HH, Katz M, Quilici C, Dunn AR. “Emergency” granulopoiesis in G-CSF-deficient mice in response to Candida albicans infection. Blood. 2000;95:3725. [PubMed] [Google Scholar]
- 36.Gregory AD, Hogue LA, Ferkol TW, Link DC. Regulation of systemic and local neutrophil responses by G-CSF during pulmonary Pseudomonas aeruginosa infection. Blood. 2007;109:3235. doi: 10.1182/blood-2005-01-015081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, Fowler KJ, Basu S, Zhan YF, Dunn AR. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. 1994;84:1737. [PubMed] [Google Scholar]
- 38.Lawrence E, Van Eeden S, English D, Hogg JC. Polymorphonuclear leukocyte (PMN) migration in streptococcal pneumonia: comparison of older PMN with those recently released from the marrow. Am J Respir Cell Mol Biol. 1996;14:217. doi: 10.1165/ajrcmb.14.3.8845171. [DOI] [PubMed] [Google Scholar]
- 39.Sato Y, van Eeden SF, English D, Hogg JC. Bacteremic pneumococcal pneumonia: bone marrow release and pulmonary sequestration of neutrophils. Crit Care Med. 1998;26:501. doi: 10.1097/00003246-199803000-00022. [DOI] [PubMed] [Google Scholar]
- 40.van Eeden SF, Kitagawa Y, Klut ME, Lawrence E, Hogg JC. Polymorphonuclear leukocytes released from the bone marrow preferentially sequester in lung microvessels. Microcirculation. 1997;4:369. doi: 10.3109/10739689709146801. [DOI] [PubMed] [Google Scholar]
- 41.Garnica MR, de Moraes LV, Rizzo LV, de Andrade HF., Jr Supplementation of CXCL12 (CXCL12) induces homing of CD11c+ dendritic cells to the spleen and enhances control of Plasmodium berghei malaria in BALB/c mice. Immunology. 2005;115:399. doi: 10.1111/j.1365-2567.2005.02178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garnica MR, Souto JT, Silva JS, de Andrade HF., Jr Stromal cell derived factor 1 synthesis by spleen cells in rodent malaria, and the effects of in vivo supplementation of SDF-1alpha and CXCR4 receptor blocker. Immunol Lett. 2002;83:47. doi: 10.1016/s0165-2478(02)00067-6. [DOI] [PubMed] [Google Scholar]
- 43.Kalinkovich A, Tavor S, Avigdor A, Kahn J, Brill A, Petit I, Goichberg P, Tesio M, Netzer N, Naparstek E, Hardan I, Nagler A, Resnick I, Tsimanis A, Lapidot T. Functional CXCR4-expressing microparticles and SDF-1 correlate with circulating acute myelogenous leukemia cells. Cancer Res. 2006;66:11013. doi: 10.1158/0008-5472.CAN-06-2006. [DOI] [PubMed] [Google Scholar]
- 44.Hotchkiss RS, Chang KC, Grayson MH, Tinsley KW, Dunne BS, Davis CG, Osborne DF, Karl IE. Adoptive transfer of apoptotic splenocytes worsens survival, whereas adoptive transfer of necrotic splenocytes improves survival in sepsis. Proc Natl Acad Sci U S A. 2003;100:6724. doi: 10.1073/pnas.1031788100. [DOI] [PMC free article] [PubMed] [Google Scholar]