

Abstract
Developments of new methods that meet the demand of high-throughput, high-fidelity screening of hit compounds are important to searching modalities of important diseases such as neurological disorders, HIV, and cancer. A surface plasmon resonance (SPR)-based method capable of continuously screening enzyme inhibitors at a single chip with antibody-amplified signal enhancement is developed. The proof of concept is demonstrated by monitoring the cleavage of chip-confined peptide substrates (a segment of the amyloid precursor protein (APP) with the Swiss mutation) by β-site APP cleaving enzyme 1 (BACE1). In the presence of a non- inhibitor, BACE1 clips the peptide substrate at the cleavage site, detaching a fragment that is homologous to the N-terminus of the amyloid beta (Aβ) peptide. Consequently, a subsequent injection of the Aβ antibody does not lead to any molecular recognition or SPR signal change at the chip. In contrast, abolishment of the BACE1 activity by a strong inhibitor leaves the peptide substrate intact, and the subsequent antibody attachment produces an easily detectable SPR signal. Compared to the widely used FRET (fluorescence resonance energy transfer) assay, the method reported here is more cost effective, as unlabeled peptide is used as the BACE1 substrate. Furthermore, the assay is more rapid (each screening cycle lasts for ca. 1.5 h) and can be continuously carried out at a single, regenerable SPR chip for more than 30 h. Consequently, excellent reproducibility (RSD% < 5%) and throughput can be attained. Two inhibitors were screened and their half maximal inhibitory concentrations (IC50) determined by the SPR method are in excellent agreements with values deduced from ELISA and mass spectrometry.
INTRODUCTION
High-throughput drug screening generally relies on rapid and multiplexed evaluations of binding affinities between a target molecule and a plethora of drug candidates extracted from natural products or available from the combinatorial libraries of synthetic compounds. In the era of genomics and proteomics, the formidable task of high-throughput drug discovery is now confronted by new challenges such as continuously changing targets, targets of ill-defined structures, and complications in kinetic and biochemical assays of newly available drug candidates.1,2 Many detection methods have been employed for drug screening, which include, but are not limited to, mass spectrometry (MS),3,4 NMR,5–7 calorimetry,8,9 absorbance and fluorescence (FRET, fluorescence anisotropy, etc.) measurements,10,11 and surface plasmon resonance (SPR)12,13. For automated and high-throughput assays, these detection methods are used in conjunction with robotic solution delivery systems that accommodate simultaneous readouts of multiple reactions. For example, MS has become a powerful tool when combined with separation-based techniques for proteomics research.3,14 The conventional thermal shift assay can be miniaturized in a high-density microplate format when differential scanning calorimetry is replaced with spectrofluorimetry.8,15 Finally, the widely used high-density microarrays (chips) are developed with fluorescence imaging of molecules labeled with different fluorophores.10,16
While the aforementioned approaches are useful for initial compound screening, determination of the relative binding affinities is insufficient to firmly identify hit compounds. This is because a strong ligand does not necessarily bind to the active site(s) of the target molecule. This problem is further exacerbated by the fact that many target molecules do not have known X-ray or NMR structures (i.e., binding sites are not known) or lack in vivo ligands.1,5,17 Thus, binding assays are typically aided by functional studies (e.g., enzymatic activity assays, which determine inhibitory potency according to the inhibition concentrations or dosages).1,17–19
The sequential proteolysis of the transmembrane APP by the β-secretase (i.e., BACE1) and γ-secretase20 results in aberrant overproduction of Aβ peptides (e.g., Aβ(1–40) and Aβ(1–42) peptides). Subsequent aggregation of Aβ peptides and accumulation of Aβ aggregates lead to neuronal cell damage and ultimately Alzheimer’s disease (AD).21,22 Therefore, inhibition of BACE1 has been considered as a possible modality to treat AD.23 ELISA (enzyme-linked immunosorbent assay) is the first reported method for assessing BACE1 inhibition, and currently the more commonly used assay is FRET.24–26 In FRET, two different fluorophores are attached to both ends of a BACE1 peptide whose cleavage by BACE1 separates the two fluorophores, causing a change in the fluorescence signal. In contrast, when the BACE1 activity is suppressed by an inhibitor, the FRET process is unhalted. Ermolieff et al. cloned memapsin2 (BACE1) and devised an FRET assay to evaluate the enzymatic activity.27 Later on, FRET between fluorophores attached to segments of APP with the Swiss mutation (KM→NL28) was used to assess the efficacies of statine-based peptidic inhibitors29 or to demonstrate the amenability of a fluorescence plate reader to high-throughput screening.24 Using a peptide substrate sandwiched by CdSe/ZnS quantum dots (QDs) and gold nanoparticles, Choi et al. recently extended FRET to BACE1 inhibitor screening in living cells.30 However, traditional FRET assays have relatively small stokes shifts and the near-UV wavelengths for fluorophore excitation could overlap with the absorption peaks of many small molecules.24 As a consequence, the choices of the donor/acceptor pairs in many applications is limited and varies from study to study.24,27,30,31 Although the use of QDs offers a unique approach to fine-tune the excitation wavelength, the instability of QDs at acidic pH where BACE1 exhibits higher activity is a limitation. Labeling both ends of a peptide substrate with fluorophores also increases assay cost and the steric hindrance imposed by the fluorophores (especially nanoparticles) could decrease the BACE1 activity. Such steric hindrance might be the reason why relatively long reaction times (≥ 3 h) are needed for FRET-based BACE1 inhibition assays.24,30
In recent years SPR has emerged as a viable alternative for studying biomolecular interactions.32–34 In contrast to fluorescence measurements, which require fluorescent tags,31 or NMR, which typically resorts to isotope labeling,5,6,35 detection of mass changes at the solution/metal interface by SPR obviates the need for labeling the ligand or receptor molecules. SPR measures the changes in dielectric constants associated with binding reactions, which increase with sizes of molecules bound to the SPR chips.36 Because many pharmaceutical drugs are small compounds,1,17 their attachments to the chip-confined target molecules can be difficult to measure. Although for drug discovery simultaneous screening of many compounds in multiple fluidic channels by imaging SPR (SPRi) is more desirable, the use of a CCD camera in SPRi further compromises the detection sensitivity.32,37–39 As a result, it is still relatively challenging for SPR and SPRi to screen small molecules.40 To overcome the sensitivity issue, efforts have been made to design more sensitive SPR instruments32,37 or to devise signal amplification schemes involving enzymatic reactions, nanoparticles, and antibody.41–44 Utilization of the localized SPR effect45,46 has also significantly improved the sensitivity for detecting oligonucleotides and small peptides. With enzymatic reactions, nucleic acids and proteins have been quantified at ultra-trace levels.47–49 Mrksich and co-workers were the first to design peptide chips and used SPR as one of several different techniques to perform quantitative determination of protein kinase activity.50 In their work, the anti-fouling (prevention of nonspecific adsorption) capability of self-assembled monolayers (SAMs) and cross-linking reactions for controlled immobilization of peptides were also elegantly demonstrated. However, phosphoimaging, instead of SPR, was used for assaying kinase inhibitors. Using a conjugate of streptavidin-biotinylated antibody for SPR signal amplification, we recently quantified sub-nanomolar levels of amyloid beta (Aβ) peptides in cerebrospinal fluids.44 Nevertheless, the combined use of signal amplification and SPR-based enzymatic assays for screening small molecule compounds has remained largely unexplored.
In this work, we mixed BACE1 with each of the three candidate compounds (Table 1) and used SPR to study their effects on inhibiting the BACE1 cleavage of the immobilized peptide substrates (cleavage sites denoted by residues in italics). The peptide substrates used for this work are not labeled and can be easily produced by an automatic peptide synthesizer. We determined the BACE1 inhibition by examining whether and how many of the Aβ antibody molecules can be attached to the peptide substrates (antibody recognition segment underlined in Table 1). We demonstrate that multiple enzymatic assays at a single SPR chip can be performed for an extended period. Consequently, the assay cost and time are reduced, and the accuracy and reproducibility of the assays are significantly improved.
Table 1.
BACE1 peptide substrates and compounds screened
| BACE1 Substrates and Compounds Screened | Sequences |
|---|---|
| Substrate 1 | CGGGKTEEISEVNLDAEFRHDSGY |
| Substrate 2 | HHHHHHKTEEISEVNLDAEFRHDSGY |
|
| |
| Compound 1 | KTEEISEVN-Sta-VAEF (Sta = statine) |
| Compound 2 | Ph-LL-4,5-dehydro-L-CHO |
| Compound 3 | DPDNEAYEMPSEEG |
EXPERIMENTAL SECTION
Chemicals and Materials
N-hydroxysuccinimide (NHS), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), ethanolamine hydrochloride, triethyleneglycol mono- 11-mercaptoundecyl ether (HSC11PEG3-OH), NaH2PO4, Na2HPO4, streptavidin (SA), biotin- maleimide and Tween 20 were acquired from Sigma (St. Louis, MO). Sodium acetate and acetic acid were purchased from Fisher Scientific (Pittsburgh, PA). N-(5-amino-1-carboxypentyl) iminodiacetic acid (aminated NTA) and imidazole were obtained from Dojindo Inc. (Rockville, MD) and Fisher Scientific Inc., respectively. Hexaethylene glycol mono-11-mercaptoundecyl acid (HSC11PEG6-COOH) was purchased from Sensopath Technologies (Bozeman, MT). The antibody (clone 6E10) that is specific to the EFRHDS segment at the N-terminus of Aβ peptides was obtained from Covance Inc. (Dedham, MA). Recombinant human BACE1 was purchased from MP Biomedicals, LLC (Solon, OH).
KTEEISEVN-Sta-VAEF (compound 1 in Table 1) and Ph-LL-4,5-dehydro-L-CHO (compound 2) were purchased from Anaspec Inc. (Fremont, CA) and BaChem Inc. (Torrance, CA), respectively. The peptide substrates for BACE1, CGGGKTEEISEVNLDAEFRHDSGY and H6KTEEISEVNLDAEFRHDSGY and compound 3 were synthesized in house on an automatic peptide synthesizer (Symphony Quartet, Protein Technologies, Inc., Tucson, AZ). It has been reported that the length of the peptide does not affect the enzymatic activity of BACE1 so long as the KTEEISEVNLDAEF sequence is included.51 Insertion of the GGG segment between the substrate and the cysteine residue or use of a hexahistidine extension helps position the peptide substrate away from the SPR chip surface so that a higher BACE1 cleavage efficiency can be attained. These peptides were purified with reversed-phase chromatography (Shimadzu AD, Columbia, MO) using a column (Jupiter-10-C18-300, 10 mm i.d. × 250 mm) from Phenomenex (Torrance, CA) and verified with an electrospray mass spectrometer (Exactive, Thermo-Fisher Inc., Santa Clara, CA).
Instrument
The SPR measurements were conducted on a BI-SPR 4000 system equipped with a temperature controller (Biosensing Instrument Inc., Tempe, AZ). Phosphate running buffer (pH 7.4, 10 mM phosphate) containing 0.01% (V/V) Tween 20 was degassed under vacuum for 30 min. The samples were preloaded into a 200 μL sample loop on an injection valve and then delivered to the flow cell by a syringe pump (Model KDS260, KD Scientific, Holliston, MA).
Procedures
Solution Preparation
All stock solutions were prepared daily with deionized water collected from a water purification system (Simplicity 185, Millipore Corp, Billerica, MA). HSC11PEG6-COOH and HSC11PEG3-OH were dissolved in anhydrous ethanol. BACE1 was prepared in acetate buffer (20 mM, pH = 4.5), because its enzymatic activity is much greater at acidic pH. The peptide substrates and the Aβ antibody were diluted with running buffer. The three compounds screened were first dissolved in DMSO and then diluted with acetate buffer.
Modification of SPR Chips
Au films coated onto BK7 glass slides were purchased from Biosensing Instrument Inc. and annealed in a hydrogen flame to eliminate surface contaminants. Coating of the Au SPR chips with mixed PEG monolayer followed our reported procedure.44 Two types of derivatized PEG chips (SA and NTA coated onto PEG) were produced in house according to published procedures.44,52 For the SA-coated PEG chip, biotin-melamide was first immobilized onto the SA chip. Flowing 10 μM substrate 1 (cf. Table 1) over the resultant SPR chip at 20 μL/min results in attachment of ca. 400 pg/mm2 of the substrate. As for the NTA- coated chip, 40 mM NiCl2 was allowed to flow over the chip at 20 μL/min for 10 min. Injecting 10 μM substrate 2 into the fluidic channels leads to a surface coverage that is slightly less than (< 10%) that of substrate 1 at the SA-coated PEG chip.
Inhibitor Screening
Compounds to be screened were mixed with 10 nM BACE1 and the resultant solutions were introduced into the fluidic channels at 3 μL/min for 1 h. Subsequently, the residual solutions were removed by flushing the channels with the running buffer at 20 μL/min. To assess the potency of the inhibitor, 20 nM Aβ antibody was injected. The activity of BACE1 at the body temperature (37°C) and acidic pH (4.5) was confirmed by HPLC.
RESULTS AND DISCUSSION
The scheme of continuous SPR screening of BACE1 inhibitors at a single chip is illustrated in Figure 1. Modifying the SPR Au chip with a mixed monolayer of PEG/carboxylated PEG reduces nonspecific adsorption of BACE1 and the antibody. Tethering the BACE1 substrates can be accomplished by a variety of cross-linking reactions. In this work, the cysteine-terminated BACE1 substrate can be anchored onto the SA chip using biotin-melaimide. Alternatively, the His-terminated substrate can be affixed to the Ni-NTA chip. The substrate fragment remaining at the chip can be conveniently removed by imidazole, and the regenerated chip can be re-grafted with the full-length BACE1 substrate (cf. steps denoted by the dashed arrows). The molecular weight of the Aβ antibody is 150000 Da, which is much greater than that of the detached peptide fragment, DAEFRHDSGY (1195 Da). Consequently, the large mass of the antibody amplifies the otherwise small or difficult-to-detect SPR signal associated with the loss of the peptide fragment.
Figure 1.

Schematic representation of continuous SPR screening of inhibitory compounds for BACE1 at a single chip. The histidine-terminated BACE1 substrate (cleaving site shown in red and antibody recognition segment in green) is tethered onto a Ni-NTA film. BACE1 in the presence of a non-inhibitor cleaves the substrate and the detachment of the antibody-recognition segment at the surface prevents the antibody from adsorbing to the chip (middle panel). The surface can be regenerated and recoated with the full-length peptide substrate (steps depicted by dashed arrows in the top panel). In the presence of a potent inhibitor, blockage of the BACE1 active site inhibits the enzymatic activity. Consequently, recognition of the intact substrate by the antibody produces a large SPR signal (bottom panel).
With two fluidic channels, any background signal (e.g., bulk refractive index change or environmental noise) can be subtracted by serially flowing the antibody solution into a reference channel (no substrate pre-immobilized) and the analytical channel (substrate pre-immobilized and exposed to BACE1). In Figure 2A, no binding signal in curve a is expected because the antibody-recognition (EFRHDS) segment had been cleaved off the chip surface by BACE1. We observed a similar response from the chip that was first exposed to a mixture of 10 nM BACE1 and compound 3 (curve b). This suggests that compound 3 is either a non-inhibitor or the concentration used is too low to inhibit the BACE1 activity. Notice that the nonspecific adsorption of BACE1 and the antibody is absent at the PEG film. Interestingly, upon mixing BACE1 with sub-nM compound 1, we detected antibody binding and found that the amount of antibody attachment is dependent on the inhibitor concentration (curves c and d). We obtained curve e at a surface that was only exposed to the phosphate running buffer and the red curve at a chip that was exposed to compound 1 dissolved in acetate buffer. That curve e and the red curve are largely congruent is indicative of little interaction between compound 1 and the immobilized peptide substrate. Furthermore, between curves d and e, the steady-state signals (at ca. 250 s after the antibody injection) are the same, suggesting that compound 1 at 120 nM completely inhibits the BACE1 activity. In contrast, 40 nM compound 1 is insufficient to completely halt the peptide cleavage by BACE1, since the steady-state signal in curve c is about 30% less than that in curve e.
Figure 2.
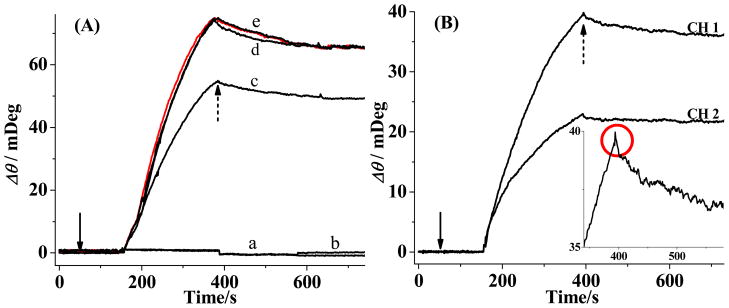
(A) Background-subtracted SPR sensorgrams corresponding to injections of 20 nM antibody at 20 μL/min into fluidic channels wherein the pre-immobilized substrate had been exposed to (a) 10 nM BACE1 only, (b) 10 nM BACE1 mixed with 120 nM compound 3, (c) 10 nM BACE1 mixed with 40 nM compound 1, (d) 10 nM BACE1 mixed with 120 nM compound 1, and (e) phosphate running buffer only. A sensorgram corresponding to pre-exposure to 120 nM compound 1 dissolved in acetate buffer is shown in red. (B) SPR sensorgrams obtained by injecting 20 nM antibody into channels wherein the substrate had been pre-exposed to 10 nM BACE1 mixed with 30 nM compound 1 (CH1) and 10 nM BACE1 mixed with 2.0 μM compound 2 (CH2). The inset is an enlarged view of the small blip indicated by the red circle. The solid and dashed arrows indicate the beginnings and endings of injections, respectively. SA-coated PEG and cysteine-terminated BACE1 (substrate 2) were used for these experiments.
The two SPR channels can also be used for simultaneous screening of two different inhibitors or two different concentrations of the same inhibitor (Figure 2B). In this configuration background subtraction cannot be performed because both fluidic channels exhibited inhibition of the BACE1 activity by the two different compounds. Consequently, a small blip at the end of the injection (inset of Figure 2B) was observed. Nevertheless, the presence of a small blip does not affect the determination of the inhibitory effect, as the net changes in the baseline SPR signals before and after the antibody injection (i.e., the total amount of antibody molecules attached to the chip surface) are the same between sensorgrams collected with and without background corrections. Notice that 30 nM compound 1 exhibits a higher inhibitory effect than 2 μM compound 2. Thus, compound 1 is a much more potent inhibitor.
We determined the optimal BACE1 concentration and reaction time for the most extensive cleavage of the BACE1 substrate. In Figure 3A, the SPR signal decreases precipitously with the BACE1 concentration between 0.1 and 5.0 nM. We did not observe any antibody attachment beyond 10.0 nM. We found a similar trend for the dependence on the reaction time, which shows no antibody attachment at 60 min and beyond. Thus 10.0 nM and 60 min are the optimal BACE1 concentration and reaction time.
Figure 3.
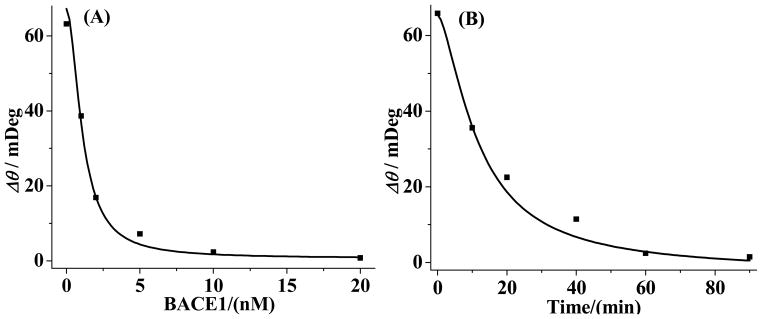
Dependence of the amount of antibody attachment on (A) BACE1 concentration and (B) BACE1 cleavage time. In (A) the BACE1 exposure time was 1 h and in (B) the concentration of BACE1 was 10 nM. The data points are averages of three replicate measurements.
For inhibitor screening, it is desirable that assays are performed rapidly and continuously. Furthermore, accuracy in determining the efficacy of inhibition would be significantly improved if comparison of two compounds or two concentrations of the same compound is made with results obtained from the same SPR chip. Acquiring a large amount of data at a single chip also makes identification of hit compounds a time-efficient and cost-effective process. These attractive features can be obtained by using the Ni-NTA-coated PEG chip. Figure 4 shows four SPR screening cycles comprising BACE1 cleavage, antibody attachment, surface regeneration, and substrate reattachment. A day’s experiment always begins with the injection of an antibody solution (cf. the injection peak indicated by the leftmost red arrow). The net SPR signal increase corresponding to the “maximal antibody coverage” established the reference value to which the extent of BACE1 inhibition can be compared. Notice that injection of 20 mM NaOH solution causes the baseline to return to its original value (blue arrow), indicating that the attached antibody molecules had been completely desorbed. Now the chip is ready for inhibitor screenings. Each cycle begins with an injection of a mixture of BACE 1 and compound to be screened (indicated by the green arrow in each cycle). In cycle 1, the signal fluctuation upon BACE1 injection is a convoluted result of the following parameter changes: (1) a more acidic acetate buffer/0.05% DMSO solution (switched from the neutral phosphate running buffer), (2) a higher temperature (elevated from 25 to 37°C), and (3) a slower flow rate (decreased from 20 to 3 μL/min). These parameters were changed to achieve the highest BACE1 activity. Similar behavior was also observed at SA chips. We observed no antibody attachment when BACE1 is uninhibited (cf. cycle 1) or a value smaller than the “maximal antibody coverage” when BACE1 is partially inhibited (cycle 2). Before the next screening cycle, the peptide fragments remaining at the sensor chip need to be removed, which can be accomplished by flushing the fluidic channels with 200 mM imidazole (injection peaks identified by the black arrows). The completely regenerated chip can then be recoated with the full-length peptide substrate (peaks denoted by the purple arrows). We found that the amount of antibody attachment is the same as the “maximal antibody coverage” when BACE1 is completely inhibited (cf. cycle 3). In this case, the chip surface can be simply regenerated with NaOH before the subsequent screening cycle (cf. the partially shown cycle 4). In cycle 4, 2 μM compound 2 mixed with BACE1 was injected first (injection commenced at the time indicated by the green arrow), which was followed by injection of the antibody solution (denoted by the red arrow). The sensorgram exhibited a peak smaller than the “maximal antibody coverage”, suggesting that compound 2 at 2 μM partially inhibits BACE1. A typical screening cycle lasts for about 1.5 h, which is faster than those in FRET-based assays (typically ~3 h).24,30 The multiple screening cycles shown in Figure 4 can be continuously run for at least 30 h without discernible signal degradation. Any small signal degradation after numerous repetitions is likely caused by either incomplete stripping of the cleaved peptide segments or failure to reattach the full-length peptide substrate to the initial surface coverage. We also found that the peptide-covered NTA chips remain viable for at least four days under a continuously flowing stream of the running buffer. The method should be readily implementable with an autosampler, which enables unmanned screening of many compounds for an extended period of time. We also envision that the methodology can be readily extended to imaging SPR (SPRi), which will further enhance the sample throughput by screening samples in multiple fluidic channels. Along this line, we should note that the signal amplification scheme in our method should compensate for the lower sensitivity inherent in most common SPRi instruments.37,53,54
Figure 4.

Continuous screening of BACE1-inhibiting compounds at a single SPR chip, with each screening cycle encircled by the brackets on top of the Figure. The flow rate and temperature for the BACE1 cleavage were 3 μL/min and 37°C, respectively, while injections of other solutions were made at 20 μL/min at 25°C. In cycles 1, 2, 3, and 4, 10 nM BACE1 was mixed with 120 nM compound 3, 20 nM compound 1, 120 nM compound 1, and 2 μM compound 2, respectively. For clarity, the entire BACE1 injection peak is shown only for cycle 1 and the large imidazole injection peaks are truncated.
Finally, we should mention that random errors given rise by different chips are largely absent in results collected from a single chip. We measured SPR signals as a function of concentrations for compounds 1 and 2 (Figure 5) and deduced their IC50 values. The two plots in Figure 5 are of the sigmoidal shapes expected from an enzyme-inhibition assay.55 The IC50 values of compound 1 (29 nM) and compound 2 (2.6 μM) are in excellent agreements with those deduced with other methods (30 nM for the former,26 measured from ELISA, and 3.0 μM for the latter,2 determined by MS). All of the RSD values of the data acquired at Ni-NTA chips are less than 5%, which are in contrast to the higher RSD values (~10%) of the data collected from multiple SA-coated PEG chips.
Figure 5.
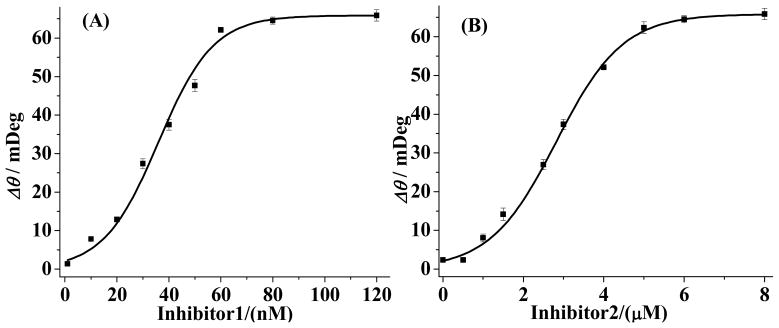
Dependence of SPR signals on concentrations of compound 1 (A) and compound 2 (B). The error bars for each data point are computed from three replicate measurements. Ni-NTA chips were used to obtain the curves.
CONCLUSION
We successfully developed an SPR-based, signal-amplified enzymatic assay for facile inhibitor screening. Compared to the commonly used FRET method, our assay obviates the use of fluorophore-labeled peptide substrates. Moreover, initiating the (inhibited) enzymatic reaction at the surface/solution interface is a better mimicry of the BACE1 cleavage of APP at the neuronal cell surface. A particularly noteworthy advantage of the renewable chip is that multiple assays can be continuously performed, which enhances sample throughput and reduces assay cost. Inhibitory concentrations (dosages) of hit compounds can also be more accurately and reproducibly deduced when a single SPR chip is used. The methodology developed herein should also be applicable to the searches of candidate compounds for inhibiting other enzymes (e.g., HIV-1 protease, which is also an aspartate-cleaving enzyme). Another attractive feature is that numerous cross-linking reactions developed for SPR afford versatile immobilization of enzyme substrates and surface regeneration. Finally, verification of the inhibition with an antibody not only ensures the fidelity of the enzymatic assay, but also enhances the assay signals for evaluating both strong and weak inhibitors.
Scheme 1.
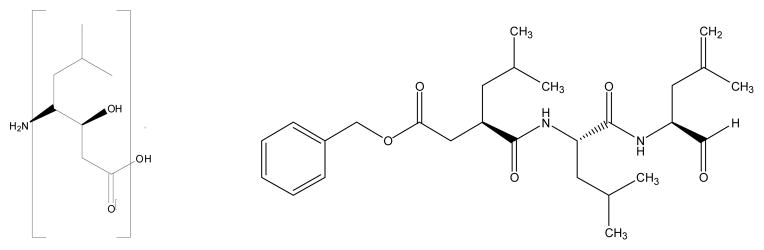
The statine moiety in compound 1 (left) and the structure of compound 2 (right)
Acknowledgments
Partial support of this work by a grant from the National Institutes of Health (SC1NS070155-01 to FZ), a National Science Foundation (NSF) grant (No. 1112105 to FZ), the NSF-CREST Program at California State University, Los Angeles (NSF HRD-0931421 to FZ) and the National Natural Science Foundation of China (No. 21175156 to JW) is gratefully acknowledged.
References
- 1.Bleicher KH, Bohm HJ, Muller K, Alanine AI. Nat Rev Drug Discov. 2003;2:369–378. doi: 10.1038/nrd1086. [DOI] [PubMed] [Google Scholar]
- 2.Wagner SL, Munoz B. J Clin Invest. 1999;104:1329–1332. doi: 10.1172/JCI8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mei H, Hsieh YS, Nardo C, Xu XY, Wang SY, Ng K, Korfmacher WA. Rapid Commun Mass Spectrom. 2003;17:97–103. doi: 10.1002/rcm.876. [DOI] [PubMed] [Google Scholar]
- 4.Reyzer ML, Hsieh YS, Ng K, Korfmacher WA, Caprioli RM. J Mass Spectrom. 2003;38:1081–1092. doi: 10.1002/jms.525. [DOI] [PubMed] [Google Scholar]
- 5.Meyer B, Peters T. Angew Chem, Int Ed. 2003;42:864–890. doi: 10.1002/anie.200390233. [DOI] [PubMed] [Google Scholar]
- 6.Lepre CA, Moore JM, Peng JW. Chem Rev. 2004;104:3641–3675. doi: 10.1021/cr030409h. [DOI] [PubMed] [Google Scholar]
- 7.Pellecchia M, Sem DS, Wuthrich K. Nat Rev Drug Discov. 2002;1:211–219. doi: 10.1038/nrd748. [DOI] [PubMed] [Google Scholar]
- 8.Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, Salemme FR. J Biomol Screen. 2001;6:429–440. doi: 10.1177/108705710100600609. [DOI] [PubMed] [Google Scholar]
- 9.Weber PC, Salemme FR. Curr Opin Struct Biol. 2003;13:115–121. doi: 10.1016/s0959-440x(03)00003-4. [DOI] [PubMed] [Google Scholar]
- 10.Pope AJ, Haupts UM, Moore KJ. Drug Discov Today. 1999;4:350–362. doi: 10.1016/s1359-6446(99)01340-9. [DOI] [PubMed] [Google Scholar]
- 11.Hertzberg RP, Pope AJ. Curr Opin Chem Biol. 2000;4:445–451. doi: 10.1016/s1367-5931(00)00110-1. [DOI] [PubMed] [Google Scholar]
- 12.Rich RL, Myszka DG. Curr Opin Biotechnol. 2000;11:54–61. doi: 10.1016/s0958-1669(99)00054-3. [DOI] [PubMed] [Google Scholar]
- 13.Karlsson R. J Mol Recognit. 2004;17:151–161. doi: 10.1002/jmr.660. [DOI] [PubMed] [Google Scholar]
- 14.Matuszewski BK, Constanzer ML, Chavez-Eng CM. Anal Chem. 2003;75:3019–3030. doi: 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- 15.Lo MC, Aulabaugh A, Jin GX, Cowling R, Bard J, Malamas M, Ellestad G. Anal Biochem. 2004;332:153–159. doi: 10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 16.Falsey JR, Renil M, Park S, Li SJ, Lam KS. Bioconjugate Chem. 2001;12:346–353. doi: 10.1021/bc000141q. [DOI] [PubMed] [Google Scholar]
- 17.Hajduk PJ, Greer J. Nat Rev Drug Discov. 2007;6:211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 18.Congreve M, Aharony D, Albert J, Callaghan O, Campbell J, Carr RAE, Chessari G, Cowan S, Edwards PD, Frederickson M, McMenamin R, Murray CW, Patel S, Wallis N. J Med Chem. 2007;50:1124–1132. doi: 10.1021/jm061197u. [DOI] [PubMed] [Google Scholar]
- 19.Wang YS, Strickland C, Voigt JH, Kennedy ME, Beyer BM, Senior MM, Smith EM, Nechuta TL, Madison VS, Czarniecki M, McKittrick BA, Stamford AW, Parker EM, Hunter JC, Greenlee WJ, Wyss DF. J Med Chem. 2010;53:942–950. doi: 10.1021/jm901472u. [DOI] [PubMed] [Google Scholar]
- 20.Wolfe MS, Xia WM, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 21.Selkoe DJ. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 22.Hardy J, Selkoe DJ. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 23.Ghosh AK, Hong L, Tang J. Curr Medicinal Chem. 2002;9:1135–1144. doi: 10.2174/0929867023370149. [DOI] [PubMed] [Google Scholar]
- 24.Kennedy ME, Wang WY, Song LX, Lee J, Zhang LL, Wong G, Wang LY, Parker E. Anal Biochem. 2003;319:49–55. doi: 10.1016/s0003-2697(03)00253-7. [DOI] [PubMed] [Google Scholar]
- 25.Pietrak BL, Crouthamel MC, Tugusheva K, Lineberger JE, Xu M, DiMuzio JM, Steele T, Espeseth AS, Stachel SJ, Coburn CA, Graham SL, Vacca JP, Shi XP, Simon AJ, Hazuda DJ, Lai MT. Anal Biochem. 2005;342:144–151. doi: 10.1016/j.ab.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 26.Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, Seubert P, Suomensaari SM, Wang SW, Walker D, Zhao J, McConlogue L, John V. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- 27.Ermolieff J, Loy JA, Koelsch G, Tang J. Biochemistry. 2000;39:12450–12456. doi: 10.1021/bi001494f. [DOI] [PubMed] [Google Scholar]
- 28.Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L. Nat Genet. 1992;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- 29.Hom RK, Fang LY, Mamo S, Tung JS, Guinn AC, Walker DE, Davis DL, Gailunas AF, Thorsett ED, Sinha S, Knops JE, Jewett NE, Anderson JP, John V. J Med Chem. 2003;46:1799–1802. doi: 10.1021/jm025619l. [DOI] [PubMed] [Google Scholar]
- 30.Choi Y, Cho Y, Kim M, Grailhe R, Song R. Anal Chem. 2012;84:8595–8601. doi: 10.1021/ac301574b. [DOI] [PubMed] [Google Scholar]
- 31.Matayoshi ED, Wang GT, Krafft GA, Erickson J. Science. 1990;247:954–958. doi: 10.1126/science.2106161. [DOI] [PubMed] [Google Scholar]
- 32.Homola J. Chem Rev. 2008;108:462–493. doi: 10.1021/cr068107d. [DOI] [PubMed] [Google Scholar]
- 33.Wang S, Boussaad S, Tao NJ. Rev Sci Instrum. 2001;72:3055–3060. [Google Scholar]
- 34.Linman MJ, Abbas A, Cheng QA. Analyst. 2010;135:2759–2767. doi: 10.1039/c0an00466a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pellecchia M, Bertini I, Cowburn D, Dalvit C, Giralt E, Jahnke W, James TL, Homans SW, Kessler H, Luchinat C, Meyer B, Oschkinat H, Peng J, Schwalbe H, Siegal G. Nat Rev Drug Discov. 2008;7:738–745. doi: 10.1038/nrd2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanken DG, Jordan CE, Frey BL, Corn RM. In: Electroanalytical Chemistry. Bard AJ, Rubinstein I, editors. Vol. 20. Marcel Dekker Inc; New York: 1998. pp. 141–225. [Google Scholar]
- 37.VanWiggeren GD, Bynum MA, Ertel JP, Jefferson S, Robotti KA, Thrush EP, Baney DA, Killeen KP. Sensor Actuator B-Chem. 2007;127:341–349. [Google Scholar]
- 38.Hickel W, Kamp D, Knoll W. Nature. 1989;339:186–186. [Google Scholar]
- 39.Linman MJ, Culver SP, Cheng Q. Langmuir. 2009;25:3075–3082. doi: 10.1021/la803835a. [DOI] [PubMed] [Google Scholar]
- 40.Tanious FA, Nguyen B, Wilson WD. Biophysical Tools for Biologists: Vol 1 in Vitro Techniques. Vol. 84. Elsevier Academic Press Inc; San Diego: 2008. pp. 53–77. [Google Scholar]
- 41.Hegnerova K, Bockova M, Vaisocherova H, Kristofikova Z, Ricny J, Ripova D, Homola J. Sensor Actuator B-Chem. 2009;139:69–73. [Google Scholar]
- 42.Yao X, Li X, Toledo F, Zurita-Lopez C, Gutova M, Momand J, Zhou F. Anal Biochem. 2006;354:220–228. doi: 10.1016/j.ab.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 43.Pelossof G, Tel-Vered R, Liu XQ, Willner I. Chem Eur J. 2012;17:8904–8912. doi: 10.1002/chem.201100601. [DOI] [PubMed] [Google Scholar]
- 44.Xia N, Liu L, Harrington MG, Wang J, Zhou F. Anal Chem. 2011;82:10151–10157. doi: 10.1021/ac102257m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haes AJ, Chang L, Klein WL, Van Duyne RP. J Am Chem Soc. 2005;127:2264–2271. doi: 10.1021/ja044087q. [DOI] [PubMed] [Google Scholar]
- 46.Haes AJ, Hall WP, Chang L, Klein WL, Van Duyne RP. Nano Lett. 2004;4:1029–1034. [Google Scholar]
- 47.Nelson BP, Grimsrud TE, Liles MR, Goodman RM, Corn RM. Anal Chem. 2001;73:1–7. doi: 10.1021/ac0010431. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Dostalek J, Knoll W. Anal Chem. 2010;83:6202–6207. doi: 10.1021/ac200751s. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y, Zhu X, Wu M, Xia N, Wang J, Zhou F. Anal Chem. 2009;81:8441–8446. doi: 10.1021/ac9014269. [DOI] [PubMed] [Google Scholar]
- 50.Houseman BT, Huh JH, Kron SJ, Mrksich M. Nat Biotechnol. 2002;20:270–274. doi: 10.1038/nbt0302-270. [DOI] [PubMed] [Google Scholar]
- 51.Sisodia SS. Proc Natl Acad Sci USA. 1992;89:6075–6079. doi: 10.1073/pnas.89.13.6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sigal GB, Bamdad C, Barberis A, Strominger J, Whitesides GM. Anal Chem. 1996;68:490–497. doi: 10.1021/ac9504023. [DOI] [PubMed] [Google Scholar]
- 53.Scarano S, Mascini M, Turner APF, Minunni M. Biosens Bioelectron. 2010;25:957–966. doi: 10.1016/j.bios.2009.08.039. [DOI] [PubMed] [Google Scholar]
- 54.Schroeder H, Adler M, Gerigk K, Muller-Chorus B, Gotz F, Niemeyer CM. Anal Chem. 2009;81:1275–1279. doi: 10.1021/ac802228k. [DOI] [PubMed] [Google Scholar]
- 55.Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]