

Abstract
Diabetic retinopathy is the fourth most common cause of blindness in adults. Current therapies, including anti-VEGF therapy, have partial efficacy in arresting the progression of proliferative diabetic retinopathy and diabetic macular edema. This review provides an overview of a novel, innovative approach to viewing diabetic retinopathy as the result of an inflammatory cycle that affects the bone marrow (BM) and the central and sympathetic nervous systems. Diabetes associated inflammation may be the result of BM neuropathy which skews haematopoiesis towards generation of increased inflammatory cells but also reduced production of endothelial progenitor cells responsible for maintaining healthy endothelial function and renewal. The resulting systemic inflammation further impacts the hypothalamus, promoting insulin resistance and diabetes, and initiates an inflammatory cascade that adversely impacts both macrovascular and microvascular complications, including diabetic retinopathy (DR). This review examines the idea of using anti-inflammatory agents that cross not only the blood-retinal barrier to enter the retina but also have the capability to target the central nervous system and cross the blood-brain barrier to reduce neuroinflammation. This neuroinflammation in key sympathetic centers serves to not only perpetuate BM pathology but promote insulin resistance which is characteristic of type 2 diabetic patients (T2D) but is also seen in T1D. A case series of morbidly obese T2D patients with retinopathy and neuropathy treated with minocycline, a well-tolerated antibiotic that crosses both the blood-retina and blood-brain barrier is presented. Our results indicates that minocycine shows promise for improving visual acuity, reducing pain from peripheral neuropathy, promoting weight loss and improving blood pressure control and we postulate that these observed beneficial effects are due to a reduction of chronic inflammation.
Keywords: diabetic retinopathy, inflammation, bone marrow, central nervous system, therapeutic approaches to diabetic retinopathy, neuroinflammation
1. Introduction
Among Americans of working age, diabetic retinopathy (DR) is the leading cause of blindness (Insitute, 2004) and ranks as the fourth most common source of vision loss in older adults (Diabetes_Group, 2000). Within the last decade, an estimated 937,000 Americans aged 40 and over have been classified as blind, with another 2.4 million suffering from low vision (Congdon et al., 2004). In 2004, retinopathy affected 3.4 percent of the US population or over 4 million individuals, with 0.75 percent or nearly 900,000 people, suffering from vision-threatening retinopathy (Kempen et al., 2004). Moreover, the number of individuals in the US with blindness or low vision is estimated to nearly double by 2020, due largely to the aging population. However, some specific groups face far higher risks of DR. A 2004 study estimated that nearly half of all American Hispanics suffered from some form of DR, with Hispanics of Mexican ancestry at particular risk (Varma et al., 2004). Diabetes is continuing to rise at an alarming rate world-wide as developing countries adopt the Western lifestyle and diet characterized by low physical activity and ingestion of foods rich in fat, refined carbohydrates and animal protein. China is no exception; the Handan Eye Study indicates in China 21.1 million adults (30 years or older) had diabetes in 2009, and of those adults, nearly 43% had developed DR and more than half of the DR cases had progressed to vision-threatening status (Wang et al., 2009).
These statistics are congruent with studies in the United States and Europe, indicating that some form of retinopathy affects 40 percent of the adult population with diabetes (Kempen et al., 2004) or 138 million people worldwide of the estimated 346 million already diagnosed with T2D alone (WHO, 2011). Despite recent advances in drug therapeutics and lifestyle management, the prevalence of T2D is estimated to nearly double by 2030 (WHO, 2011), and, with it, the prevalence of DR.
Pharmacotherapy using anti-vascular endothelial growth factor (VEGF) has shown some efficacy in addressing DR in patients with proliferative diabetic retinopathy or macular edema. In trials pertaining to PDR, treatment with bevacizumab has resulted in decreasing leakage in neovascular lesions. Mirshahi et al. conducted a large clinical studies in individuals with bilateral PDR with high-risk characteristics (Mirshahi et al., 2008). Eyes received scatter laser treatment according to the ETDRS protocol and one eye also received 1.25 mg bevacizumab injection while the other eye received sham injection. 90% of the intervention eyes had complete regression of neovascularization versus 25% in the sham group; however, after initial beneficial response, there was no difference in the two groups after 4 months. Clinical trials of bevacizumab for treatment of PDR show increased risk of TRD following a rapid retinal neovascular regression (Arevalo et al., 2008; Moradian et al., 2008). Moreover, anti-VEGF therapy does little to address the underlying causes that contribute to the occlusion and degeneration of retinal capillaries (Kern, 2007).
Recent research has identified chronic inflammation resulting from diabetes as a potential cause of damage to the retina via pro-inflammatory proteins, including cyclooxygenase-2 (COX-2), interleukin-1 (IL-1), and tumor necrosis factor-alpha (TNF-α) (Joussen et al., 2004; Kern, 2007). Still other studies have implicated changes to bone marrow-derived cells (BMDC), cells particularly adversely influenced by hyperglycaemia and unable to function to deemed to promote revascularisation and remodelling. Moreover, hyperglycaemia contributes to the production of pro-inflammatory cells (Loomans et al., 2009). Furthermore, studies have consistently established causal links between diet-induced obesity (DIO), a pro-inflammatory response and hypothalamic inflammation (Choi et al., 2010; De Souza et al., 2005; Thaler et al.). This review focuses on the links between the bone marrow, the central nervous system (CNS), diabetes and DR and provides an innovative, mechanistic persective to the pathogenesis of DR, particularly in light of the promising results from our case series showing the efficacy of an anti-inflammatory therapy in addressing aspects of diabetes. By suggesting a novel approach to treating T2D and DR targeting the CNS, this strategy improved visual acuity and symptoms associated with painful diabetic neuropathy. It may be determined that use of minocycline may serve to treat all vascular complications.
2. Mechanism implicated in the pathophysiology of retinopathy
Epidemiological and prospective data have revealed that the stressors of the diabetic vasculature persist beyond the point when good glycaemic control had been achieved (Diabetes_Group, 2000) and is referred to as ‘metabolic memory’. The first suggestion of this phenomenon came from a study by Engerman and Kern in the 1980s. Alloxan-induced diabetic dogs were divided into three groups according to glycaemic control: poor control for five years, good control for five years, and poor control for two-and-a-half years followed by good control for two-and-a-half years. Retinopathy developed in the latter group, despite subsequent sustained good glycaemic control having been achieved (Engerman and Kern, 1987).
More recently, studies by Kowluru and coworkers have shown that hyperglycaemia-induced elevations in inflammatory mediators in retinal microvascular cells resist reversal after re-institution of normal glucose conditions (Kowluru et al., 2010).
The standard parameter of glycaemic control, hemoglobin A1C is an advanced glycation endproduct (AGE). This particular cross-link formation of AGEs serves as an excellent tool to evaluate glucose control. Similar chemical changes in extracellular matrices (ECM) lead to pathological thickening of vascular basement membranes, including those of the retinal capillaries. (Bailey, 2001; Gardiner et al., 2003). Accumulation of AGEs has many deleterious effects on basement membrane physiology and cellular function (Boyd-White and Williams, 1996; Monnier et al., 1986; Wells-Knecht et al., 1995). For example, retinal vascular endothelial cells and pericytes are co-dependent on complex, two-way communication via paracrine growth factors secreted into their shared basement to ensure continued survival of both cell types. AGE formation on this basement membrane alters retinal vascular cell function and compromises cell survival by cross-linking or otherwise inactivating vital heparin-binding growth factors and integrin-recognition domains (Paul and Bailey, 1999).
Accelerated death of endothelial cells, pericytes and microvascular smooth muscle cells is known to occur in diabetic retinopathy (Mizutani et al., 1996; Stitt et al., 1994). This is believed due to the AGE-modification of basement membrane removing key survival factors from the matrix (Stitt et al., 2004). The accumulation of AGEs and associated cross-links on the arginine and lysine residues of ECM is a recognized phenomenon during diabetes (Fu et al., 1994).
Significantly, this hyperglycaemic damage targets endothelial cells as well as pericytes. The death of both cell types is a hallmark of diabetic retinopathy and leads to the formation of naked basement membrane tubes. These non-perfused, degenerated tubes/capillaries can be recognized histologically as ‘acellular capillaries’ in diabetic patients, dogs, rats and mice (Engerman and Kern, 1995; Joussen et al., 2004; Kern and Engerman, 1996a, b; Mizutani et al., 1996; Tang et al., 2003), Figure 1. In turn, acellular capillaries result in lesions that produce irreversible retinal ischemia through their inability to support blood flow. This vessel destruction leading to retinal ischemia and increased expression of angiogenic growth factors ultimately triggers retinal neovascularization. While all these histological features are well established, the mechanisms underlying diabetes-induced retinal cell injury and death are incompletely understood. Degeneration of retinal capillaries is recognized to play an important role in the pathogenesis of DR (Davis et al., 1997), with many investigators believing that the resulting retinal ischemia stimulates pre-retinal neovascularization. Acute ischemia/reperfusion of the retina also has been recognized to cause formation of these acellular capillaries, even in non-diabetic rats and mice (Zheng et al., 2005).
Figure 1. Acellular capillaries.

Trypsin digested image of diabetic rat retina showing acellular capillary (red arrow) which is the result of loss of both pericytes and endothelial cells. Acellular capillaries are the pathologic hallmark of diabetic retinopathy and their quantification is a useful way to assess the extent of retinopathy.
Endothelial progenitor cells (EPCs) play a role in maintenance of the normal vasculature (Figure 2) and defects in EPCs may contribute to capillary degeneration. In Figure 2, a trypsin digested rat retina was stained with CD133, CD14 and lectin. EPCs are present in the vasculature in the diabetic retina and typically appear at the bifurcations of the retinal capillaries and are seen as clusters as well as single cells.
Figure 2. EPC staining in rat retina.

(A) Representative trypsin digested rat retina stained with CD133 i) CD14 (ii) and lectin (iii) showing presence of EPCs (white arrow) in the retinal vasculature of a diabetic rat. These cells appeared at the bifurcations of the retinal capillaries (B) with occasional clusters (white arrow) (C) or a single cell (D).
The collective evidence indicates that the loss of retinal microvascular cells, a critical early step in diabetic retinopathy, may not only be due to increased cell death but also to altered repair mechanisms. In the retina, a defect in EPCs could prevent reparation of endothelial injury early on, leading to acellular capillary lesions and retinal ischemia.
As we show in Figure 3, CD34+ cells are dysfunctional in diabetics and have inability to home to areas of injury to orchestrate the repair process. In Figure 3, a model of ischemia reperfusion (I/R) injury that demonstrates a key feature of diabetic retinopathy, acellular capillaries, was used to examine the function of CD34+ cells. Mouse eyes were subjected to I/R injury and CD34+ were injected into the vitreous after injury. For this study we isolated CD34+ cells from diabetic patients with vascular complications or age- and gender-matched controls. Immunofluorescence studies show that control cells but not diabetic cells home to areas of injury.
Figure 3. Human peripheral blood CD34+ cells from diabetic patients are dysfunctional in vascular homing and association and cannot repair damaged retinal vessels.

Mouse eyes subjected to ischemia/reperfusion insult underwent injection (into the vitreous) with CD34+ cells isolated from long-term diabetic patients or age- and gender-matched non-diabetic controls. Two days after injection of the cells, the mouse eyes were harvested and examined by immunofluorescent laser scanning confocal microscopy. (A) CD34+ cells (identified by green fluorescence) from diabetic patients are found near vessels (visualized by red fluorescence), albeit in a random distribution (Zheng et al., 2007). (B) Most cells from non-diabetic control patients are associated with vessels. The yellow color is the result of co-localized fluorescence and indicates the CD34+ cells are associating and possibly some of the cells actually incorporating into vessel walls (Caballero et al., 2007). Images are compressed z-series (approximately 30μm total thickness). Scale bar=50μm.
This defect in migration is due in part to a diabetes-associated reduction in phosphorylation and intracellular redistribution of vasodilator-stimulated phosphoprotein (VASP), a critical actin motor protein required for cell migration that also controls vasodilation and platelet aggregation (Li Calzi et al., 2008). The effect of the chemokine stromal derived factor-1(SDF-1) on VASP redistribution is markedly reduced in diabetics (Figure 4) whereas VASP redistributes to filopodia-like structures following SDF-1 treatment in controls. Thus, SDF-1 promotes cytoskeletal changes through site- and cell type-specific VASP phosphorylation in healthy CD34+ cell but in diabetes, blunted responses to this factor and other growth factors may lead to reduced vascular repair and tissue perfusion.
Figure 4. VASP redistribution is impaired in diabetic EPCs.

Immunofluorescence images of EPCs from normal (A, B) and diabetic (C, D) donors left untreated (A, C) or treated with 100nM SDF-1 for 4 hours (B, D). Normal EPCs show a dramatic VASP redistribution following treatment (A, B) while EPCs from a diabetic donor do not show any response to SDF-1 (C, D).
Endothelial injury, acellular capillaries and subsequent retinal ischemia result in the compensatory release of chemokines and growth factors such as VEGF, SDF-1, and monocyte chemoattractant protein-1 (MCP-1 or CCL2) by the ischemia injured retina. Studies have already revealed correlations between the vitreous levels of SDF-1, MCP-1 and VEGF and the degree of macular edema and retinopathy in patients with DR (Butler et al., 2005). However, many additional factors have been shown to be increased in the vitreous of diabetics including IGF-1 (Grant et al., 1986); IL1B, IL6, IL8, CCL2, and EDN1 (Zhou et al., 2012) and these factors likely impact also DR outcomes.
Restoration of EPC function would result in repair of areas of capillary injury (acellular capillaries). This would prevent the development of ischemia and the subsequent ‘compensatory’ retinal expression of chemokines and growth factors that accumulate in the vitreous. Central to understanding the role of EPCs in vascular repair is an understanding of how BMDC become endothelial cells.
3. Microvascular disease and bone marrow
In animal studies EPC-enriched haematopoietic stem cells (HSCs), injected into the eyes of mice, promoted revascularisation, selectively targeting retinal astrocytes, cells that serve as a template for both developmental and injury-associated retinal angiogenesis (Otani et al., 2002). Furthermore, BMDC contribute to vascularisation, providing reparative functions throughout the body, from tissue perfusion during wound healing to promoting endothelialisation of the ischaemic retina (Grant et al., 2002).
3.1 Bone marrow, vasculature endothelium and its repair
In the embryonic blood island, a central HSC is surrounded by haematopoietic and endothelial precursors (Flamme and Risau, 1992). This physical association between endothelial precursors and HSCs, as well as the realisation that these cells share antigenic determinants such as Flk-1/KDR/VEGFR2, Tie-2, and CD34 (Flamme and Risau, 1992; Weiss and Orkin, 1996), suggested that these cells are derived from a common precursor, the haemangioblast. Since HSCs isolated from the peripheral circulation are able to provide sustained haematopoietic function (Brugger et al., 1995), it was reasonable that circulating cells may also function as EPCs.
In fact, Asahara et al. demonstrated that cells isolated on the basis of the expression of the haematopoietic stem cell marker CD34 not only express CD31, Flk-1, and E-selectin after 7 days in culture, but also express endothelial NOS, produce nitric oxide, and home to areas of angiogenesis in vivo (Asahara et al., 1997). Their results were confirmed by Shi et al. (1998) with CD34+ cells that were 95% pure. Other investigators have demonstrated that the number of circulating CD34+ cells and potentially their ability to direct endothelial repair is increased by treatment with HMG CoA reductase inhibitors (Dimmeler et al., 2001) as well as with erythropoietin (Bahlmann et al., 2004). In addition, injection of healthy nondiabetic CD34+ cells into diabetic mice accelerates revascularisation of ischaemic limbs (Schatteman et al., 2000) and wound healing (Sivan-Loukianova et al., 2003). Only a subset of CD34+ cells, those expressing VEGFR-2 and CD133 (Peichev et al., 2000), likely represent circulating cells capable of differentiating into endothelial cells. CD34+ cells, as well as the resultant colonies and/or cells that express endothelial markers as a result of culturing peripheral mononuclear cells under the appropriate conditions, are typically identified and called EPCs.
Interestingly, HMG CoA reductase inhibitors (Dimmeler et al., 2001; Llevadot et al., 2001) and erythropoietin (Bahlmann et al., 2004) not only increase the number of CD34+ in circulation but also have been shown to enhance EPC chemotactic activity and enhance their ability to form capillary tubes on matrigel.
3.2. Bone marrow and nervous system
Communication between the bone marrow, CNS, and peripheral nervous system is critical in order to affect rapid immune responses and to sustain vascular health. Trans-synaptically connected neurons can be traced using pseudo rabies virus (PRV) expressing green fluorescent protein and when PRV is injected into bone marrow of mice, green fluorescent protein (gfp)+ neurons can be detected in the brain (Card et al.). The sympathetic nervous system (SNS) regulates the release of BM cells into the circulation (Mendez-Ferrer et al., 2008).
3.3 Bone marrow neuropathy and DR
In models of both T2D and T1D, diabetes is associated with a reduced number of EPCs in the circulation. Moreover, the circadian rhythmicity of release of EPCs from the BM is altered, and the development of BM neuropathy is responsible for these events (Busik et al., 2009). BM neuropathy precedes the development of DR (Busik et al., 2009), thus connecting DR for the first time to peripheral nerve dysfunction in the BM. These concepts represent a paradigm shift in the pathogenesis of DR. BM neuropathy is central to the loss of protective vascular progenitors.
Previous studies showed a decrease in tyrosine hydroxylase (TH+) nerves in diabetic rats, suggesting that diabetes is associated with loss of adrenergic neurotransmitters, secondary to peripheral neuropathy affecting the BM (Katayama et al., 2006). Based on experiments with chemically (6-OH-DA) sympathectomized adult mice and the α1-adrenergic antagonist prazosin, Maestroni et al. established that regulated production of monocytes was under an inhibitory noradrenergic tone (Maestroni et al., 1992), suggesting that loss of this noradrenergic influence, as is seen in rodents with diabetic peripheral neuropathy, would result in the generation of more monocytes (Rameshwar and Gascon, 1992), which is exactly what we observe in diabetes.
HSCs reside in two basic places in the marrow: 1) adherent to stromal cells at the endosteal bone surface (primitive HSCs such as LTR-HSCs) or 2) at the sinusoids where they are adherent to endothelial cells (more mature HSC). BM stromal cells are derived from mesenchymal stem cells and have a ‘fibroblast-like phenotype (Sorrell et al., 2009). Neurotransmitters and neuropeptides stimulate BM stromal cells to produce cytokines/factors that regulate HSC self-renewal, proliferation and differentiation (Takahashi et al., 2003).
Acting on BM stromal cells, neuropeptides released by sensory neurons regulate anti-inflammatory cytokine production by these cells, including interleukin-10 (IL-10). Thus, the loss of key neuropeptides would result in loss of IL-10, contributing to a proinflammatory BM environment in diabetes (Sakagami et al., 1993). Taken together, these findings suggest that loss of neurotransmitters and neuropeptides promotes changes in the BM microenvironment that leads to an increased numbers of monocytes and a phenotypic switch of these cells to a more pathological phenotype.
4. Chronic inflammation and its impacts on DR
Metabolic and physiologic abnormalities in the retina stemming from diabetes suggest inflammation plays a direct role in the development of DR (Kern, 2007). We observe an increase in inflammatory cells throughout the retina of the diabetic as demonstrated by CD45+ immunohistochemistry of retinal cross sections compared to control mice (Figure 5). One key component of the inflammatory process involves the attraction and adhesion of leukocytes to the vascular wall. This process, leukostasis, is particularly increased in the retinas of diabetic mice (Vincent and Mohr, 2007), while in rats leukostasis is associated with retinal endothelial cell injury and death (Joussen et al., 2001). In Figure 6, we show flat mounts of trypsin digested retinas from diabetic and control mice. The diabetic vasculature shows the presence of CD133 cells with positive staining for the monocyte marker CD14. The choice of these phenotypic markers was based on CD133 being the earliest stem cell marker expressed by circulating EPCs (Quirici et al., 2001) while it has been previously reported that CD14 may serve as therapeutic alternative for diabetic patients with compromised CD34 function (Awad et al., 2006). Also CD34 is often expressed by capillary endothelium (Fina et al., 1990) and late differentiating EPCs (Thill et al., 2008; Yoder et al., 2007), which would make it difficult to separate circulating EPCs from capillary endothelium. Thus the use of CD133 as a selection marker for EPCs enabled us to easily identify this distinct population of EPCs in the retinal vasculature.
Figure 5. Diabetic retina contains higher numbers of CD45+cells.

Immunohistochemical analysis of CD45+ cells in retinal cross sections from control (A) and mice with one month of STZ-induced diabetes was performed. (C) Quantitation of the number of CD45+ cells/section is shown and demonstrates increased number in diabetic compared to controls.
Figure 6. Immature progenitors are present in the diabetic retina.
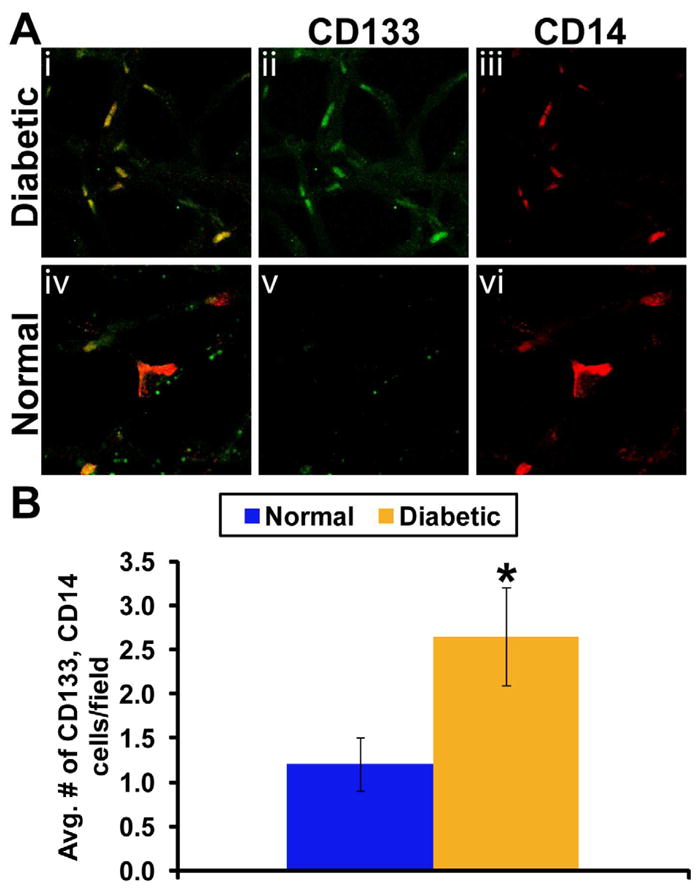
(A) Trypsin digested diabetic retinal vasculature (i) shows presence of CD133+ cells in diabetic vasculature (ii) with positive staining for CD14 marker (iii). Surprizingly, vehicle control rat retina (iv) did not show CD133+ cells (v) in 60% of the trypsin-digests although monocytic CD14+ expression were detected (vi). N=6 each vehicle control or diabetic treatment. (B) Individual count of number of CD133+ and CD14+ cells showing two fold increase in CD133+ cells (p<0.001).
Both T1D and T2D represent forms of chronic inflammation, triggered by metabolic surplus that engages the same signaling pathways present in classic inflammation. (Hotamisligil, 2006) distinguishes this form of inflammation from the classic response to injury or immune system challenge with the term ‘metaflammation’ for metabolically-triggered inflammation. This chronic metaflammation is characterized by abnormal cytokine production, as well as the activation of a network of inflammatory signaling pathways, in particular the IKK/NFκB pathway that, in non-diabetics, provides protection against insulin resistance (Ye, 2008). Moreover, diabetes is associated with an altered inflammatory response (Chang et al., 1996; Graves et al., 2005; Hotamisligil et al., 1993; Hu et al., 2004; Lalla et al., 2007; Liu et al., 2006; Naguib et al., 2004; Pickup, 2004; Pradhan et al., 2001; Yan et al., 2009). Diabetics demonstrate increased serum levels of inflammatory markers, including C-reactive protein (CRP) and Interleukin-6 (IL-6), as well as pro-inflammatory cytokines such as TNF-α (Shoelson et al., 2006).
4.1 Diet-induced obesity, inflammation and DR pathogenesis
The first definitive link between obesity, T2D and chronic inflammation emerged from studies revealing the over-expression of TNF-α in the adipose tissue of obese mice (Hotamisligil et al., 1993). A proinflammatory cytokine, TNF-α, is similarly over-expressed in both the adipose and muscle tissue of obese humans (Hotamisligil et al., 1995; Kern and Engerman, 1995). Strikingly, (Hotamisligil et al., 1993) and (Feinstein et al., 1993) both induced insulin resistance via TNF-α in rats in vivo and in vitro by (Kanety et al., 1995). Moreover, after initiation of a high-fat diet (HFD), the endothelium expresses cell adhesion molecules to bind leukocytes. In contrast, circulating leukocytes in healthy, non-obese patients do not adhere to the endothelium (Blake and Ridker, 2002). Over the past decade, studies have consistently documented the impact of an HFD and of diet-induced obesity (DIO) on the expression of proinflammatory cytokines and, perhaps more importantly, on inflammatory responsive proteins in the hypothalamus (De Souza et al., 2005; Posey et al., 2009).
Central to the pathogenesis of DR is increased oxidative stress and subsequent activation of inflammatory pathways including TNF-α, IL-1α and NFk-β activation (Kowluru and Chan, 2007). Inflammatory cytokines increase iNOS and the resulting pathologically high levels of nitric oxide (NO) diffuse to adjacent tissues where they can adversely impact retinal tissues (Caldwell et al., 2005). Receptors for cytokines are found on multiple cell types in the retina including endothelial cells, neuronal cells, Müller cells, microglia, and astrocytes. Both circulating cytokines, entering the retina via disruption of retinal vasculature, and cytokines released by cells within the retina (neurons, microglia, and astrocytes) contribute to the inflammatory process. While the role of brain inflammation in T2D is well established, T1D is also associated with increased expression of pro-inflammatory factors, Interleukin 1 beta (IL-1β), Interleukin 2 (IL-2), IL-6, TNF-α, receptor for advanced glycation end products (RAGE), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), compared to age matched control brains (Gebicke-Haerter et al., 2001; Sima et al., 2009). In addition, TNF-α and IL-1β can induce COX-2 activity in perivascular macrophages of the blood brain barrier (BBB) and generate prostaglandin E2, which enters the brain and stimulates PVN neurons regulating ACTH release and sympathetic drive (Beishuizen and Thijs, 2003). Increased expression of JAM-1 induces leukocytes accumulation in the microvasculature which leads to enhancement of cell transmigration across tight junctions and increased production of cytokines (Persidsky et al., 2006).
4.2 Hypothalamic inflammation
In DIO, the earliest markers of hypothalamic inflammation are present a mere eight weeks after beginning a HFD (Calegari et al.) and, in rodent models, within 24 hours of HFD feeding (Thaler et al.). This inflammation is marked by activation, recruitment and proliferation of microglia, immune cells of the brain that act similarly to macrophages (Sofroniew, 2009). This hypothalamic inflammation results in disruption of pancreatic β-cell function and a dysfunctional increase in insulin secretion.
Neuroinflammation also plays a role in T1D. Vargus et al induced diabetes using streptozotocin (STZ) and showed increased expressions of Ang II, ICAM-1, LFA-1 and CD8 positive cells in diverse zones of cerebrum and cerebellum of diabetic rats (week 8). Treatment of diabetic animals with losartan or enalapril reduced the expression of those molecules. These authors concluded that the presence of Ang II-mediated brain inflammatory events in diabetes is probably mediated by AT1 receptors. These results are consistent with our observations that activated microglia are present in increased numbers in the paraventricular nucleus (PVN) of T1D mice compared to controls [Grant MB, unpublished results].
4.3 Endocrine effects of hypothalamic inflammation
Various hypothalamic nuclei regulate satiety and metabolic and fluid homeostasis (Masuo et al.). Thus, hypothalamic inflammation causes resistance to both leptin (Enriori et al., 2007; Ozcan et al., 2009) and insulin (De Souza et al., 2005; Posey et al., 2009), resulting in further increases in adipose tissue and insulin resistance (Choi et al., 2010).
4.4 Insulin resistance in diabetes and DR
A typical Western diet can thus cause a cycle of increasing hypothalamic inflammation, which leads to insulin (and leptin) resistance, leading to weight gain (Velloso et al., 2008). Even independent of obesity, hypothalamic inflammation can impair insulin release from β cells, impair peripheral insulin action, and potentiate hypertension (Kang et al., 2009; Purkayastha et al.), which is pertinent to T1D as well as T2D.
In T2D, patients enter into a vicious cycle where their insulin resistance increases adiposity, which, in turn, also spurs more hypothalamic inflammation that increases insulin resistance. Even if patients initiate low-fat, moderate carbohydrate diets, their insulin resistance can make losing weight difficult and lessen the odds of patients remaining compliant with a restricted intake of fats and carbohydrates (Fletcher et al., 2006). Decreased sensitivity to insulin and resulting obesity can ultimately lead to T2D, which leads to still higher levels of hypothalamic inflammation and insulin resistance (Shoelson et al., 2006). However, patients who lost massive amounts of body mass following bariatric surgery showed lowered amounts of hypothalamic inflammation after weight loss, as detected via fMRI (Ochner et al.), as well as decreased concentrations of IL-6 (van de Sande-Lee et al., 2011).
Recently, there has been great excitement regarding the almost immediate improvement in blood glucose (remission of T2D) in patients undergoing Roux-en-Y gastric bypass. Pournaras et al showed that exclusion of glucose passage through the proximal small bowel results in enhanced insulin and gut hormone responses in patients after gastric bypass (Pournaras et al.). Thus this strategy to restore metabolic control will likely favorably impact CNS inflammation in T2D.
Obviously, significant weight loss, resulting in decreased adiposity and improved glucose control, provides the ideal means for treating T2D. Unfortunately, some obese and morbidly obese patients with T2D remain poor candidates for procedures like the Roux-en-Y gastric bypass used in van de Sande-Lee et al. due to potentially high rates of complications or their status as high risk cases for anaesthesia (van de Sande-Lee et al., 2011).
5. Interventions addressing DR
Hyperglycaemia is at the center of DR pathogenesis as large-scale T1D and T2D clinical trials have been conducted worldwide and have demonstrated that early intensive glycaemic control can reduce the incidence and progression of micro and macrovascular complications (Association, 1998; Diabetes_Group, 2000). The benefits of tight control have been documented in Type 1 in the Diabetes Control and Complication Trial (DCCT) study (DCCT, 1993) and the United Kingdom Prospective Diabetes Study (UKPDS) (UKPDS, 1998) demonstrated that controlling blood glucose levels reduced the risk of diabetic retinopathy and nephropathy in T2D.
The beneficial effects of intensive glycaemic control achieved by early intervention can persist for several years, a phenomenon that first described as ‘metabolic memory’ by the Diabetes Control and Complications Trial and Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) collaboration (Group., 2003).
While prevention of hyperglycaemia is the cornerstone of prevention of DR, once DR occurs, the impact of tight control remains important. As mentioned above, tight glucose control equally benefits patients with T1D or T2D while the strategies to achieve this in T1D includes insulin pumps and continuous glucose sensors, the judicious use of multiple agents from the armamentarium of drugs available make this equally possible in T2D. The key role of diet and exercise is equally effective and important for both T1D and T2D. However, tight control is not possible in all patients, thus supplemental strategies are needed to prevent progression of this disease and to treat it. Several therapeutic approaches show promise in addressing DR by decreasing systemic inflammation, with the full range best covered in Kern’s 2007 review (Kern, 2007). However, given this article’s focus on the links between BM, the CNS and inflammation, this review will here focus only on those interventions that touch on the BM and CNS inflammation.
5.1 Interventions addressing inflammation
Four pharmacologic interventions have displayed some efficacy in reducing inflammatory response in patients, thus improving DR in human and animal studies. Of these four, the first, intravitreal corticosteroids, treat DR locally, in a manner similar to current anti-VEGF therapy. The second, aldose reductase inhibitors, have demonstrated inconsistent outcomes across animal and human studies. The third, aspirin and salicylates, shows some efficacy, albeit with side effects. Minocycline, an antibiotic with anti-inflammtory properties, is currently proving successful in animal studies (Yune et al., 2007) and, most recently, in our case series of obese, diabetic patients with DR.
5.2 Corticosteroids
Known to exert profound anti-inflammatory effects on a variety of conditions, corticosteroids have proven effective as a localized therapy for inflammation in DR. Intravitreal injections show promise in reversing or preventing vascular permeability in DR (Dubey, 2006; Felinski and Antonetti, 2005). Currently, glucocorticoids prove particularly useful in preserving the BBB in the treatment of brain tumors and show similar effects on retinal vasculature, making them a promising treatment option for aspects of DR management.
5.3 Aldose reductase inhibitors
The polyol pathway enzyme, aldose reductase, inhibits expression of ICAM-1, COX-2, and leukostasis via inhibition of pro-inflammatory NF-κB activity (Ramana et al., 2004). As a result, aldose reductase inhibitors have been extensively studied for their inhibition of aldose reductase under hyperglycemic conditions (Kern, 2007). However, this class of drugs has shown only mixed results, at best, in inhibiting DR in animal models (Kojima et al., 1985) and, moreover, has proven unsuccessful in treating diabetic patients (Arauz-Pacheco et al., 1992; Group, 1990). Currently, further studies remain to establish the possibility that aldose reductase inhibitors provide beneficial effects due to their anti-inflammatory actions (Kern, 2007).
5.4 Aspirin and salicylates
Researchers beginning in the late nineteenth century discovered high doses of salicylates dramatically lowered the glucose levels of diabetic patients (Ebstein, 1876), an effect confirmed a quarter-century later in the impact of salicylates on reducing sugar secreted in the urine of diabetics (Williamson, 1901). The effectiveness of salicylates in treating T2D was rediscovered accidentally in the 1950s when a physician administered a high dose of aspirin to treat arthritis associated with rheumatic fever and discovered his patient no longer required daily insulin injections (Reid et al., 1957). When the patient’s joint symptoms resolved, the aspirin was discontinued, and a subsequent glucose tolerance test showed elevated blood glucose. Intrigued, Reid and his colleagues administered aspirin to a small series of diabetics, whose blood glucose returned to normal, non-diabetic levels in every patient (Reid et al., 1957). Unfortunately, follow-up studies focused on the mechanism of salicylates in reducing insulin secretion with inconclusive results (Shoelson et al., 2006).
Only recently have studies begun to focus on the efficacy of salicylates in reducing inflammation associated with the development of obesity and T2D. Aspirin, at a typical dose of 650mg, inhibits NF-κB (Kopp and Ghosh, 1994), as well as leukocyte adhesion (Pierce et al., 1996). Hundal et al. demonstrated that the same high dose of aspirin fomented substantial reductions in fasting and postprandial glucose (Hundal et al., 2002). Moreover, aspirin, initiated with onset of induced diabetes in dogs over a 5-year trial, significantly inhibited retinal aneurysm and the formation of acellular capillaries but proved less successful in diminishing pericyte ghosts and the frequency of micoraneurysms. Notably, over the course of this trial, aspirin was administered at only 20mg/kg of body weight, a level significantly lower than the 650mg that proved efficacious in earlier human studies but judged to represent the maximum tolerable chronic dose to avoid severe side effects (Kern and Engerman, 2001). However, the therapeutic benefits of aspirin in reducing or preventing DR in T2D patients are currently offset by the effects of the requisite high dosage, including gastrointestinal irritation and unacceptably high risks of bleeding (Hundal et al., 2002).
5.5 Minocycline
A second generation, chemically modified tetracycline, minocycline is commonly prescribed for its antimicrobial action but also displays significant anti-inflammatory effects (Amin et al., 1996; Yrjanheikki et al., 1999). Moreover, minocycline shows promise in decreasing the microglial activation in the hypothalamus (Lai and Todd, 2006) that accompanies the inflammatory cycle connecting the BMDC and CNS to insulin resistance and T2D (Rothwell et al., 1999). Animal studies of the efficacy of minocycline in treating DR in mice show that long-term administration of the drug successfully prevented retinal capillary degeneration in diabetic mice (Vincent and Mohr, 2007). Minocycline has been successfully used to treat painful diabetic neuropathy (Dray, 2008). Moreover, minocycline’s ability to decrease hypothalamic inflammation could provide a particularly effective strategy in treating DR by improving patients’ blood glucose control.
5.5.1 Efficacy in humans
This potential efficacy was recently tested by our laboratory through a case series of patients with obesity, insulin resistance, T2D, and DR. As an anti-inflammatory agent, minocycline freely crosses the BBB, is neuro-protective and inhibits microglial activation in the brain (Zhu et al., 2002). Minocycline also produces beneficial effects in acute and chronic brain disorders, including stroke, traumatic brain injury and neurodegenerative diseases (Kreuter et al., 2004; Sanchez et al., 2001; Tikka and Koistinaho, 2001).
5.5.2 Case series: Minocycline improved visual acuity and neuropathic pain in diabetic subjects
Nine T2D patients with either mild to moderate DR came from three different settings: three patients from a diabetes and weight management clinic, three from a weight management clinic at the University of Florida’s College of Medicine, and three from clinician referrals at the University of Florida’s Shands Hospital. Notably, all nine morbidly obese patients began the study with minocycline prescribed on a compassionate basis, secondary to their inadequate response to weight management programs and the use of multiple oral hypoglycaemic and anti-hypertensive agents. The short duration of the study limited the interpretation of retinal changes; however all patients had mild to moderate non-proliferative diabetic retinopathy at onset and also experienced blurred vision and reduced visual acuity with episodes of hyperglycaemia. At the time of minocycline initiation, all nine patients were also already taking atorvastatin (Lipitor®) or other HMG CoA reductase inhibitors. All patients had a diagnosis of Type 2 diabetes for a minimum of ten years but not more than 21 years. Patients also had body mass indices (BMI) between 37 and 60 kg/m2. An adult with a BMI between 25 and 29 kg/m2 is classified as overweight, with obese adults having a BMI greater than 30 kg/m2, and morbidly obese adults as having a BMI greater than 40 kg/m2. Consistent with links established by epidemiology studies, all patients also had hypertension. All nine patients also experienced symptoms consistent with peripheral neuropathy, confirmed on physical examination as decreased sensation to light touch or pin prick, although no patients underwent nerve conduction studies to document degree of neuropathy. Although the four patients with the highest initial BMIs had been previously diagnosed with sleep apnea, all nine patients complained of sleep disturbances. Hypertension was managed with either beta-blockers, centrally acting alpha-agonists, ACE inhibitors or angiotensin receptor blockers, with a median blood pressure of 168 ± 16/93 ± 10 mmHg.
Patients were started on 100 mg twice daily of minocycline for ten months. Patients also received counseling on continuing the high protein, moderate fat and low carbohydrate diet—with carbohydrate intake limited to 30 grams or less at each meal—which they had been on at the time of the initiation of glucose, months prior to initiating minocycline. Every month, patients’ weight loss and blood pressure was recorded. Blood glucose control, established via HbA1c levels, was measured every three months during the study’s ten months.
All nine patients achieved consistent weight loss of an average of 37.9 ± 4.0 kg over a ten-month period, decreasing their BMI by an average of 30 ± 7 %. Patients not in either a diabetes or weight management clinic achieved the same weight loss as patients receiving more aggressive counseling on diet, exercise, and behavioral modifications. Patients’ weight loss was also accompanied by improvement in HbA1c, with a reduction of an average of 22 ± 12%. All patients reported improved visual acuity during the study and improvement of neuropathic pain of their extremities.
No patients reported adverse effects from minocycline. On the contrary, patients on minocycline reported mild appetite suppression and earlier satiety. Following their weight loss, all patients noted improved quality of sleep. Patients also reported improved mood and increased ability to exercise, due in part to their substantial weight loss but also to minocycline’s apparent effects in decreasing neuromuscular pain resulting from inflammation (McMahon and Wells, 2004; Zhang et al., 2009). These same effects, coupled with weight loss, resulted in patients’ subjective improvement of pain symptoms associated with peripheral neuropathy.
6. Implications of anti-inflammatory strategies
All patients uniformly reported improved visual acuity. These results are likely due to improved blood glucose and blood pressure control but may also be due to decreased retinal and CNS inflammation. The mechanism(s) by which minocycline produces these dramatic beneficial outcomes remains speculative at the present time. However, based on existing evidence, we believe that its effects may, in part, be mediated by its anti-inflammatory actions on the brain for three reasons. Firstly, other anti-inflammatory drugs have only limited ability to penetrate the BBB, such as salicylate and doxycycline, exhibit limited effects on diabetes and obesity although they are known to reduce plasma inflammatory markers (Goldfine et al., 2008; Shoelson and Goldfine, 2009). Secondly, minocycline has been shown to inhibit microglial activation and inhibition of brain inflammatory cytokines (Homsi et al., 2010; Morgado et al.). Thirdly, activation of brain microglial cells and increases in brain inflammation are observed in nutrition-associated obesity (Tapia-Gonzalez et al.).
In the treatment of DR, especially for the thirty percent of patients who fail to respond to anti-VEGF therapy, anti-inflammatory interventions show particular promise. With its long history as a well-tolerated antibiotic and its minimal side effects, minocycline can potentially ameliorate DR. At the same time, its use can potentially improve blood glucose control, result in weight loss, and thus prevent complications of T2D before they occur. Several questions remain to be resolved through a large-scale trial of minocycline in diabetic patients with DR, including side effects that stem from its chronic use.
6.2 Conclusions and future directions
The future holds promise for use of single agents as well as combination therapy targeting conventional as well as novel signaling pathways (Afzal et al., 2007; Kramerov et al., 2008). Viewing DR as an expression of neuroinflammation, allows the use of agents such as minocycline to simultaneous address micro and macrovascular complications. Complications from diabetes include not only vascular complications but also Alzheimer’s disease, which is correlated with diabetes (Arvanitakis et al., 2004a; Arvanitakis et al., 2004b; Luchsinger et al., 2001). In a recent Dutch study, T2D doubled the risk of patients for developing dementia, with patients receiving insulin having a four-fold risk, compared to non-diabetics (Ott and Kivipelto, 1999).
This novel approach to treating DR via neuroinflammation counters the shortcomings of existing treatments. While evidence shows that anti-inflammatory agents, such as tetracycline, can prove efficacious in treating DR by reducing inflammation locally, tetracycline’s inability to cross the BBB inhibits its ability to reduce neuroinflammation. Given the low cost of minocycline and its potential to replace substantial pharmacologic regimens, including insulin sensitisers, anti-hyperglycaemic agents, anti-hypertensive agents, and anti-VEGF therapies, this approach to treating DR via central inflammation deserves further investigation.
In conclusion, conceptually we are proposing that BM neuropathy is central to the inciting events of DR. The concepts proposed represent a paradigm shift in the pathogenesis of DR (Figure 7). The BM is a rich source of reparative cells for the retina (Grant et al., 2002; Otani et al., 2002). However, these key cellular components become dysfunctional in diabetes. In combination with BM neuropathy that promotes the generation of an excess of monocytes, the loss of reparative cells with the simultaneous increase in detrimental inflammatory cells, such as monocytes, promote the development of endothelial dysfunction.
Figure 7. Bone marrow-CNS connection in diabetes.

Our hypothesis suggests a strong connection between the bone marrow and CNS and retina. An imbalance in the bone marrow leads to too many monocytes being produced and too few reparative endothelial progenitors. The inflammatory cells extravasate and promote local activation of resident microglia in the retina and brain contributing to both CNS and retinal pathology. Specific CNS centers that regulate the sympathetic nervous system (SNS) make direct connections to the bone marrow while the bone marrow, via the cells it releases, communicates back to these specific SNS centers. CNS inflammation may play a central role, not only in disease process such as neurogenic hypertension and metabolic syndrome, but also in perpetuating the inflammation that plays a critical role in the pathogenesis of diabetic retinopathy. (BN = basal amygdala, GIC = granular insular cortex, MC = middle commissure, PVN = paraventricular nucleus of hypothalamus)
The communication between the BM, CNS and peripheral nervous system is critical to affect rapid immune responses and to sustain vascular health. The development of BM neuropathy sets up systemic inflammation by shifting haematopoiesis towards the generation of inflammatory cells. Importantly, BM neuropathy precedes the development of DR, thus connecting DR for the first time to peripheral nerve dysfunction in the BM. The altered bone marrow population can, in turn, perpetuate not only retina, but also CNS inflammation. Inflammation in these key regions of the SNS that regulate the BM perpetuate the dysfunction. Thus, strategies that lead to specific reduction of monocyte subpopulations may represent an optimal strategy for DR management, as well as management of generalized inflammation implicated in both microvascular and macrovascular disease in diabetes.
Article Highlights.
Blocking systemic inflammation may prove an effective means of treating DR.
High-fat diets promote inflammation in the hypothalamus.
High-fat diets may play a direct role in the development of DR.
Anti-inflammatory therapies also address other complications of Type 2 diabetes.
Acknowledgments
NIH/NCRR Clinical and Translational Science Award to the University of Florida UL1 RR029890; EY 007739, EY012601, DK 096221 to MBG and HL110170 to MBG and MKR.
This study did not receive nor was it sponsored by any private, non-profit, or any institutional research funding.
Abbreviations
- AGE
advanced glycation end products
- VEGF
vascular endothelial growth factors
- BBB
blood-brain barrier
- BM
bone marrow
- BMDC
bone marrow-derived cell
- CRP
C-reactive protein
- CNS
central nervous system
- COX-2
cyclooxygenase-2
- DR
diabetic retinopathy
- DIO
diet induced obesity
- EPCs
endothelial progenitor cells
- HSCs
haematopoietic stem cells
- HFD
high-fat diet
- IL-1
interleukin-1
- IL-6
interleukin-6
- IL-10
interleukin-10
- MCP-1 or CCL2
monocyte chemoattractant protein-1
- PRV
pseudo rabies virus
- SDF-1
stromal derived factor-1
- TNF-α
tumor necrosis factor alpha
- T1D
Type 1 diabetes
- T2D
Type 2 diabetes
Footnotes
Contributions
MKR and MBG conceived the case series. ADB, AS and QL generated data for figures. ADB edited the manuscript. MBG provided follow-up and outcomes. LCS modeled the data. DC provided dietary counseling. SC edited the manuscript and generated data for figure. SLC edited the manuscript and generated data for figure. YD and MBG contributed to the writing of the paper.
None of the authors have any competing interests.
No ethical or Institutional Review Board (IRB) approval was required for this study.
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: no support from any organisation for the submitted work; no financial relationships with any organisations that might have an interest in the submitted work in the previous three years, no other relationships or activities that could appear to have influenced the submitted work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afzal A, Shaw LC, Ljubimov AV, Boulton ME, Segal MS, Grant MB. Retinal and choroidal microangiopathies: therapeutic opportunities. Microvasc Res. 2007;74:131–144. doi: 10.1016/j.mvr.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Amin AR, Attur MG, Thakker GD, Patel PD, Vyas PR, Patel RN, Patel IR, Abramson SB. A novel mechanism of action of tetracyclines: effects on nitric oxide synthases. Proc Natl Acad Sci U S A. 1996;93:14014–14019. doi: 10.1073/pnas.93.24.14014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arauz-Pacheco C, Ramirez LC, Pruneda L, Sanborn GE, Rosenstock J, Raskin P. The effect of the aldose reductase inhibitor, ponalrestat, on the progression of diabetic retinopathy. J Diabetes Complications. 1992;6:131–137. doi: 10.1016/1056-8727(92)90024-f. [DOI] [PubMed] [Google Scholar]
- Arevalo JF, Fromow-Guerra J, Sanchez JG, Maia M, Berrocal MH, Wu L, Saravia MJ, Costa RA. Primary intravitreal bevacizumab for subfoveal choroidal neovascularization in age-related macular degeneration: results of the Pan-American Collaborative Retina Study Group at 12 months follow-up. Retina. 2008;28:1387–1394. doi: 10.1097/IAE.0b013e3181884ff4. [DOI] [PubMed] [Google Scholar]
- Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004a;61:661–666. doi: 10.1001/archneur.61.5.661. [DOI] [PubMed] [Google Scholar]
- Arvanitakis Z, Wilson RS, Schneider JA, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and progression of rigidity and gait disturbance in older persons. Neurology. 2004b;63:996–1001. doi: 10.1212/01.wnl.0000138432.16676.4b. [DOI] [PubMed] [Google Scholar]
- Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- Association AD. Implications of the United Kingdom Prospective Diabetes Study. Diabetes Care. 1998;21:2180–2184. doi: 10.2337/diacare.21.12.2180. [DOI] [PubMed] [Google Scholar]
- Awad O, Dedkov EI, Jiao C, Bloomer S, Tomanek RJ, Schatteman GC. Differential healing activities of CD34+ and CD14+ endothelial cell progenitors. Arterioscler Thromb Vasc Biol. 2006;26:758–764. doi: 10.1161/01.ATV.0000203513.29227.6f. [DOI] [PubMed] [Google Scholar]
- Bahlmann FH, De Groot K, Spandau JM, Landry AL, Hertel B, Duckert T, Boehm SM, Menne J, Haller H, Fliser D. Erythropoietin regulates endothelial progenitor cells. Blood. 2004;103:921–926. doi: 10.1182/blood-2003-04-1284. [DOI] [PubMed] [Google Scholar]
- Bailey AJ. Molecular mechanisms of ageing in connective tissues. Mech Ageing Dev. 2001;122:735–755. doi: 10.1016/s0047-6374(01)00225-1. [DOI] [PubMed] [Google Scholar]
- Beishuizen A, Thijs LG. Endotoxin and the hypothalamo-pituitary-adrenal (HPA) axis. J Endotoxin Res. 2003;9:3–24. doi: 10.1179/096805103125001298. [DOI] [PubMed] [Google Scholar]
- Blake GJ, Ridker PM. Tumour necrosis factor-alpha, inflammatory biomarkers, and atherogenesis. Eur Heart J. 2002;23:345–347. doi: 10.1053/euhj.2001.2905. [DOI] [PubMed] [Google Scholar]
- Boyd-White J, Williams JC., Jr Effect of cross-linking on matrix permeability. A model for AGE-modified basement membranes. Diabetes. 1996;45:348–353. doi: 10.2337/diab.45.3.348. [DOI] [PubMed] [Google Scholar]
- Brugger W, Heimfeld S, Berenson RJ, Mertelsmann R, Kanz L. Reconstitution of hematopoiesis after high-dose chemotherapy by autologous progenitor cells generated ex vivo. N Engl J Med. 1995;333:283–287. doi: 10.1056/NEJM199508033330503. [DOI] [PubMed] [Google Scholar]
- Busik JV, Tikhonenko M, Bhatwadekar A, Opreanu M, Yakubova N, Caballero S, Player D, Nakagawa T, Afzal A, Kielczewski J, Sochacki A, Hasty S, Li Calzi S, Kim S, Duclas SK, Segal MS, Guberski DL, Esselman WJ, Boulton ME, Grant MB. Diabetic retinopathy is associated with bone marrow neuropathy and a depressed peripheral clock. J Exp Med. 2009;206:2897–2906. doi: 10.1084/jem.20090889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JM, Guthrie SM, Koc M, Afzal A, Caballero S, Brooks HL, Mames RN, Segal MS, Grant MB, Scott EW. SDF-1 is both necessary and sufficient to promote proliferative retinopathy. J Clin Invest. 2005;115:86–93. doi: 10.1172/JCI22869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell RB, Bartoli M, Behzadian MA, El-Remessy AE, Al-Shabrawey M, Platt DH, Liou GI, Caldwell RW. Vascular endothelial growth factor and diabetic retinopathy: role of oxidative stress. Curr Drug Targets. 2005;6:511–524. doi: 10.2174/1389450054021981. [DOI] [PubMed] [Google Scholar]
- Calegari VC, Torsoni AS, Vanzela EC, Araujo EP, Morari J, Zoppi CC, Sbragia L, Boschero AC, Velloso LA. Inflammation of the hypothalamus leads to defective pancreatic islet function. J Biol Chem. 286:12870–12880. doi: 10.1074/jbc.M110.173021. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Card JP, Kobiler O, Ludmir EB, Desai V, Sved AF, Enquist LW. A dual infection pseudorabies virus conditional reporter approach to identify projections to collateralized neurons in complex neural circuits. PLoS One. 6:e21141. doi: 10.1371/journal.pone.0021141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KM, Lehrhaupt N, Lin LM, Feng J, Wu-Wang CY, Wang SL. Epidermal growth factor in gingival crevicular fluid and its binding capacity in inflamed and non-inflamed human gingiva. Arch Oral Biol. 1996;41:719–724. doi: 10.1016/s0003-9969(96)00024-6. [DOI] [PubMed] [Google Scholar]
- Choi SJ, Kim F, Schwartz MW, Wisse BE. Cultured hypothalamic neurons are resistant to inflammation and insulin resistance induced by saturated fatty acids. Am J Physiol Endocrinol Metab. 2010;298:E1122–1130. doi: 10.1152/ajpendo.00006.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congdon N, O’Colmain B, Klaver CC, Klein R, Munoz B, Friedman DS, Kempen J, Taylor HR, Mitchell P. Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol. 2004;122:477–485. doi: 10.1001/archopht.122.4.477. [DOI] [PubMed] [Google Scholar]
- Davis TM, Stratton IM, Fox CJ, Holman RR, Turner RC. U.K. Prospective Diabetes Study 22. Effect of age at diagnosis on diabetic tissue damage during the first 6 years of NIDDM. Diabetes Care. 1997;20:1435–1441. doi: 10.2337/diacare.20.9.1435. [DOI] [PubMed] [Google Scholar]
- DCCT. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, Saad MJ, Velloso LA. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005;146:4192–4199. doi: 10.1210/en.2004-1520. [DOI] [PubMed] [Google Scholar]
- Diabetes_Group. Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. N Engl J Med. 2000;342:381–389. doi: 10.1056/NEJM200002103420603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmeler S, Aicher A, Vasa M, Mildner-Rihm C, Adler K, Tiemann M, Rutten H, Fichtlscherer S, Martin H, Zeiher AM. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest. 2001;108:391–397. doi: 10.1172/JCI13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray A. Neuropathic pain: emerging treatments. Br J Anaesth. 2008;101:48–58. doi: 10.1093/bja/aen107. [DOI] [PubMed] [Google Scholar]
- Dubey AK. Intravitreal injection of triamcinolone acetonide for diabetic macular edema: principles and practice. Indian J Ophthalmol. 2006;54:290. doi: 10.4103/0301-4738.27969. author reply 290–291. [DOI] [PubMed] [Google Scholar]
- Ebstein W. Zur therapie des diabetes mellitus, insbesondere uber die anewendeng des salicylsuren natron bei demselben. Klin Wochschr. 1876;13:337–340. [Google Scholar]
- Engerman RL, Kern TS. Progression of incipient diabetic retinopathy during good glycemic control. Diabetes. 1987;36:808–812. doi: 10.2337/diab.36.7.808. [DOI] [PubMed] [Google Scholar]
- Engerman RL, Kern TS. Retinopathy in animal models of diabetes. Diabetes Metab Rev. 1995;11:109–120. doi: 10.1002/dmr.5610110203. [DOI] [PubMed] [Google Scholar]
- Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, Billes SK, Glavas MM, Grayson BE, Perello M, Nillni EA, Grove KL, Cowley MA. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab. 2007;5:181–194. doi: 10.1016/j.cmet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Feinstein R, Kanety H, Papa MZ, Lunenfeld B, Karasik A. Tumor necrosis factor-alpha suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substrates. J Biol Chem. 1993;268:26055–26058. [PubMed] [Google Scholar]
- Felinski EA, Antonetti DA. Glucocorticoid regulation of endothelial cell tight junction gene expression: novel treatments for diabetic retinopathy. Curr Eye Res. 2005;30:949–957. doi: 10.1080/02713680500263598. [DOI] [PubMed] [Google Scholar]
- Fina L, Molgaard HV, Robertson D, Bradley NJ, Monaghan P, Delia D, Sutherland DR, Baker MA, Greaves MF. Expression of the CD34 gene in vascular endothelial cells. Blood. 1990;75:2417–2426. [PubMed] [Google Scholar]
- Flamme I, Risau W. Induction of vasculogenesis and hematopoiesis in vitro. Development. 1992;116:435–439. doi: 10.1242/dev.116.2.435. [DOI] [PubMed] [Google Scholar]
- Fletcher BR, Calhoun ME, Rapp PR, Shapiro ML. Fornix lesions decouple the induction of hippocampal arc transcription from behavior but not plasticity. J Neurosci. 2006;26:1507–1515. doi: 10.1523/JNEUROSCI.4441-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu MX, Wells-Knecht KJ, Blackledge JA, Lyons TJ, Thorpe SR, Baynes JW. Glycation, glycoxidation, and cross-linking of collagen by glucose. Kinetics, mechanisms, and inhibition of late stages of the Maillard reaction. Diabetes. 1994;43:676–683. doi: 10.2337/diab.43.5.676. [DOI] [PubMed] [Google Scholar]
- Gardiner TA, Anderson HR, Stitt AW. Inhibition of advanced glycation end-products protects against retinal capillary basement membrane expansion during long-term diabetes. J Pathol. 2003;201:328–333. doi: 10.1002/path.1429. [DOI] [PubMed] [Google Scholar]
- Gebicke-Haerter PJ, Spleiss O, Ren LQ, Li H, Dichmann S, Norgauer J, Boddeke HW. Microglial chemokines and chemokine receptors. Prog Brain Res. 2001;132:525–532. doi: 10.1016/S0079-6123(01)32100-3. [DOI] [PubMed] [Google Scholar]
- Goldfine AB, Silver R, Aldhahi W, Cai D, Tatro E, Lee J, Shoelson SE. Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin Transl Sci. 2008;1:36–43. doi: 10.1111/j.1752-8062.2008.00026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant MB, May WS, Caballero S, Brown GA, Guthrie SM, Mames RN, Byrne BJ, Vaught T, Spoerri PE, Peck AB, Scott EW. Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization. Nat Med. 2002;8:607–612. doi: 10.1038/nm0602-607. [DOI] [PubMed] [Google Scholar]
- Grant MB, Schmetz I, Russell B, Harwood HJ, Jr, Silverstein J, Merimee TJ. Changes in insulin-like growth factors I and II and their binding protein after a single intramuscular injection of growth hormone. The Journal of clinical endocrinology and metabolism. 1986;63:981–984. doi: 10.1210/jcem-63-4-981. [DOI] [PubMed] [Google Scholar]
- Graves DT, Naguib G, Lu H, Leone C, Hsue H, Krall E. Inflammation is more persistent in type 1 diabetic mice. J Dent Res. 2005;84:324–328. doi: 10.1177/154405910508400406. [DOI] [PubMed] [Google Scholar]
- Group SR. A randomized trial of sorbinil, an aldose reductase inhibitor, in diabetic retinopathy. Sorbinil Retinopathy Trial Research Group. Arch Ophthalmol. 1990;108:1234–1244. doi: 10.1001/archopht.1990.01070110050024. [DOI] [PubMed] [Google Scholar]
- W.T.f.t.D.C.a.C.T.E.o.D.I.a.C.R Group. Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: the Epidemiology of Diabetes Interventions and Complications (EDIC) study. JAMA. 2003;290:2159–2167. doi: 10.1001/jama.290.16.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homsi S, Piaggio T, Croci N, Noble F, Plotkine M, Marchand-Leroux C, Jafarian-Tehrani M. Blockade of acute microglial activation by minocycline promotes neuroprotection and reduces locomotor hyperactivity after closed head injury in mice: a twelve-week follow-up study. J Neurotrauma. 2010;27:911–921. doi: 10.1089/neu.2009.1223. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95:2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- Hu F, Lu R, Huang B, Liang M. Free radical scavenging activity of extracts prepared from fresh leaves of selected Chinese medicinal plants. Fitoterapia. 2004;75:14–23. doi: 10.1016/j.fitote.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Hundal RS, Petersen KF, Mayerson AB, Randhawa PS, Inzucchi S, Shoelson SE, Shulman GI. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest. 2002;109:1321–1326. doi: 10.1172/JCI14955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insitute NE. Statement on the prevalence of diabetic retinopathy and age-related macular degeneration among Hispanic/Latino Americans. 2004. Press release. [Google Scholar]
- Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol. 2001;158:147–152. doi: 10.1016/S0002-9440(10)63952-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joussen AM, Poulaki V, Le ML, Koizumi K, Esser C, Janicki H, Schraermeyer U, Kociok N, Fauser S, Kirchhof B, Kern TS, Adamis AP. A central role for inflammation in the pathogenesis of diabetic retinopathy. Faseb J. 2004;18:1450–1452. doi: 10.1096/fj.03-1476fje. [DOI] [PubMed] [Google Scholar]
- Kanety H, Feinstein R, Papa MZ, Hemi R, Karasik A. Tumor necrosis factor alpha-induced phosphorylation of insulin receptor substrate-1 (IRS-1). Possible mechanism for suppression of insulin-stimulated tyrosine phosphorylation of IRS-1. J Biol Chem. 1995;270:23780–23784. doi: 10.1074/jbc.270.40.23780. [DOI] [PubMed] [Google Scholar]
- Kang HW, Kim D, Kim HJ, Kim CH, Kim YS, Park MJ, Kim JS, Cho SH, Sung MW, Jung HC, Lee HS, Song IS. Visceral obesity and insulin resistance as risk factors for colorectal adenoma: a cross-sectional, case-control study. Am J Gastroenterol. 2009;105:178–187. doi: 10.1038/ajg.2009.541. [DOI] [PubMed] [Google Scholar]
- Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, Frenette PS. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124:407–421. doi: 10.1016/j.cell.2005.10.041. [DOI] [PubMed] [Google Scholar]
- Kempen JH, O’Colmain BJ, Leske MC, Haffner SM, Klein R, Moss SE, Taylor HR, Hamman RF. The prevalence of diabetic retinopathy among adults in the United States. Arch Ophthalmol. 2004;122:552–563. doi: 10.1001/archopht.122.4.552. [DOI] [PubMed] [Google Scholar]
- Kern TS. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res. 2007;2007:95103. doi: 10.1155/2007/95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern TS, Engerman RL. Vascular lesions in diabetes are distributed non-uniformly within the retina. Exp Eye Res. 1995;60:545–549. doi: 10.1016/s0014-4835(05)80069-7. [DOI] [PubMed] [Google Scholar]
- Kern TS, Engerman RL. Capillary lesions develop in retina rather than cerebral cortex in diabetes and experimental galactosemia. Arch Ophthalmol. 1996a;114:306–310. doi: 10.1001/archopht.1996.01100130302013. [DOI] [PubMed] [Google Scholar]
- Kern TS, Engerman RL. A mouse model of diabetic retinopathy. Arch Ophthalmol. 1996b;114:986–990. doi: 10.1001/archopht.1996.01100140194013. [DOI] [PubMed] [Google Scholar]
- Kern TS, Engerman RL. Pharmacological inhibition of diabetic retinopathy: aminoguanidine and aspirin. Diabetes. 2001;50:1636–1642. doi: 10.2337/diabetes.50.7.1636. [DOI] [PubMed] [Google Scholar]
- Kojima K, Matsubara H, Harada T, Mizuno K, Suzuki M, Hotta N, Kakuta H, Sakamoto N. Effects of aldose reductase inhibitor on retinal microangiopathy in streptozotocin-diabetic rats. Jpn J Ophthalmol. 1985;29:99–109. [PubMed] [Google Scholar]
- Kopp E, Ghosh S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science. 1994;265:956–959. doi: 10.1126/science.8052854. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Chan PS. Oxidative stress and diabetic retinopathy. Exp Diabetes Res. 2007;2007:43603. doi: 10.1155/2007/43603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru RA, Zhong Q, Kanwar M. Metabolic memory and diabetic retinopathy: role of inflammatory mediators in retinal pericytes. Exp Eye Res. 2010;90:617–623. doi: 10.1016/j.exer.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramerov AA, Saghizadeh M, Caballero S, Shaw LC, Li Calzi S, Bretner M, Montenarh M, Pinna LA, Grant MB, Ljubimov AV. Inhibition of protein kinase CK2 suppresses angiogenesis and hematopoietic stem cell recruitment to retinal neovascularization sites. Mol Cell Biochem. 2008;316:177–186. doi: 10.1007/s11010-008-9831-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuter M, Langer C, Kerkhoff C, Reddanna P, Kania AL, Maddika S, Chlichlia K, Bui TN, Los M. Stroke, myocardial infarction, acute and chronic inflammatory diseases: caspases and other apoptotic molecules as targets for drug development. Arch Immunol Ther Exp (Warsz) 2004;52:141–155. [PubMed] [Google Scholar]
- Lai AY, Todd KG. Hypoxia-activated microglial mediators of neuronal survival are differentially regulated by tetracyclines. Glia. 2006;53:809–816. doi: 10.1002/glia.20335. [DOI] [PubMed] [Google Scholar]
- Lalla E, Kaplan S, Yang J, Roth GA, Papapanou PN, Greenberg S. Effects of periodontal therapy on serum C-reactive protein, sE-selectin, and tumor necrosis factor-alpha secretion by peripheral blood-derived macrophages in diabetes. A pilot study. J Periodontal Res. 2007;42:274–282. doi: 10.1111/j.1600-0765.2006.00945.x. [DOI] [PubMed] [Google Scholar]
- Li Calzi S, Purich DL, Chang KH, Afzal A, Nakagawa T, Busik JV, Agarwal A, Segal MS, Grant MB. Carbon monoxide and nitric oxide mediate cytoskeletal reorganization in microvascular cells via vasodilator-stimulated phosphoprotein phosphorylation: evidence for blunted responsiveness in diabetes. Diabetes. 2008;57:2488–2494. doi: 10.2337/db08-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Bal HS, Desta T, Krothapalli N, Alyassi M, Luan Q, Graves DT. Diabetes enhances periodontal bone loss through enhanced resorption and diminished bone formation. J Dent Res. 2006;85:510–514. doi: 10.1177/154405910608500606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llevadot J, Murasawa S, Kureishi Y, Uchida S, Masuda H, Kawamoto A, Walsh K, Isner JM, Asahara T. HMG-CoA reductase inhibitor mobilizes bone marrow--derived endothelial progenitor cells. J Clin Invest. 2001;108:399–405. doi: 10.1172/JCI13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomans CJ, van Haperen R, Duijs JM, Verseyden C, de Crom R, Leenen PJ, Drexhage HA, de Boer HC, de Koning EJ, Rabelink TJ, Staal FJ, van Zonneveld AJ. Differentiation of bone marrow-derived endothelial progenitor cells is shifted into a proinflammatory phenotype by hyperglycemia. Mol Med. 2009;15:152–159. doi: 10.2119/molmed.2009.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchsinger JA, Tang MX, Stern Y, Shea S, Mayeux R. Diabetes mellitus and risk of Alzheimer’s disease and dementia with stroke in a multiethnic cohort. Am J Epidemiol. 2001;154:635–641. doi: 10.1093/aje/154.7.635. [DOI] [PubMed] [Google Scholar]
- Maestroni GJ, Conti A, Pedrinis E. Effect of adrenergic agents on hematopoiesis after syngeneic bone marrow transplantation in mice. Blood. 1992;80:1178–1182. [PubMed] [Google Scholar]
- Masuo K, Lambert GW, Esler MD, Rakugi H, Ogihara T, Schlaich MP. The role of sympathetic nervous activity in renal injury and end-stage renal disease. Hypertens Res. 33:521–528. doi: 10.1038/hr.2010.35. [DOI] [PubMed] [Google Scholar]
- McMahon JM, Wells DJ. Electroporation for gene transfer to skeletal muscles: current status. BioDrugs. 2004;18:155–165. doi: 10.2165/00063030-200418030-00002. [DOI] [PubMed] [Google Scholar]
- Mendez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452:442–447. doi: 10.1038/nature06685. [DOI] [PubMed] [Google Scholar]
- Mirshahi A, Roohipoor R, Lashay A, Mohammadi SF, Abdoallahi A, Faghihi H. Bevacizumab-augmented retinal laser photocoagulation in proliferative diabetic retinopathy: a randomized double-masked clinical trial. Eur J Ophthalmol. 2008;18:263–269. doi: 10.1177/112067210801800215. [DOI] [PubMed] [Google Scholar]
- Mizutani M, Kern TS, Lorenzi M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest. 1996;97:2883–2890. doi: 10.1172/JCI118746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnier VM, Vishwanath V, Frank KE, Elmets CA, Dauchot P, Kohn RR. Relation between complications of type I diabetes mellitus and collagen-linked fluorescence. N Engl J Med. 1986;314:403–408. doi: 10.1056/NEJM198602133140702. [DOI] [PubMed] [Google Scholar]
- Moradian S, Ahmadieh H, Malihi M, Soheilian M, Dehghan MH, Azarmina M. Intravitreal bevacizumab in active progressive proliferative diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 2008;246:1699–1705. doi: 10.1007/s00417-008-0914-4. [DOI] [PubMed] [Google Scholar]
- Morgado C, Pereira-Terra P, Cruz CD, Tavares I. Minocycline completely reverses mechanical hyperalgesia in diabetic rats through microglia-induced changes in the expression of the potassium chloride co-transporter 2 (KCC2) at the spinal cord. Diabetes Obes Metab. 13:150–159. doi: 10.1111/j.1463-1326.2010.01333.x. [DOI] [PubMed] [Google Scholar]
- Naguib G, Al-Mashat H, Desta T, Graves DT. Diabetes prolongs the inflammatory response to a bacterial stimulus through cytokine dysregulation. J Invest Dermatol. 2004;123:87–92. doi: 10.1111/j.0022-202X.2004.22711.x. [DOI] [PubMed] [Google Scholar]
- Ochner CN, Kwok Y, Conceicao E, Pantazatos SP, Puma LM, Carnell S, Teixeira J, Hirsch J, Geliebter A. Selective reduction in neural responses to high calorie foods following gastric bypass surgery. Ann Surg. 253:502–507. doi: 10.1097/SLA.0b013e318203a289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani A, Kinder K, Ewalt K, Otero FJ, Schimmel P, Friedlander M. Bone marrow-derived stem cells target retinal astrocytes and can promote or inhibit retinal angiogenesis. Nat Med. 2002;8:1004–1010. doi: 10.1038/nm744. [DOI] [PubMed] [Google Scholar]
- Ott EA, Kivipelto J. Influence of chromium tripicolinate on growth and glucose metabolism in yearling horses. J Anim Sci. 1999;77:3022–3030. doi: 10.2527/1999.77113022x. [DOI] [PubMed] [Google Scholar]
- Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG, Jr, Ozcan U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9:35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Paul RG, Bailey AJ. The effect of advanced glycation end-product formation upon cell-matrix interactions. Int J Biochem Cell Biol. 1999;31:653–660. doi: 10.1016/s1357-2725(99)00023-0. [DOI] [PubMed] [Google Scholar]
- Peichev M, Naiyer AJ, Pereira D, Zhu Z, Lane WJ, Williams M, Oz MC, Hicklin DJ, Witte L, Moore MA, Rafii S. Expression of VEGFR-2 and AC133 by circulating human CD34(+) cells identifies a population of functional endothelial precursors. Blood. 2000;95:952–958. [PubMed] [Google Scholar]
- Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol. 2006;1:223–236. doi: 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- Pickup JC. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care. 2004;27:813–823. doi: 10.2337/diacare.27.3.813. [DOI] [PubMed] [Google Scholar]
- Pierce JW, Read MA, Ding H, Luscinskas FW, Collins T. Salicylates inhibit I kappa B-alpha phosphorylation, endothelial-leukocyte adhesion molecule expression, and neutrophil transmigration. J Immunol. 1996;156:3961–3969. [PubMed] [Google Scholar]
- Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, Pennathur S, Baskin DG, Heinecke JW, Woods SC, Schwartz MW, Niswender KD. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab. 2009;296:E1003–1012. doi: 10.1152/ajpendo.90377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pournaras DJ, Aasheim ET, Bueter M, Ahmed AR, Welbourn R, Olbers T, le Roux CW. Effect of bypassing proximal gut on gut hormones involved with glycemic control weight loss. Surg Obes Relat Dis. doi: 10.1016/j.soard.2012.01.021. [DOI] [PubMed] [Google Scholar]
- Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. Jama. 2001;286:327–334. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- Purkayastha S, Zhang H, Zhang G, Ahmed Z, Wang Y, Cai D. Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proc Natl Acad Sci U S A. 108:2939–2944. doi: 10.1073/pnas.1006875108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirici N, Soligo D, Caneva L, Servida F, Bossolasco P, Deliliers GL. Differentiation and expansion of endothelial cells from human bone marrow CD133(+) cells. Br J Haematol. 2001;115:186–194. doi: 10.1046/j.1365-2141.2001.03077.x. [DOI] [PubMed] [Google Scholar]
- Ramana KV, Friedrich B, Srivastava S, Bhatnagar A, Srivastava SK. Activation of nuclear factor-kappaB by hyperglycemia in vascular smooth muscle cells is regulated by aldose reductase. Diabetes. 2004;53:2910–2920. doi: 10.2337/diabetes.53.11.2910. [DOI] [PubMed] [Google Scholar]
- Rameshwar P, Gascon P. Release of interleukin-1 and interleukin-6 from human monocytes by antithymocyte globulin: requirement for de novo synthesis. Blood. 1992;80:2531–2538. [PubMed] [Google Scholar]
- Reid J, Macdougall AI, Andrews MM. Aspirin and diabetes mellitus. Br Med J. 1957;2:1071–1074. doi: 10.1136/bmj.2.5053.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PM, Gutnikov SA, McKinney PA, Schober E, Ionescu-Tirgoviste C, Neu A. Seasonality of birth in children with diabetes in Europe: multicentre cohort study. European Diabetes Study Group. Bmj. 1999;319:887–888. doi: 10.1136/bmj.319.7214.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakagami Y, Girasole G, Yu XP, Boswell HS, Manolagas SC. Stimulation of interleukin-6 production by either calcitonin gene-related peptide or parathyroid hormone in two phenotypically distinct bone marrow-derived murine stromal cell lines. J Bone Miner Res. 1993;8:811–816. doi: 10.1002/jbmr.5650080706. [DOI] [PubMed] [Google Scholar]
- Sanchez M, Rene OBA, Ona V, Li H, Mingwei MD, Friedlander M. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery. 2001;48:1399–1401. doi: 10.1097/00006123-200106000-00051. [DOI] [PubMed] [Google Scholar]
- Schatteman GC, Hanlon HD, Jiao C, Dodds SG, Christy BA. Blood-derived angioblasts accelerate blood-flow restoration in diabetic mice. J Clin Invest. 2000;106:571–578. doi: 10.1172/JCI9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Goldfine AB. Getting away from glucose: fanning the flames of obesity-induced inflammation. Nat Med. 2009;15:373–374. doi: 10.1038/nm0409-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sima AA, Zhang W, Muzik O, Kreipke CW, Rafols JA, Hoffman WH. Sequential abnormalities in type 1 diabetic encephalopathy and the effects of C-Peptide. Rev Diabet Stud. 2009;6:211–222. doi: 10.1900/RDS.2009.6.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivan-Loukianova E, Awad OA, Stepanovic V, Bickenbach J, Schatteman GC. CD34+ blood cells accelerate vascularization and healing of diabetic mouse skin wounds. J Vasc Res. 2003;40:368–377. doi: 10.1159/000072701. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrell JM, Baber MA, Caplan AI. Influence of adult mesenchymal stem cells on in vitro vascular formation. Tissue Eng Part A. 2009;15:1751–1761. doi: 10.1089/ten.tea.2008.0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt AW, Anderson HR, Gardiner TA, Archer DB. Diabetic retinopathy: quantitative variation in capillary basement membrane thickening in arterial or venous environments. Br J Ophthalmol. 1994;78:133–137. doi: 10.1136/bjo.78.2.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt AW, Frizzell N, Thorpe SR. Advanced glycation and advanced lipoxidation: possible role in initiation and progression of diabetic retinopathy. Curr Pharm Des. 2004;10:3349–3360. doi: 10.2174/1381612043383124. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Mitsui K, Yamanaka S. Role of ERas in promoting tumour-like properties in mouse embryonic stem cells. Nature. 2003;423:541–545. doi: 10.1038/nature01646. [DOI] [PubMed] [Google Scholar]
- Tang J, Mohr S, Du YD, Kern TS. Non-uniform distribution of lesions and biochemical abnormalities within the retina of diabetic humans. Curr Eye Res. 2003;27:7–13. doi: 10.1076/ceyr.27.2.7.15455. [DOI] [PubMed] [Google Scholar]
- Tapia-Gonzalez S, Garcia-Segura LM, Tena-Sempere M, Frago LM, Castellano JM, Fuente-Martin E, Garcia-Caceres C, Argente J, Chowen JA. Activation of microglia in specific hypothalamic nuclei and the cerebellum of adult rats exposed to neonatal overnutrition. J Neuroendocrinol. 23:365–370. doi: 10.1111/j.1365-2826.2011.02113.x. [DOI] [PubMed] [Google Scholar]
- Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, Zhao X, Sarruf DA, Izgur V, Maravilla KR, Nguyen HT, Fischer JD, Matsen ME, Wisse BE, Morton GJ, Horvath TL, Baskin DG, Tschop MH, Schwartz MW. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 122:153–162. doi: 10.1172/JCI59660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thill M, Strunnikova NV, Berna MJ, Gordiyenko N, Schmid K, Cousins SW, Thompson DJ, Csaky KG. Late outgrowth endothelial progenitor cells in patients with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49:2696–2708. doi: 10.1167/iovs.07-0955. [DOI] [PubMed] [Google Scholar]
- Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol. 2001;166:7527–7533. doi: 10.4049/jimmunol.166.12.7527. [DOI] [PubMed] [Google Scholar]
- UKPDS. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:837–853. [PubMed] [Google Scholar]
- van de Sande-Lee S, Pereira FR, Cintra DE, Fernandes PT, Cardoso AR, Garlipp CR, Chaim EA, Pareja JC, Geloneze B, Li LM, Cendes F, Velloso LA. Partial reversibility of hypothalamic dysfunction and changes in brain activity after body mass reduction in obese subjects. Diabetes. 2011;60:1699–1704. doi: 10.2337/db10-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma R, Paz SH, Azen SP, Klein R, Globe D, Torres M, Shufelt C, Preston-Martin S. The Los Angeles Latino Eye Study: design, methods, and baseline data. Ophthalmology. 2004;111:1121–1131. doi: 10.1016/j.ophtha.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Velloso LA, Araujo EP, de Souza CT. Diet-induced inflammation of the hypothalamus in obesity. Neuroimmunomodulation. 2008;15:189–193. doi: 10.1159/000153423. [DOI] [PubMed] [Google Scholar]
- Vincent JA, Mohr S. Inhibition of caspase-1/interleukin-1beta signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes. 2007;56:224–230. doi: 10.2337/db06-0427. [DOI] [PubMed] [Google Scholar]
- Wang FH, Liang YB, Zhang F, Wang JJ, Wei WB, Tao QS, Sun LP, Friedman DS, Wang NL, Wong TY. Prevalence of diabetic retinopathy in rural China: the Handan Eye Study. Ophthalmology. 2009;116:461–467. doi: 10.1016/j.ophtha.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Weiss MJ, Orkin SH. In vitro differentiation of murine embryonic stem cells. New approaches to old problems. J Clin Invest. 1996;97:591–595. doi: 10.1172/JCI118454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells-Knecht KJ, Zyzak DV, Litchfield JE, Thorpe SR, Baynes JW. Mechanism of autoxidative glycosylation: identification of glyoxal and arabinose as intermediates in the autoxidative modification of proteins by glucose. Biochemistry. 1995;34:3702–3709. doi: 10.1021/bi00011a027. [DOI] [PubMed] [Google Scholar]
- WHO. WHO Diabetes Fact Sheet N312. Diabetes 2011 [Google Scholar]
- Williamson RT. On the Treatment of Glycosuria and Diabetes Mellitus with Sodium Salicylate. Br Med J. 1901;1:760–762. doi: 10.1136/bmj.1.2100.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SF, Ramasamy R, Schmidt AM. Receptor for AGE (RAGE) and its ligands-cast into leading roles in diabetes and the inflammatory response. J Mol Med (Berl) 2009;87:235–247. doi: 10.1007/s00109-009-0439-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J. Regulation of PPARgamma function by TNF-alpha. Biochem Biophys Res Commun. 2008;374:405–408. doi: 10.1016/j.bbrc.2008.07.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder MC, Mead LE, Prater D, Krier TR, Mroueh KN, Li F, Krasich R, Temm CJ, Prchal JT, Ingram DA. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109:1801–1809. doi: 10.1182/blood-2006-08-043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci U S A. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yune TY, Lee JY, Jung GY, Kim SJ, Jiang MH, Kim YC, Oh YJ, Markelonis GJ, Oh TH. Minocycline alleviates death of oligodendrocytes by inhibiting pro-nerve growth factor production in microglia after spinal cord injury. J Neurosci. 2007;27:7751–7761. doi: 10.1523/JNEUROSCI.1661-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZY, Zhang Z, Fauser U, Schluesener HJ. Improved outcome of EAN, an animal model of GBS, through amelioration of peripheral and central inflammation by minocycline. J Cell Mol Med. 2009;13:341–351. doi: 10.1111/j.1582-4934.2008.00333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng MH, Chen J, Kirilak Y, Willers C, Xu J, Wood D. Porcine small intestine submucosa (SIS) is not an acellular collagenous matrix and contains porcine DNA: possible implications in human implantation. J Biomed Mater Res B Appl Biomater. 2005;73:61–67. doi: 10.1002/jbm.b.30170. [DOI] [PubMed] [Google Scholar]
- Zhou J, Wang S, Xia X. Role of intravitreal inflammatory cytokines and angiogenic factors in proliferative diabetic retinopathy. Curr Eye Res. 2012;37:416–420. doi: 10.3109/02713683.2012.661114. [DOI] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BYS, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wuk DC, Gullans S, Ferrante RJ, Przedborskikq S, Kristal BS, Friedlander M. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]