

Abstract
Rationale
Insulin-like growth factor binding protein-3 (IGFBP-3) modulates vascular development by regulating endothelial progenitor cell (EPC) behavior, specifically stimulating EPC cell migration. This study was undertaken to investigate the mechanism of IGFBP-3 effects on EPC function and how IGFBP-3 mediates cytoprotection following vascular injury.
Objective
To examine the mechanism of IGFBP-3 mediated repair following vascular injury.
Methods and Results
We utilized two complementary vascular injury models: laser occlusion of retinal vessels in adult GFP chimeric mice and oxygen induced retinopathy (OIR) in mouse pups. Intravitreal injection of IGFBP-3 expressing plasmid into lasered GFP chimeric mice stimulated homing of endothelial precursor cells (EPCs), while reversing ischemia induced increases in macrophage infiltration. IGFBP-3 also reduced the retinal ceramide/sphingomyelin ratio that was increased following laser injury. In the OIR model, IGFBP-3 prevented cell death of resident vascular endothelial cells and EPCs, while simultaneously increasing astrocytic ensheathment of vessels. For EPCs to orchestrate repair, these cells must migrate into ischemic tissue. This migratory ability is mediated, in part, by endogenous nitric oxide (NO) generation. Thus, we asked whether IGFBP-3’s migratory effects were due to stimulation of NO generation. IGFBP-3 increased eNOS expression in human EPCs leading to NO generation. IGFBP-3 exposure also led to the redistribution of vasodilator stimulated phosphoprotein (VASP), a NO regulated protein critical for cell migration. IGFBP-3 mediated NO generation required HDL receptor activation and stimulation of PI3K/Akt pathway.
Conclusion
These studies support consideration of IGFBP-3 as a novel agent to restore the function of injured vasculature and restore NO generation.
Keywords: Hematopoietic Stem Cells, Nitric oxide, Insulin-like growth factor binding protein-3, Vascular Repair
INTRODUCTION
Ischemic tissues, by definition, require improved or greater oxygen delivery. Most current pharmacological therapies of ischemic retinopathies try to inhibit pathological neovascularization, but do not correct the underlying lack of blood supply. A more judicious approach would be to develop a pharmacologic intervention to repair or create more normal vessels, thus converting ischemic tissue to non-ischemic tissue. The insulin-like growth factor (IGF) system has been implicated in vascular physiology, including in the retina, as unregulated levels of IGF-1 can lead to inadequate vascularization, as well as pathological ocular neovascularization 1-6. The effects of IGF-1 are mediated by the IGF-1 receptor (IGF-1R) and modulated by complex interactions with six different IGF binding proteins (IGFBPs), which function not only as transporter proteins and storage pools for IGF-1 within tissues, but also as signaling molecules 7, 8. IGFBP-3 has IGF-1 independent effects and autocrine and paracrine actions affecting cell mobility and survival 9, 10. Moreover, IGFBP-3 levels are increased by hypoxia 5.
Recently, we showed IGFBP-3 could serve as a modulator of vascular development by potently regulating bone marrow-derived cell (BMDC) function, specifically, stimulating endothelial precursors to differentiate into endothelial cells and promoting their migration 11. Studies using the oxygen induced retinopathy (OIR) model showed that IGFBP-3 prevented oxygen induced vaso-obliteration and vascular regression in vivo 11 (during phase 1 of the model) and subsequently mitigated pre-retinal neovascularization (phase 2 of the model) 10, 11. Lofqvist et al. showed that IGFBP-3 deficient mice undergoing the OIR model have increased retinal vessel loss compared to wild type controls10. They also showed that premature infants with retinopathy of prematurity (ROP) had lower serum levels of IGFBP-3 than infants with no ROP, thus concluding that increased serum IGFBP-3 improved clinical outcome. These studies collectively support the role of IGFBP-3 as a critical regulator of vascular development.
In this study, we examined possible mechanism(s) by which IGFBP-3 modulated EPC behavior and mediated cytoprotection following vascular injury. For these studies, we utilized both in vitro systems and complementary in vivo vascular injury models: laser occlusion of retinal vessels in adult GFP chimeric mice and OIR in mouse pups. These two models were selected to examine the effects of IGFBP-3 on two distinct types of vasculature. The adult laser model represented the effects of IGFBP-3 on a stable vascular bed and the hyperoxia induced retinal injury model represented the effects of IGFBP-3 on an immature, unstable vascular bed, which was still undergoing active endothelial cell proliferation and migration12.
METHODS
Bone marrow transplantation procedure
The generation of the C57BL/6J.gfp chimeric mice was previously described13.
Laser injury to vessels to induce experimental neovascularization
Two groups of C57BL/6J.gfp chimeric mice: laser only (n=15) and laser with IGFBP-3 injection (n=18) were subjected to laser injury in their right eye as previously described13. A third group of chimeric mice received only IGFBP-3 injection into their right eye (n=16).
The IGFBP-3 plasmid, under control of a proliferating endothelial cell specific promoter, 14 was packaged into liposomes as previously described14. Immediately following laser treatment to the right eye, a total of 2 μl of IGFBP-3 plasmid (2 μg/μl) packaged in liposomes was delivered intravitreally into the right eye of the appropriate groups.
OIR model studies
We used the OIR model developed by Smith, et al 15. Mice were intravitreally injected with 0.5 μl (2 mg/ml) into one eye with either the IGFBP-3 plasmid or the cloning vector on postnatal day 1 (P1). Some animals were euthanized upon removal from hyperoxia at P12, while others were sacrificed at P17. At both time points, eyes from mice injected with the plasmid expressing IGFBP-3 (n=6) were compared to eyes from mice injected with empty cloning vector (n=6) or the uninjected eye of the same animal (n=6). Mice were euthanized and eyes were enucleated and fixed for analysis as previously reported16. Details of immunohistochemistry procedures are in the supplemental methods section17.
Lipid analysis by nESI-MS/MS
Lipid species were detected using lipid class-specific precursor ion and neutral loss MS/MS scans as previously described18, 19 and fatty acid constituents of individual lipid species were verified by product ion mode MS/MS in negative ion mode.
Refer to the supplemental methods section for details of the following studies: determination of NO production by DAF-FM fluorescence imaging, eNOS activity and in vitro functional studies in rat mesenteric and cerebral arteries.
RESULTS
IGFBP-3 promotes retinal vascular repair by increasing BMDC homing following mechanical disruption by laser in GFP chimeric mice
In GFP chimeric mice receiving intravitreal injection of IGFBP-3 plasmid, GFP+ cells robustly participated in vascular remodeling (Figure 1A-C) with GFP+ cells differentiating into endothelial cells (Online Figure 1A-I). Greater GFP+ cell incorporation was clearly evident in IGFBP-3 plasmid injected eyes than in non-injected control eyes (Figure 1A-C compared with Figure 1D-F). When the vasculature of GFP chimeric mice was injured by laser, significant numbers of circulating GFP+ cells took on the appearance of activated macrophages (Online Figure 2). In the animals receiving both laser and IGFBP-3 plasmid (Online Figure 2 D-F), the distribution of GFP+ cells exhibited a similar vascular pattern as seen with IGFBP-3 alone (Online Figure 2 A-C), but the number of activated macrophages was greatly reduced compared to laser alone treated mice. We also confirmed by mRNA expression that intraocular injection of the IGFBP-3 expressing plasmid resulted in high IGFBP-3 mRNA levels in laser treated mice for up to 1 week post laser injury (Online Figure 3).
Figure 1. IGFBP-3 enhances GFP+ BMDCs incorporation into vasculature.

(A-C) IGFBP-3 injected chimeric mice showing GFP+ cells participating in vascular remodeling and incorporating into existing blood vessels (B) GFP cells stained with anti-GFP antibody (conjugated to Alexa 488) (C) red stain is rhodamine agglutinin (blood vessel specific). (D-F) control chimeric mouse retina depicting little GFP+ incorporation into the retinal vasculature. (E) GFP cells stained with anti-GFP antibody (conjugated to Alexa 488) (F) red stain is rhodamine agglutinin.
IGFBP-3 reduces inflammation following laser occlusion by reducing the retinal ceramide/sphingomyelin ratio
Sphingolipids are lipid components of the membrane known to be highly expressed in the retina. The most abundant sphingolipid in the membrane, sphingomyelin, can be converted to ceramide under certain conditions, such as inflammation and the ceramide/sphingomyelin ratio can be used as an indicator of the inflammatory/pro-apoptotic state. Laser treatment induced a 3.34-fold increase in ceramide/sphingomyelin ratio, consistent with pro-inflammatory and pro- apoptotic effects. IGFBP-3 returned ceramide/sphingomyelin ratio to control (untreated) levels (Figure 2). We found no difference in ceramide/sphingomyelin levels in the IGFBP-3 only injected animals (Online Figure 4B) compared to controls (Online Figure 4A). We analyzed another member of the sphingolipid family, sphingosine-1-phosphate (S1P). The levels of retinal S1P were low in all groups and there was no difference between the groups, suggesting that any elevation of SIP following injury had returned to baseline levels by three weeks (data not shown).
Figure 2. IGFBP-3 reduces the retinal ceramide/sphingomyelin ratio.

Retinal lipid extracts from control, laser, or laser plus IGFBP-3 treated mice were analyzed for ceramide and sphingomyelin molecular species as their [M+H]+ ions by precursor ion scanning of m/z 264.4 and m/z 184, respectively, after alkaline hydrolysis of glycerophospholipids. The data were normalized to internal standards and ratios of total retinal ceramide/sphingomyelin were calculated. The results are mean±SD of 3 independent experiments. *P<0.05 vs. control is statistically significant.
IGFBP-3 reversed lipid profile changes induced by laser occlusion
As lipid peroxidation and lipid metabolism changes are highly associated with a number of retinal pathologies, we next addressed the effect of laser treatment on the structural component of retinal membranes, specifically membrane phospholipids. Laser treatment induced a significant increase in total retinal incorporation of DHA (22:6n3) into glycerophosphatidylcholine (GPCho) and glycerophosphatidylinositol (GPIns). All GPCho and GPIns changes were completely reversed by IGFBP-3 treatment (Online Figure 5 and 6, respectively).
IGFBP-3 reduces endothelial cell death following hyperoxia-induced injury
We next used the OIR model, which exhibits extensive endothelial cell loss by apoptosis, to evaluate whether IGFBP-3 could mediate cytoprotection following injury by reducing cell death. We injected IGFBP-3 plasmid into the vitreous of mouse pups (P1) and then placed pups in hyperoxia on P7 for 5 days following the standard OIR model. The retinas of mice injected with IGFBP-3 had significantly reduced endothelial cell death (Figure 3 D-F) in both the hyperoxic (phase1) [Mid-peripheral P>0.05, Peripheral P<0.05] and hypoxic phases (phase 2) of the OIR model compared to controls (A-C) [Mid-peripheral P<0.05; Peripheral P<0.05) (One-way ANOVA)]. This reduction in endothelial cell death was only evident in the mid-peripheral and peripheral retina as the oxygen induced vascular regression associated with the OIR model results in very little vasculature in the central retina. IGFBP-3 did not reduce pericyte or astrocyte cell death (data not shown).
Figure 3. IGFBP-3 has vascular stabilizing effects by preventing endothelial cell death.

(A-C) shown in green are TUNEL+ cells; blue indicates GS lectin+ endothelial cells; white arrows point to TUNEL+/GS lectin+ endothelial cells. In comparison to controls (A-C) IGFBP-3 injected eyes (D-F) had significantly reduced endothelial cell death during the hyperoxic phase of the OIR model.
IGFBP-3 increases astrocytic ensheathment of neovasculature following hyperoxia-induced injury
We next asked whether IGFBP-3 could stabilize vessels during hyperoxia by increasing astrocytic ensheathment of retinal blood vessels. Compared with control plasmid injected eyes (Figure 4 A-D), IGFBP-3 injected eyes led to astrocytes with far greater S-100/GFP immunoreactivity (shown as red in Figure 4 E-H). The S-100 ensheathment of the underlying vasculature (shown as blue) was much more complete and the astrocytes had larger, thicker processes. Quantification of the frequency of astrocytic ensheathment showed that S-100 astrocytic ensheathment of retinal vasculature was significantly increased in both P12 (Figure 4 I-J) and P17 (Figure 4 K-L) IGFBP-3 injected eyes in comparison to control plasmid injected eyes (p<0.05).
Figure 4. IGFBP-3 increases astrocytic ensheathment of neovasculature following hyperoxia induced injury.

In comparison to control (A-D) the S-100/GFAP ensheathment of underlying vasculature in the IGFBP-3 injected eyes (E-H) was much more complete and the astrocytes showed larger, thicker processes. (I-L) shows representative fields of view from the mid-peripheral retina using a 20× objective during normal development and during exposure to the oxygen induced retinopathy model. The ensheathment of GS Lectin+ vascular endothelial cells (blue) by S-100+ astrocytes (red) were determined. S-100 ensheathment of underlying vasculature in the IGFBP-3 injected eyes (J) was much more complete than in control injected eyes (I) at P12. S-100 ensheathment of underlying vasculature in the IGFBP-3 injected eyes (L) was much more complete than in control injected eyes (K) at P17. (M) Astrocyte ensheathment of blood vessels was found to be more significant (P<0.05) in both P12 and P17 IGFBP-3 injected eyes in comparison to controls.
IGFBP-3 generates NO via the SRB1
IGFBP-3 has both pro-mitotic and pro-apoptotic effects. Such diverse biological effects can be mediated by activation of scavenger receptors20-24. We asked whether IGFBP-3 may be initiating its vascular protective effect by binding to the scavenger receptor class B, type1 (SR- B1), which is the HDL receptor25, 26. Activation of this receptor results in NO generation27. In HUVECs, IGFBP-3 at a physiological concentration (100 ng/ml) increased NO release as seen by an increase in DAF-FM fluorescence (Figure 5). HDL at the physiological concentration of 1 mg/ml similarly increased NO generation, as did the combination of IGFBP-3 and HDL (P<0.05 for all treatments). In HUVECs, pre-treatment with SR-B1 blocking antibody (SRB1-Ab, 1:100) prevented NO release by IGFBP-3 (P<0.02) and, as expected, decreased the NO production in response to HDL P<0.0001 as well as to the combination of both agents (P<0.01). Similar results were observed in HMVEC-L whereas in the RGC-5 cells that do not express SR-B1, IGFBP-3 mediated NO release was not altered with co-administration of HDL and was not affected by the addition of SRB1-Ab.
Figure 5. IGFBP-3 activates the SR-Bl receptor leading to NO generation in endothelial cells.

Determination of NO release in response to IGFBP-3 and HDL by DAF-FM fluorescence imaging. NO release in response to IGFBP-3 and/or HDL in HUVEC, HMEC and RGC-5 cells. Treatment with SRB1-Ab significantly decreased NO release by either IGFBP-3 or HDL or the combination of IGFBP-3 and HDL. (A-C) shown in the right were respective scales for fluorescence in three different cell types. Images obtained were cells that were either not treated (control) or treated as labeled. (D) Changes in fluorescence with different treatments were expressed as percent increase over the control.*P<0.05 and **P<0.02 compared with IGFBP-3; ***P<0.01 compared with HDL+IGFBP-3; ****P<0.0001 compared with HDL. #P<0.05 compared with IGFBP-3 or HDL alone; ##P<0.01 compared with HDL+IGFBP-3; ###P<0.001 compared with HDL or IGFBP-3. Representative results from three independent experiments are shown.
IGFBP-3 increased NO production in human CD34+ cells (Figure 6) and in contrast to the response in HUVECs and HMVEC-L, the response to IGFBP-3 was greater than the response to HDL. When both agents were added simultaneously to CD34+ cells, the response was intermediate, less than with IGFBP-3, but greater than with HDL (Figure 6). We next confirmed the effect of IGFBP-3 on NO generation was due to increasing phosphorylation of eNOS at Ser1177 (Online Figure 7 A&B) in human CD34+ cells.
Figure 6. IGFBP-3 increased NO production in CD34+ cells and the response to IGFBP-3 was greater than the response observed with HDL.
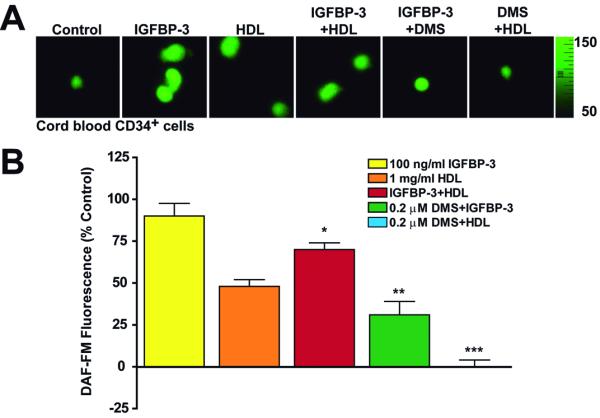
Determination of NO release by DAF-FM fluorescence imaging in human cord blood CD34+ cells. (A) shown in the right is the scale for fluorescence. Images obtained were cells that were either not treated (control) or treated as labeled. (B) changes in fluorescence with different treatments were expressed as percent increase over the control.*P<0.01 compared with IGFBP-3 or HDL alone; **P<0.001 compared with IGFBP-3; ***P<0.0001 compared with HDL. Representative results from three independent experiments are shown.
Our studies thus far show that IGFBP-3 stimulates NO release independent of HDL in two distinct endothelial cell types and in endothelial progenitors and that this effect is mediated by the SR-B1 receptor. To examine the signaling pathways by which SR-B1 activation by IGFBP-3 induces NO generation, we used selective pharmacological inhibitors to block PI3K or Akt. IGFBP- 3 mediated NO generation that was sensitive to blockade of SR-B1 or eNOS (by L-NAME) and was significantly decreased by either pretreatment with the PI3K inhibitor LY294002 (P<0.0001) or the Akt inhibitor, triciribin (P<0.01) (Figure 7 A&B). We further confirmed these observations using a biochemical assay of eNOS activity involving determination of the conversion of L-arginine to L- citrulline in HUVECs. IGFBP-3 (100 ng/ml) stimulated eNOS activity was significantly reduced by pretreatment with SRB1-Ab or LY294002 or triciribin (P<0.05) (Figure 7C). We next examined the effect of IGFBP-3 on phosphorylation of eNOS at ser1177 in HUVECs. IGFBP-3 increased eNOS phosphorylation which was blocked by pretreatment with SRB1-Ab or triciribin significantly (Online Figure 8). These results support that IGFBP-3 induces eNOS activation and NO generation which is dependent upon the SRB1-PI3K-Akt signaling pathway.
Figure 7. IGFBP-3 activates nitric oxide synthase and causes NO release by activating SR-B1/PI3-kinase/Akt pathway.

(A) NO production in response to IGFBP-3 was measured by DAF- FM fluorescence in HUVECs and effects of different pharmacological blockers were evaluated (color scale for fluorescence). Images obtained were cells that were either not treated (time control) or treated as labeled. (B) Changes in fluorescence with different treatments were expressed as percent increase over the time control. NO release by IGFBP-3 was significantly decreased by pretreatment with SR-B1 blocking antibody (SRB1-Ab), LY294002, triciribin or L- NAME. *P<0.01 and ***P<0.0001 compared with IGFBP-3. (C) eNOS activity expressed as L- NAME inhibitable conversion of [14C]L-arginine to [14C]L-citrulline was stimulated by 100 ng/ml IGFBP-3. Pretreatment with SRB1-Ab, 30 μM LY294002 or 30 μM triciribin significantly decreased IGFBP-3 induced eNOS activation (*P<0.05). Representative results from three independent experiments are shown.
IGFBP-3 has also been shown to increase the activity of the sphingosine kinase (SphK-1), the enzyme responsible for the generation of the potent angiogenic factor sphingosine-1- phosphate (S1P) 7. Addition of the sphingosine kinase (SphK) inhibitor, N,N-Dimethylsphingosine (DMS), resulted in a reduction in NO generation in response to IGFBP-3, as well as HDL, supporting that S1P generation was contributing to the effects of both HDL and IGFBP-3 on NO generation. CD34+ cells, HUVECs, and HMVEC-L express S1P receptors, primarily S1PR1, and express both SphK1 and SphK2 (Online Figure 9 A&B).
IGFBP-3 causes vasodilatation via SRB1-dependent NO release
One of the primary functions of NO in the vasculature is to stimulate vasodilatation and numerous vascular protective agents stimulate NO generation. To determine whether IGFBP-3 mediates its protective effects by promoting NO mediated vasodilatation, we performed studies using intact arterial segments from two different vascular beds and assessed whether the NO released by IGFBP-3 produced a functional response in vitro. Rat mesenteric arterial segments (diameter 276±11 microns, n=12) pre-constricted with phenylephrine, were completely relaxed to carbachol, indicating the presence of functional endothelium. HDL (1 mg/ml) produced marked relaxation in the phenylephrine pre-contracted arteries. IGFBP-3 (100 ng/ml) produced a relaxant effect, however, weaker than HDL (Figure 8 A&B). Phenylephrine pre-contraction was decreased by HDL and IGFBP-3 to 58±8% (n=4 p<0.0001) and 81±7% (n=4, P<0.001) of the control, respectively. After IGFBP-3 treatment, addition of HDL resulted in an additional relaxation causing a total decrease in pre-contraction to 51±7% (n=3) (Figure 8D). Conversely, addition of IGFBP-3 after HDL treatment did not cause any further relaxation (data not shown). Furthermore, in a separate set of experiments, we observed IGFBP-3 did not produce any relaxation in arteries pretreated with L-NAME, a commonly used inhibitor of eNOS. (Figure 8 C&D).
Figure 8. IGFBP-3 decreases arterial tone by increasing NO release in rat arteries.

Representative tracings of tension (mN) developed (constriction) in wire-mounted rat mesenteric arterial segments to phenylephrine and the relaxation (dilatation) produced by (A) 1 mg/ml HDL and (B) 100 ng/ml IGFBP-3 with further additions of HDL and 10 μM carbachol. (C) Pretreatment with L-NAME (100 μM) resulted in blockade of relaxation response to IGFBP-3. (D) Summary of the effects of HDL and IGFBP-3 on constriction to phenylephrine. Data presented were percent constriction of phenylephrine. *P<0.001 compared to control. In the presence of L-NAME no relaxation was observed with IGFBP-3. (E) IGFBP-3 caused dilation of rat cerebral arteries by attenuating pressure induced constriction at 70 mmHg. Pressure induced constriction, expressed as percent decrease in the intraluminal diameter, was significantly decreased in the presence of intraluminal IGFBP-3 (100 ng/ml, *P<0.04) and this decrease was blocked when IGFBP-3 was applied with SR-B1 function blocking antibody (SRB1-Ab) or 300 μM L-NAME.
In another set of experiments we pressurized rat posterior cerebral arteries (passive diameter 175±5 microns, n=15) in vitro at 70 mm Hg and evaluated the effect of IGFBP-3 on pressure induced constriction (decrease in intraluminal diameter) by applying it intraluminally. Pressurized arteries showed stable constriction in response to pressure and this constriction was significantly decreased by intraluminal IGFBP-3 (100 ng/ml). In the presence of intraluminal SRB1-Ab or L-NAME, dilation by IGFBP-3 (decrease in constriction) was not observed (Figure 8E), suggesting that IGFBP-3 can cause arterial dilatation via SR-B1 dependent endothelial NO release in the arterial wall.
IGFBP-3 mediated NO generation stimulates VASP redistribution
Previously, we demonstrated that a critical function of NO is to promote EPC migration by regulating the distribution of the cytoskeletal protein VASP17. VASP plays a pivotal role in promoting actin filament elongation at the leading edge of the cell by forming an active molecular motor complex that propels motility. Migration of BMDCs into areas of ischemia is paramount to their ability to initiate and orchestrate repair11. IGFBP-3 stimulated migration was mediated by NO generation and blocked by the addition of the NO scavenger 2-(4- carboxyphenol)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (data not shown). To examine this further, we asked whether treatment with IGFBP-3 resulted in redistribution of VASP. Compared to control conditions (Online Figure 10A), IGFBP-3 treatment resulted in redistribution of to the lamellipodia VASP fluorescence to the periphery (Online Figure 10 C), which was entirely eliminated by pre-treatment with an NO scavenger (Online Figure 10E).
DISCUSSION
In this study, we identified cellular and signaling mechanisms responsible for IGFBP-3’s vascular protective effects. We examined the effects of IGFBP-3 on two distinct types of vasculature, a stable vascular bed (adult chimeric mice undergoing laser occlusion) and an immature, unstable vascular bed undergoing active endothelial cell proliferation and migration (the hyperoxia induced retinal injury model).
In the adult model, we show IGFBP-3 enhances repair by recruiting BMDCs to sites of laser occlusion within the ischemic retina. Even in the absence of retinal injury, overexpression of IGFBP-3 by the resident retinal endothelium promoted extravasation of BMDCs from the circulation into the perivascular region and their incorporation into areas of vascular remodeling. The reparative effects of IGFBP-3 are not limited to promoting BMDC homing, as IGFBP-3 reduced endothelial cell death and inflammation. During repair of the retina, IGFBP-3 increased astrocyte ensheathment, facilitating astrocyte-endothelial cell interactions, likely leading to enhanced barrier properties of the neovasculature and enhanced neuro-vascular coupling. Astrocytes serve as a template for both developmental and injury associated angiogenesis28.
NO is an essential signaling molecule that promotes angiogenesis and regulates vascular tone and vascular remodeling29. Endogenous NO generation by BMDC is critical for their migration, which in turn is required for their reparative function. IGFBP-3 increased eNOS phosphorylation leading to increased NO generation and subsequent VASP redistribution promoting cell migration. Previously, we showed that IGFBP-3 promotes cell migration11. In this study, we showed that following IGFBP-3 treatment redistribution of VASP occurs to focal adhesions and pseudopodia, which we quantified (Online Figure 9). The rapid increase in VASP (within 15 minutes) suggests that VASP redistribution occurred, rather than a change in VASP protein expression.
Our studies support that SRB1 mediates IGFBP-3 induced NO generation and that this occurs independent of HDL. Activation of eNOS by phosphorylation at Ser1177 by IGFBP-3 is dependent on activation of SR-B1 and the downstream signaling pathway involving P13K and Akt activity. We further show that SR-B1 dependent release of NO by IGFBP-3 modulates vascular reactivity in intact artery preparations from rat mesenteric and cerebral vascular beds resulting in decreased tone or dilation and this effect is potentially vascular protective. Based on these studies we postulate that SR-B1 activation by IGFBP-3 in the retinal vasculature generates NO, which is critical for vasodilatation, facilitating increased blood flow thereby infiltration of BMDCs to the sites of ischemia. NO released by IGFBP-3 in circulating EPCs and resident endothelial cells may modulate the function of BMDCs in an autocrine as well as a paracrine manner hence, contributing to vascular repair.
In agreement with Granata et al in HUVECs7, our studies in CD34+ cells support the notion that IGFBP-3 mediated NO generation is also dependent on activation of SphK1, as we observed that NO generation is blocked by SphK inhibition. S1P mediated NO generation occurs by activation of endothelial differentiation gene receptors (EDGRs), also known as the sphingosine-1-phosphate receptors (S1PRs). S1P, much like IGFBP-3, increases NO generation, thus, promoting migration of cells 30. Moreover S1P, like IGFBP-3, has direct vascular protective effects. In the blood, S1P is associated with lipoproteins including low density lipoprotein, very low density lipoprotein, and HDL with the majority of S1P being bound to HDL31,32. Our studies support that in EPCs, both IGFBP-3 and HDL activate SphK1 as inhibiting SphK1 with DMS resulted in loss of NO generation in response to either agents (Figure 6).
While we observed no increase in S1P in vivo in response to increase in IGFBP-3, we performed our measurements 3 weeks following retinal injury and we suspect that any acute rise in S1P would have returned to baseline levels by this point. Despite our inability to detect S1P changes in the retina, we detected changes in other sphingolipids that persisted at 3 weeks. The laser treated eyes had a higher ceramide/sphingomyelin ratio consistent with an inflammatory, pro-apoptotic state. However, this increase in ceramide/sphingomyelin ratio was completely normalized by IGFBP-3. This suggests IGFBP-3 mediates vascular protection through inhibition of inflammation, allowing IGFBP-3 to promote the recruitment of the reparative BMDC population, EPCs, rather than an inflammatory population. As observed in our studies using the adult laser injury model, IGFBP-3 reduces the number of inflammatory cells, specifically macrophages. We also found a striking increase in DHA containing lysolipids in laser treated eyes, indicating oxidative damage to the retina. These changes were completely normalized by increasing expression of IGFBP-3. In addition, there was an increase in DHA containing phospholipids in laser treated eyes and IGFBP-3 treatment returned the phospholipid profile to normal levels, further supporting the role of IGFBP-3 in the repair of retinal vasculature.
While this study has identified a new receptor system that mediates the effects of IGFBP- 3 with respect to NO generation, IGFBP-3 has been shown to bind to several other receptors including the TGF-β receptor V 33, transferrin receptor 34 and low-density lipoprotein receptor (LDLR)-related protein (LRP). However when IGFBP-3 binds to LRP-1, it is degraded35. Hence, the diverse physiological effects of IGFBP-3 are likely to be mediated by distinct receptor systems.
In summary, our data support the hypothesis that IGFBP-3 mediates functional revascularization in the retina by promoting the homing of beneficial EPCs, while reducing the number of detrimental inflammatory cells, such as macrophages. Once EPCs are routed to areas of damage, IGFBP-3 enhances their incorporation and differentiation into endothelial cells, which facilitates vessel formation11. IGFBP-3 serves to protect the resident endothelium and EPC from cell death and increases astrocyte ensheathment to enhance vessel barrier properties and autoregulatory capacity36. Our studies suggest that these beneficial effects may be mediated by increased NO generation which occurs via IGFBP-3 binding of SR-B1 and subsequent activation of the P13K–Akt pathway. Our studies also support a second mechanism by which IGFBP-3 stimulates NO generation by activation of Sphk-1. We postulate that IGFBP-3 may modulate interactions between the scavenger receptor system and the S1PR system and may serve to regulate both physiological and pathological angiogenesis and vascular remodeling. Our findings support that IGFBP-3 in the circulation and in tissues may represent an endogenous vascular protective protein and following vascular injury increased levels of IGFBP-3 could promote proper revascularization.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Guoqin Niu for preparing the liposomes, Doug Smith and Neal Benson for invaluable assistance with cell sorting and flow cytometry and Dr. Koji Hosaka for technical assistance during laser procedures.
Sources of Funding This work was supported by grants to MBG from NIH (EY007739 and EY012601), JDRF and the AHA, to JB from NIH/NEI (EY-016077) and to TCL from ISLG (CG100045) and the NHMRC of Australia (#402824, 464859).
Non-standard Abbreviations and Acronyms
- (IGFBP-3)
Insulin-like growth factor binding protein-3
- (BMDC)
Bone marrow derived cell
- (EPC)
Endothelial progenitor cell
- (HSC)
Hematopoietic stem cells
- (SR-B1)
Scavenger Receptor class B, type 1
Footnotes
Disclosure None.
REFERENCES
- 1.Grant MB, Schmetz I, Russell B, Harwood HJ, Jr., Silverstein J, Merimee TJ. Changes in insulin-like growth factors I and II and their binding protein after a single intramuscular injection of growth hormone. J Clin Endocrinol Metab. 1986;63(4):981–984. doi: 10.1210/jcem-63-4-981. [DOI] [PubMed] [Google Scholar]
- 2.Lofqvist C, Andersson E, Sigurdsson J, Engstrom E, Hard AL, Niklasson A, Smith LE, Hellstrom A. Longitudinal postnatal weight and insulin-like growth factor I measurements in the prediction of retinopathy of prematurity. Arch Ophthalmol. 2006;124(12):1711–1718. doi: 10.1001/archopht.124.12.1711. [DOI] [PubMed] [Google Scholar]
- 3.Spoerri PE, Ellis EA, Tarnuzzer RW, Grant MB. Insulin-like growth factor: receptor and binding proteins in human retinal endothelial cell cultures of diabetic and non-diabetic origin. Growth Horm IGF Res. 1998;8(2):125–132. doi: 10.1016/s1096-6374(98)80102-0. [DOI] [PubMed] [Google Scholar]
- 4.Moralez A, Busby WH, Jr., Clemmons D. Control of insulin-like growth factor binding protein-5 protease synthesis and secretion by human fibroblasts and porcine aortic smooth muscle cells. Endocrinology. 2003;144(6):2489–2495. doi: 10.1210/en.2002-220896. [DOI] [PubMed] [Google Scholar]
- 5.Firth SM, Baxter RC. Cellular actions of the insulin-like growth factor binding proteins. Endocr Rev. 2002;23(6):824–854. doi: 10.1210/er.2001-0033. [DOI] [PubMed] [Google Scholar]
- 6.Sakai K, Busby WH, Jr., Clarke JB, Clemmons DR. Tissue transglutaminase facilitates the polymerization of insulin-like growth factor-binding protein-1 (IGFBP-1) and leads to loss of IGFBP-1’s ability to inhibit insulin-like growth factor-I-stimulated protein synthesis. J Biol Chem. 2001;276(12):8740–8745. doi: 10.1074/jbc.M008359200. [DOI] [PubMed] [Google Scholar]
- 7.Granata R, Trovato L, Garbarino G, Taliano M, Ponti R, Sala G, Ghidoni R, Ghigo E. Dual effects of IGFBP-3 on endothelial cell apoptosis and survival: involvement of the sphingolipid signaling pathways. Faseb J. 2004;18(12):1456–1458. doi: 10.1096/fj.04-1618fje. [DOI] [PubMed] [Google Scholar]
- 8.Liu B, Lee KW, Li H, Ma L, Lin GL, Chandraratna RA, Cohen P. Combination therapy of insulin-like growth factor binding protein-3 and retinoid X receptor ligands synergize on prostate cancer cell apoptosis in vitro and in vivo. Clinical Cancer Research. 2005;11:4851–4856. doi: 10.1158/1078-0432.CCR-04-2160. [DOI] [PubMed] [Google Scholar]
- 9.Le Jan S, Le Meur N, Cazes A, Philippe J, Le Cunff M, Leger J, Corvol P, Germain S. Characterization of the expression of the hypoxia-induced genes neuritin, TXNIP and IGFBP3 in cancer. FEBS Lett. 2006;580(14):3395–3400. doi: 10.1016/j.febslet.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 10.Lofqvist C, Chen J, Connor KM, Smith AC, Aderman CM, Liu N, Pintar JE, Ludwig T, Hellstrom A, Smith LE. IGFBP3 suppresses retinopathy through suppression of oxygen-induced vessel loss and promotion of vascular regrowth. Proc Natl Acad Sci U S A. 2007;104(25):10589–10594. doi: 10.1073/pnas.0702031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang KH, Chan-Ling T, McFarland EL, Afzal A, Pan H, Baxter LC, Shaw LC, Caballero S, Sengupta N, Li Calzi S, Sullivan SM, Grant MB. IGF binding protein- 3 regulates hematopoietic stem cell and endothelial precursor cell function during vascular development. Proc Natl Acad Sci U S A. 2007;104(25):10595–10600. doi: 10.1073/pnas.0702072104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med. 1995;1(10):1024–1028. doi: 10.1038/nm1095-1024. [DOI] [PubMed] [Google Scholar]
- 13.Sengupta N, Caballero S, Mames RN, Butler JM, Scott EW, Grant MB. The role of adult bone marrow-derived stem cells in choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44(11):4908–4913. doi: 10.1167/iovs.03-0342. [DOI] [PubMed] [Google Scholar]
- 14.Shaw LC, Pan H, Afzal A, Li Calzi S, Spoerri PE, Sullivan SM, Grant MB. Proliferating endothelial cell-specific expression of IGF-I receptor ribozyme inhibits retinal neovascularization. Gene Ther. 2006;13(9):752–760. doi: 10.1038/sj.gt.3302718. [DOI] [PubMed] [Google Scholar]
- 15.Smith LE, Wesolowski E, McLellan A, Kostyk SK, D’Amato R, Sullivan R, D’Amore PA. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35(1):101–111. [PubMed] [Google Scholar]
- 16.Chan-Ling T. Glial, vascular, and neuronal cytogenesis in whole-mounted cat retina. Microsc Res Tech. 1997;36(1):1–16. doi: 10.1002/(SICI)1097-0029(19970101)36:1<1::AID-JEMT1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 17.Li Calzi S, Purich DL, Chang KH, Afzal A, Nakagawa T, Busik JV, Agarwal A, Segal MS, Grant MB. Carbon monoxide and nitric oxide mediate cytoskeletal reorganization in microvascular cells via vasodilator-stimulated phosphoprotein phosphorylation: evidence for blunted responsiveness in diabetes. Diabetes. 2008;57(9):2488–2494. doi: 10.2337/db08-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pulfer M, Murphy RC. Electrospray mass spectrometry of phospholipids. Mass Spectrom Rev. 2003;22(5):332–364. doi: 10.1002/mas.10061. [DOI] [PubMed] [Google Scholar]
- 19.Han X, Gross RW. Shotgun lipidomics: multidimensional MS analysis of cellular lipidomes. Expert Rev Proteomics. 2005;2(2):253–264. doi: 10.1586/14789450.2.2.253. [DOI] [PubMed] [Google Scholar]
- 20.Seetharam D, Mineo C, Gormley AK, Gibson LL, Vongpatanasin W, Chambliss KL, Hahner LD, Cummings ML, Kitchens RL, Marcel YL, Rader DJ, Shaul PW. High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B type I. Circ Res. 2006;98(1):63–72. doi: 10.1161/01.RES.0000199272.59432.5b. [DOI] [PubMed] [Google Scholar]
- 21.Tauber JP, Cheng J, Gospodarowicz D. Effect of high and low density lipoproteins on proliferation of cultured bovine vascular endothelial cells. J Clin Invest. 1980;66(4):696–708. doi: 10.1172/JCI109907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sawamura T, Kume N, Aoyama T, Moriwaki H, Hoshikawa H, Aiba Y, Tanaka T, Miwa S, Katsura Y, Kita T, Masaki T. An endothelial receptor for oxidized low- density lipoprotein. Nature. 1997;386(6620):73–77. doi: 10.1038/386073a0. [DOI] [PubMed] [Google Scholar]
- 23.Chen CH, Jiang T, Yang JH, Jiang W, Lu J, Marathe GK, Pownall HJ, Ballantyne CM, McIntyre TM, Henry PD, Yang CY. Low-density lipoprotein in hypercholesterolemic human plasma induces vascular endothelial cell apoptosis by inhibiting fibroblast growth factor 2 transcription. Circulation. 2003;107(16):2102–2108. doi: 10.1161/01.CIR.0000065220.70220.F7. [DOI] [PubMed] [Google Scholar]
- 24.Imanishi T, Hano T, Matsuo Y, Nishio I. Oxidized low-density lipoprotein inhibits vascular endothelial growth factor-induced endothelial progenitor cell differentiation. Clin Exp Pharmacol Physiol. 2003;30(9):665–670. doi: 10.1046/j.1440-1681.2003.03894.x. [DOI] [PubMed] [Google Scholar]
- 25.Yuhanna IS, Zhu Y, Cox BE, Hahner LD, Osborne-Lawrence S, Lu P, Marcel YL, Anderson RG, Mendelsohn ME, Hobbs HH, Shaul PW. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat Med. 2001;7(7):853–857. doi: 10.1038/89986. [DOI] [PubMed] [Google Scholar]
- 26.Trigatti B, Rigotti A, Krieger M. The role of the high-density lipoprotein receptor SR-BI in cholesterol metabolism. Curr Opin Lipidol. 2000;11(2):123–131. doi: 10.1097/00041433-200004000-00004. [DOI] [PubMed] [Google Scholar]
- 27.Nofer JR, van der Giet M, Tolle M, Wolinska I, von Wnuck Lipinski K, Baba HA, Tietge UJ, Godecke A, Ishii I, Kleuser B, Schafers M, Fobker M, Zidek W, Assmann G, Chun J, Levkau B. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J Clin Invest. 2004;113(4):569–581. doi: 10.1172/JCI18004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otani A, Kinder K, Ewalt K, Otero FJ, Schimmel P, Friedlander M. Bone marrow- derived stem cells target retinal astrocytes and can promote or inhibit retinal angiogenesis. Nat Med. 2002;8(9):1004–1010. doi: 10.1038/nm744. [DOI] [PubMed] [Google Scholar]
- 29.Sessa WC. Molecular control of blood flow and angiogenesis: role of nitric oxide. J Thromb Haemost. 2009;7(Suppl 1):35–37. doi: 10.1111/j.1538-7836.2009.03424.x. [DOI] [PubMed] [Google Scholar]
- 30.Rikitake Y, Hirata K, Kawashima S, Ozaki M, Takahashi T, Ogawa W, Inoue N, Yokoyama M. Involvement of endothelial nitric oxide in sphingosine-1-phosphate- induced angiogenesis. Arterioscler Thromb Vasc Biol. 2002;22(1):108–114. doi: 10.1161/hq0102.101843. [DOI] [PubMed] [Google Scholar]
- 31.Kimura T, Sato K, Kuwabara A, Tomura H, Ishiwara M, Kobayashi I, Ui M, Okajima F. Sphingosine 1-phosphate may be a major component of plasma lipoproteins responsible for the cytoprotective actions in human umbilical vein endothelial cells. J Biol Chem. 2001;276(34):31780–31785. doi: 10.1074/jbc.M104353200. [DOI] [PubMed] [Google Scholar]
- 32.Murata N, Sato K, Kon J, Tomura H, Yanagita M, Kuwabara A, Ui M, Okajima F. Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem J. 2000;352(Pt 3):809–815. [PMC free article] [PubMed] [Google Scholar]
- 33.Leal SM, Liu Q, Huang SS, Huang JS. The type V transforming growth factor beta receptor is the putative insulin-like growth factor-binding protein 3 receptor. J Biol Chem. 1997;272(33):20572–20576. doi: 10.1074/jbc.272.33.20572. [DOI] [PubMed] [Google Scholar]
- 34.Weinzimer SA, Gibson TB, Collett-Solberg PF, Khare A, Liu B, Cohen P. Transferrin is an insulin-like growth factor-binding protein-3 binding protein. J Clin Endocrinol Metab. 2001;86(4):1806–1813. doi: 10.1210/jcem.86.4.7380. [DOI] [PubMed] [Google Scholar]
- 35.Lee SH, Takahashi M, Honke K, Miyoshi E, Osumi D, Sakiyama H, Ekuni A, Wang X, Inoue S, Gu J, Kadomatsu K, Taniguchi N. Loss of core fucosylation of low-density lipoprotein receptor-related protein-1 impairs its function, leading to the upregulation of serum levels of insulin-like growth factor-binding protein 3 in Fut8-/- mice. J Biochem. 2006;139(3):391–398. doi: 10.1093/jb/mvj039. [DOI] [PubMed] [Google Scholar]
- 36.Newman EA. New roles for astrocytes: regulation of synaptic transmission. Trends Neurosci. 2003;26(10):536–542. doi: 10.1016/S0166-2236(03)00237-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.