

Abstract
PCBP2 is a member of the poly(C)-binding protein (PCBP) family, which plays an important role in posttranscriptional and translational regulation by interacting with single-stranded poly(C) motifs in target mRNAs. Several PCBP family members have been reported to be involved in human malignancies. Here, we show that PCBP2 is upregulated in human glioma tissues and cell lines. Knockdown of PCBP2 inhibited glioma growth in vitro and in vivo through inhibition of cell-cycle progression and induction of caspase-3–mediated apoptosis. Thirty-five mRNAs were identified as putative PCBP2 targets/interactors using RIP-ChIP protein-RNA interaction arrays in a human glioma cell line, T98G. Four-and-a-half LIM domain 3 (FHL3) mRNA was downregulated in human gliomas and was identified as a PCBP2 target. Knockdown of PCBP2 enhanced the expression of FHL3 by stabilizing its mRNA. Overexpression of FHL3 attenuated cell growth and induced apoptosis. This study establishes a link between PCBP2 and FHL3 proteins and identifies a new pathway for regulating glioma progression.
Introduction
Poly(C)-binding proteins (PCBPs) are characterized by their high-affinity and sequence-specific interactions with polycytosine (poly(C)). In mammalian cells, these PCBPs belong to 1 of 2 subsets: hnRNP K/J, or the α-complex proteins (e.g., PCBP1-4) (1, 2). hnRNP K, PCBP1, and PCBP2 have been studied in the greatest detail. The latter 2 proteins are also known as αCP1 and αCP2, or hnRNPE1 and hnRNPE2 (3). Recently, 2 other members of the αCP family were discovered: PCBP3 (αCP3) and PCBP4 (αCP4) (4).
PCBPs regulate gene expression at various levels, including transcription, mRNA processing, mRNA stabilization, and translation. It has been suggested that PCBPs, specifically hnRNPK and PCBP1, play a critical role in carcinogenesis. Transcriptional activation of the oncogenes c-SRC and c-Myc was increased by hnRNP K (5, 6), suggesting that hnRNPK may cooperate with additional oncoproteins to overexpress genes that promote cancerous growth. PCBP1 has been reported to be downregulated in metastatic cervical cancer (7) and metastatic breast cancer cells (8). Knockdown of PCBP1 in the prostate cancer cell line LNCaP increased androgen receptor (AR) protein targeting to the 3′ untranslated region (UTR) of AR transcripts (9). The recent finding that transforming growth factor-β–mediated (TGFβ-mediated) phosphorylation of PCBP1 induced epithelial-mesenchymal transdifferentiation (EMT) (10), and the evidence that PCBP1 can downregulate metastasis-associated PRL-3 phosphatase translation (11) indicates that PCBP1 could be a tumor suppressor.
PCBP2 mapped to 12q13.12-q13.13 and PCBP1 mapped to 2p12-p13 (12) are highly homologous proteins with 82% amino acid identity. It has been suggested that PCBP2 also plays an important role in human malignancies. However, in addition to its overexpression during leukomegenesis (13) and its underexpression in oral cancer (14), PCBP2 is one of the least studied proteins in human cancers among PCBPs. Most of the reports on PCBP2 have focused on its posttranscriptional and translational controls in RNA viruses. For example, PCBP2 has been reported to participate in the replication and translation of many RNA viruses, including poliovirus (15), coxsackievirus (16), and rhinovirus (17). PCBP2 can also bind to the 5′ UTR (18) and 3′ UTR (19) of the HCV gene. PCBP2 was induced after viral infection and interacted with MAVS, which showed that PCBP2 was a negative regulator of MAVS-mediated antiviral signaling (20).
Recent discoveries have revealed that PCBP2 is a regulator of tumor development. Molinaro et al. found that 2′,5′-oligoadenylate synthetase (OAS) activation may occur in prostate cancer cells in vivo when stimulated by Raf kinase inhibitor protein (RKIP) and PCBP2 (21). In leukemic blasts, PCBP2 expression was induced by BCR/ABL through constitutive activation of MAPKERK1/2 (22) and its regulation of CEBPA mRNA was influenced by the decoy activity of miR-328 (23). Given that more target mRNAs of PCBP2 were found by RIP-ChIP and biotin pull-down, we believe that the binding of PCBP2 to its target mRNAs may play important functions in human cancers.
Here, we showed the upregulation of PCBP2 in human glioma tissues and cell lines. Knockdown of PCBP2 inhibits glioma cell growth both in vitro and in nude mice. To elucidate how PCBP2 functions as an RNA-binding protein with its target mRNAs in gliomas, we identified 35 mRNAs that preferentially bind PCBP2 by RIP-ChIP analysis. We also showed that four- and-a-half LIM domain 3 (FHL3) is downregulated in gliomas in which mRNA is a novel binding target of PCBP2.
Results
To examine the expression pattern of PCBP2 in gliomas, total RNA and proteins were extracted from 26 primary glioma samples, which contain 9 grade II, 9 grade III, and 8 grade IV glioma tissues and 2 normal brain tissues. Real-time PCR and Western blot were then performed to analyze the gene expression profiles. We also determined mRNA and protein levels of PCBP2 in 5 different human glioma cell lines (T98G, U87MG, U251, A172, and CCF-STTG1) and 2 normal human astrocyte cell lines (NHA and HA). The results showed that the mRNA and protein expression of PCBP2 was upregulated in both glioma tissues and cell lines compared with normal brain tissues (Figure 1). A strong positive signal of PCBP2 was found in almost all glioma tissues, whereas an obvious loss of PCBP2 in normal brain tissue was observed (Figure 1C). A gradually stronger PCBP2 expression was found from grade II samples to grade IV samples (Figure 1, A–C).
Figure 1. PCBP2 is upregulated in glioma tissues and cell lines compared with normal brain tissues and normal human astrocytes.

(A) Real-time PCR analysis of PCBP2 in 2 normal brain tissues (N1, N2) and 26 glioma tissues (T1–T9 grade II, T10–T18 grade III, and T19–T26 grade IV). Columns represent means and bars represent SD. (B) Representative Western blot showing PCBP2 protein levels in 2 normal brain tissues and 27 glioma tissues. β-Actin was used as a loading control. (C) Immunohistochemical staining of PCBP2 in glioma (Grade II, Grade III, Grade IV) and pericarcinous tissues (Normal) using anti-human PCBP2 antibodies. There was no staining with rabbit IgG. Original magnification, ×400. (D) Real-time PCR analysis of PCBP2 in 2 normal human astrocyte cell lines (NHA and HA) and 5 glioma cell lines (T98G, A172, U251, U87MG, and CCF-STTG1). (E) Representative Western blot showing PCBP2 protein levels in 2 NHA cell lines and the 5 indicated glioma cell lines.
We also found significantly elevated levels of PCBP2 in T98G, U87MG, and U251 glioma cell lines (Figure 1, D and E). Therefore, these 3 cell lines were used as models for further study of PCBP2 function in gliomas. To investigate the role of PCBP2 in glioma development, we synthesized 2 siRNAs (PCBP2 siRNA and PCBP2 siRNA*) that specifically target PCBP2, as previously reported (24, 25). After transfection for 48 to 72 hours in T98G, U87MG, and U251 glioma cell lines, siRNA-mediated knockdown of PCBP2 reduced its protein level by at least 80% compared with the control samples (Figure 2A and Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI61820DS1). Glioma cells transfected with PCBP2 siRNA had a reduced number of metabolically active cells compared with those transfected with the control siRNA. The reduction became more apparent at 72 hours, as demonstrated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Figure 2B). As shown in Figure 2C by flow cytometry, the percentage of cells in the G1 phase was significantly higher in glioma cells transfected with PCBP2 siRNA than in the controls (T98G: 79.9 ± 4.7% vs. 60 ± 3.4%; U87MG: 82.1 ± 4.3% vs. 62.6 ± 6.7%; U251: 72 ± 1.6% vs. 55.9 ± 4.3%). Furthermore, a marked reduction of the cell population in the S phase was observed after transfection with PCBP2 siRNA (T98G: 15.6 ± 3.5% vs. 32.9 ± 1.1%; U87MG: 15.4 ± 1.7% vs. 26.1 ± 1.8%; U251: 21.9 ± 0.2% vs. 36.9 ± 1.5%). However, no significant difference in the G2/M population was found (T98G: 4.5 ± 0.5% vs. 7.2 ± 4.5%, P = 0.25; U87MG: 2.5 ± 2.6% vs. 11.5 ± 8.5%, P = 0.15; U251: 6.0 ± 1.8% vs. 7.1 ± 2.9%, P = 0.36). The results suggested that knockdown of PCBP2 induced a G1-S arrest in glioma cell lines. We also performed a TUNEL assay in the 3 glioma cell lines and found increased DNA damage and fragmentation in glioma cells transfected with PCBP2 siRNA compared with cells transfected with the control siRNA. A quantitative analysis also revealed a 7- to 10-fold increase in apoptosis induction in glioma cells transfected with PCBP2 siRNA (Figure 2F). PCBP2 siRNA* produced effects similar to those of PCBP2 siRNA (Supplemental Figure 6, C–E). In PCBP2-depleted glioma cells, the protein levels of cyclin-dependent kinase inhibitors P27, P21, and P16 were increased (Figure 2D), and the tumor suppressor protein pRB was in a lower phosphorylation state than the control (Figure 2E). In the control glioma cells, we were hardly able to detect processed caspase-3 and its substrate PARP. In response to PCBP2 siRNA or siRNA*, caspase-3 was partially processed into the intermediated p20 form and its active p17 subunit. PARP was also progressively processed (Figure 2G). To ascertain the role of caspase-3 in PCBP2 knockdown–induced apoptosis, we tested the effects of a pancaspase inhibitor zVAD-fmk. Pretreatment of glioma cells with zVAD-fmk blocked PCBP2 siRNA- or siRNA*-induced cell apoptosis (Supplemental Figure 2, A and B). Taken together, the above results illustrate that PCBP2 knockdown inhibits glioma cell growth through inhibition of cell-cycle progression and induction of caspase-3–mediated apoptosis.
Figure 2. Inhibition of glioma cell growth in vitro by knockdown of PCBP2.

(A) Western blot of PCBP2 expression in 3 glioma cell lines (T98G, U87MG, and U251) transiently transfected with the control siRNA or PCBP2 siRNA for 48 to 72 hours using Lipofectamine 2000. β-Actin was used as a loading control. (B) MTT assay on the same 3 glioma cell lines after transfection with the control siRNA or PCBP2 siRNA as above. Data are presented as the mean ± SD and are representative of 3 wells. *P < 0.05 compared with the control siRNA by a 2-tailed Student’s t test. (C) Approximately 72 hours after transfection, the 3 glioma cell lines were analyzed by flow cytometry. The proportions of cells in the G1, G2, and S phases of the cell cycle are depicted in the histograms. *P < 0.05 compared with the control siRNA by a 2-tailed Student’s t test. (D and E) Representative Western blot showing P27, P21, and P16 protein levels (D) and expression levels of differentially phosphorylated pRb (E) in the transfected glioma cell lines. P16 was deleted in U87MG and U251 cells. (F) Nuclear TUNEL staining for apoptotic cells in the glioma cell lines after approximately 72 hours of transfection. The ratio of TUNEL-positive cells was calculated (n = 5) and plotted on the histogram. *P < 0.05 compared with the control siRNA by a 2-tailed Student’s t test. (G) Representative Western blot showing (cleaved) caspase-3 and its substrate (cleaved) poly (ADP-ribose) polymerase (PARP) in the transfected glioma cell lines.
To test the effect of decreased PCBP2 levels in glioma cells in vivo, we packaged an analogous recombinant adenovirus encoding an shRNA that targeted PCBP2, and as a positive control, an adenovirus encoding an shRNA against survivin (Figure 3A and ref. 26). T98G cell suspensions were injected s.c. into nude mice and the tumor developments were observed at the indicated time points. When the tumor size reached approximately 7–15 mm in diameter, 8 mice in each group received the first intratumoral injection of adenovirus, each at 109 pPFUs per tumor. We recorded the tumor sizes on the first day of adenoviral injection (day 1). We repeated intratumoral injections of the same adenoviruses on days 2, 5, and 6, as shown in Figure 3C. As a result, all of the mice in the adenovirus (Ad) control shRNA group developed large tumors beyond 3,000 mm3 on average by day 15. In contrast, the mice in the Ad shPCBP2 and Ad shSurvivin groups developed much smaller tumors (Figure 3B). On days 9, 12, and 15, the mice in the Ad shPCBP2 or Ad shSurvivin group had significantly smaller tumor sizes compared with the mice in the Ad control shRNA group (Figure 3C). No significant differences in tumor size between the Ad shPCBP2 and Ad shSurvivin groups were observed at any point in this experiment. Having observed that PCBP2 knockdown can inhibit glioblastoma tumor growth in this preliminary subcutaneous xenograft model, we then proceeded to test the effects of PCBP2 knockdown in an orthotopic tumor growth model. Approximately 5 × 105 U87MG-Luc cells stably expressing either shNT (control vector) or shPCBP2 (PCBP2 shRNA) were injected intracranially (Supplemental Figure 7A), and the tumor sizes were quantified by bioluminescence imaging after 20 days (Figure 3D). Quantitation of the results from 5 mice in each group confirmed that inhibition of PCBP2 expression markedly suppressed orthotopic tumor formation in mice.
Figure 3. Inhibition of glioma cell growth in vivo by knockdown of PCBP2.

(A) Representative Western blot showing PCBP2 and survivin protein levels in T98G cells after infection with Ad shRNA. β-Actin was used as a loading control. (B) Subcutaneous tumors isolated from nude mice 2 weeks after adenovirus injection (n = 8 for each group). Scale bar: 1 cm. (C) Tumor growth curves in nude mice (n = 8 for each group). Each intratumoral injection of adenovirus is indicated with an arrow. The x-axis represents days after adenovirus injection. The y-axis represents tumor volume (V = L × W2 × π/6; V, volume; L, length; W, width). *P < 0.05 compared with Ad control shRNA by a 2-tailed Student’s t test. (D) shNT-U87MG and shPCBP2-U87MG stable transfectants (5 × 105) were each injected intracranially into 5 nude mice and then imaged after 20 days. Tumor size was quantified by in vivo bioluminescence imaging. The graph illustrates the distribution of intracranial tumor signals between the 2 groups (y-axis represents photon flux values).
Because PCBP2 is an RNA-binding protein, we sought to determine which target mRNAs were affected by PCBP2 knockdown in glioma cells. We expected PCBP2 to regulate a few hundred mRNAs involved in multiple processes, such as cell-cycle maintenance and apoptosis control. We conducted an RIP-ChIP analysis to identify novel PCBP2 targets and gain better insight into the biological processes regulated by this protein in glioma cells. We chose to use the T98G cell line as a model system for 3 reasons. First, this cell line had the strongest PCBP2 expression among those described above, and PCBP2 was shown to play a prominent role in glioma growth (Figures 1–3). Second, T98G is one of the most commonly used and tumorigenic glioblastoma cell lines for research. Third, we found that the transfection efficiency and level of transgene expression were exceptionally high in T98G cells compared with the other glioma cell lines. Previous studies have been performed using either a tag strategy or antibodies against endogenous PCBP2 in K562 cells (27, 28). To increase the confidence in our results, we conducted 3 parallel experiments with GFP as a negative control (29). Because GFP is not an RNA-binding protein and is not expressed in mammalian cells, we assumed that the RNAs pulled down by GFP represented the background signal. We conducted the RIP-ChIP experiments using a biotin-tag strategy. The biotin acceptor peptide (BAP) attached to the protein of interest was biotinylated in vivo by the BirA enzyme, and streptavidin-sepharose beads were then used to pull down and isolate the RNP complexes. Because high protein recovery dictates the success of RIP-ChIP analysis, we first performed a precipitation Western analysis to determine the levels of both BAP-GFP and BAP-PCBP2 recovered via streptavidin-sepharose bead precipitation (Supplemental Figure 3, A and B). After the precipitation, the RNAs were recovered, amplified, and labeled to probe human 44K Agilent arrays. Each set of experiments included 4 microarrays, including 3 technical replicates of BAP-PCBP2 versus the control BAP-GFP for each dye orientation (Figure 4A) from the microarray experiments conducted at the ShanghaiBio Corporation. We conservatively established a minimum of 2-fold enrichment on average, with an FDR-adjusted P value of less than 0.05, and identified 35 mRNAs that met the threshold in all experiments (Table 1). We used gene ontology to investigate the nature of the PCBP2-associated mRNAs identified by RIP-ChIP. Among the biological processes that showed enrichment were cell cycle (5.3% gene frequency), cell death (10.5% gene frequency), and cell proliferation (15.8% gene frequency). Approximately 54% of our mRNAs (19 genes) were related to the 15 biological processes (Supplemental Figure 4). We then used the characteristic subpathway network (CSPN) to find 9 proteins encoded by the PCBP2-associated mRNAs involved in cell cycle or apoptosis (Figure 4B). CSPN was used to reveal the phenotype-specific pathway interactions with formerly defined pathways and protein-protein interaction (PPI) data (30). To validate the results from the microarray analysis, we selected 9 genes from the BAP-PCBP2–associated mRNA population for confirmatory studies. α-Globin and γ-globin mRNAs served as the positive and negative controls, respectively, because the interaction of α-globin mRNA with PCBP2 has been previously characterized (31) and γ-globin mRNA lacks PCBP2-binding sites. Real-time PCR confirmed the enrichment of the 9 RNA species in the BAP-PCBP2–associated population compared with the BAP-GFP control (Figure 4C).
Figure 4. RIP-ChIP analysis of PCBP2 and identification of preferentially associated mRNAs.

(A) Schematic outline of the RIP-ChIP analysis to identify PCBP2-associated mRNAs. T98G cells were transfected transiently with BAP-tagged constructs. BAP-tagged proteins were biotinylated in vivo by the cotransfected hBirA enzyme. RNPs were recovered via precipitation with streptavidin-sepharose beads. Finally, RNAs were purified and analyzed on microarrays. The BAP-GFP control was used in 3 independent sets of experiments. (B) Detection of the “protein-protein interaction” and “pathway” relationships between the proteins encoded by the PCBP2-associated mRNAs via the CSPN. In our pathways of interest (highlighted in gray), we found 9 proteins encoded by the PCBP2-associated mRNAs (highlighted in red) and their interacting protein partners (highlighted in blue). (C) Real-time PCR validation of PCBP2-associated mRNAs identified by RIP-ChIP. We selected 9 mRNAs to be validated by real-time PCR. We also verified the enrichment of α-globin, a previously identified PCBP2 target, in the BAP-PCBP2–associated mRNA population. Blue and red bars represent the enrichment observed with the microarrays and real-time PCR, respectively. Apo, apoptosis; CC, cell cycle.
Table 1.
The 35 mRNAs enriched in the BAP-PCBP2–associated RNA population over BAP-GFP
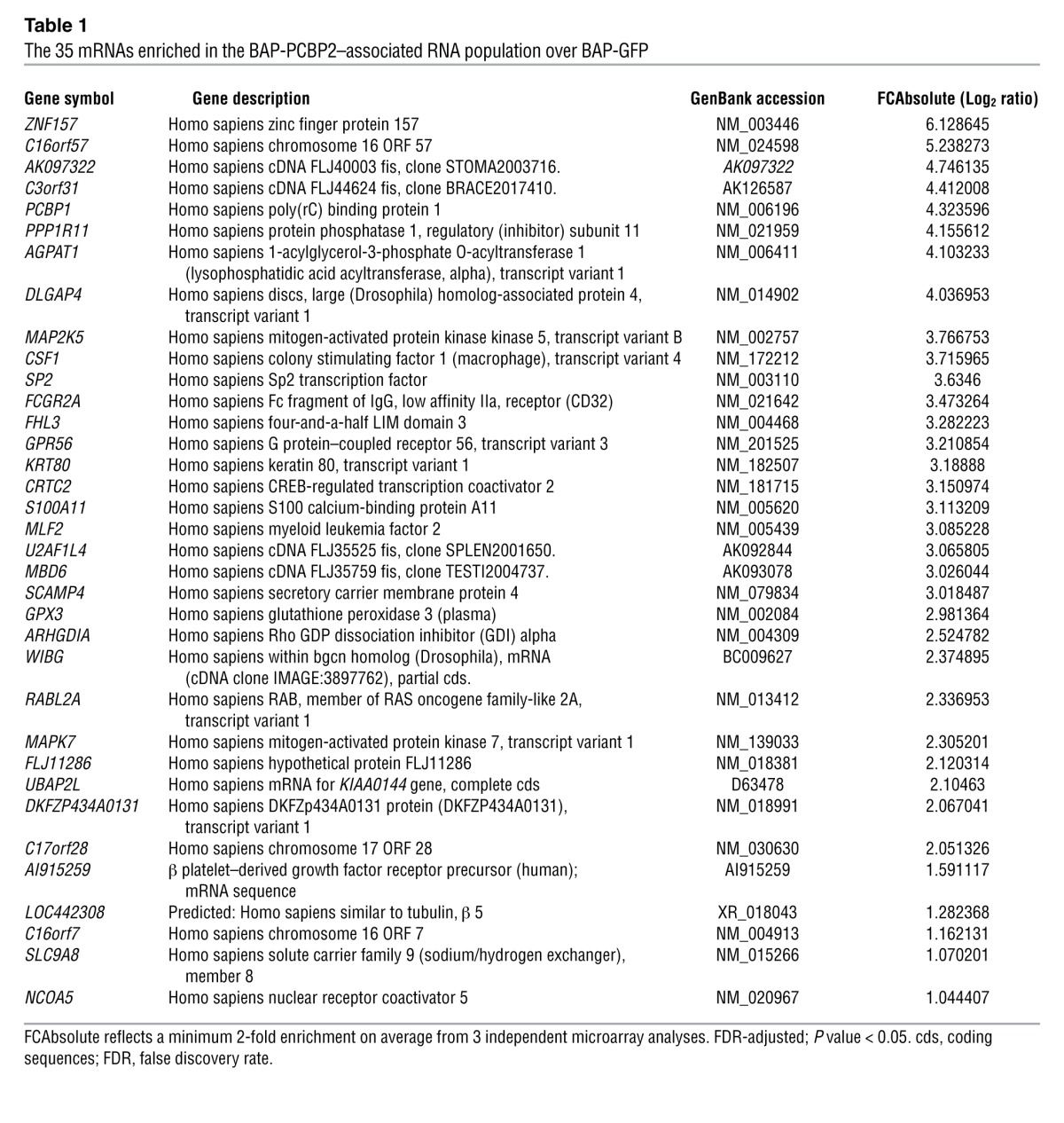
In many cases, PCBP2 functions through binding to the 3′ untranslated region (UTR) or 5′ UTR of its target mRNAs (6, 32). Defined PCBP2 binding sites contain short patches of C- or CU-rich sequences (25, 33). After we identified the mRNAs associated with PCBP2 by RIP-ChIP, we decided to test which regions of the mRNAs’ 3′ UTR or 5′ UTR were associated with PCBP2. The candidate mRNAs included CTRC2, ARHGDIA, C16orf57, SP2, FHL3, MAPK7, MAP2K5, FCGR2A, and CSF1, which are involved in cell cycle or apoptosis regulation according to CSPN analysis (Figure 4B). A biotin pull-down assay was applied for confirmation. With the exception of MAPK7, fragments located in the 3′ UTR or 5′ UTR of the other 8 candidate mRNAs were inserted into the pGEM-3zf vector containing a T7 promoter. The fragments were predicted to contain potential binding sites by bioinformatics analysis (Figure 5A). The biotin-labeled RNA probes corresponding to each fragment were synthesized in vitro with the T7 RNA polymerase. The pGEM-3zf vector sequence (nonsense sequence) and the α-globin 3′ UTR were used as negative and positive controls, respectively. The T98G lysates pulled down by the probes were analyzed by Western blot using an antibody against PCBP2. PCBP2 was not detected in the nonsense sequence pull-down complex. In contrast, the α-globin 3′ UTR showed a strong binding of PCBP2 (Figure 5B). Among the other probes, CTRCT-3′ UTR, ARHGDIA-3′A, CSF1-3′B, FHL3-3′A, SP2-3′A, SP2-3′B, C16orf57-3′A, and MAP2K5-5′B presented significant binding of PCBP2 on par with α-globin. MAP2K5-5′A showed very weak binding, but the result was not reproducible. The only mRNA that did not bind PCBP2 was FCGR2A (Figure 5B). To further confirm the specificity of the binding between PCBP2 and the RNA probes, a competition assay was performed. Each biotin-labeled RNA was incubated with 2- or 3-fold volume T98G cell lysate to ensure that the probe was in excess, and then 5- or 10-fold molar excess of an unlabeled homologous probe was added to compete with the biotin-labeled probe for PCBP2 binding. The unlabeled probes exhibited strong competition at 10-fold molar excess (Figure 5C). Based on these findings, we concluded that the major binding determinant of PCBP2 resides in the CTRCT-3′ UTR, ARHGDIA-3′A, CSF1-3′B, FHL3-3′A, SP2-3′A, SP2-3′B, C16orf57-3′A, and MAP2K5-5′B fragments. Notably, FHL3-3′A showed the strongest binding to PCBP2 among all of the fragments (Figure 5D). The sequences of these fragments are indicated in Supplemental Figure 5.
Figure 5. Binding of biotin-labeled candidate mRNAs to PCBP2.

(A) Schematic of the 8 indicated candidate mRNAs. The specific fragments used as templates for the synthesis of biotin-labeled RNAs are indicated with underlines, and the nucleotide positions amplified by PCR are shown. (B) Biotin pull-down analysis of complexes formed in vitro using biotin-labeled candidate mRNA segments (shown in A) and T98G whole-cell lysates. The α-globin-3′ UTR and a nonsense sequence were included as the positive and negative controls, respectively. (C) A specific association between PCBP2 and its candidate target mRNAs (identified in B) was confirmed by a competition assay. Five- or 10-fold excess of unlabeled RNA was added to compete with the biotin-labeled RNA for interaction with PCBP2, and 2- or 3-fold excess of cell lysates was incubated with biotin-labeled RNA. (D) Detection of the binding affinities of the target mRNAs to PCBP2 by biotin pull-down analysis using all of the biotin-labeled RNAs identified above.
Given that PCBP2 associates with FHL3 mRNA in glioma cell lines, we hypothesized that PCBP2 may regulate FHL3 expression. To test this hypothesis, we assessed the protein expression of FHL3 in 9 glioma tissues and 5 glioma cell lines. We found that although FHL3 expression was not significantly changed in the glioma cell lines compared with normal astrocytes (Figure 6C), it was decreased in all of the tumor tissues (Figure 6A). In agreement with our Western blot data, immunohistochemical analysis revealed significant FHL3 protein expression in normal brain tissues and low-grade glioma tissues, but not in high-grade glioma tissues (Figure 6B). These results suggest that PCBP2 may be negatively regulating FHL3 mRNA translation in gliomas. To address this possibility, we investigated the effect of PCBP2 knockdown on FHL3 protein expression in the glioma cell lines. As shown in Figure 6D, downregulation of PCBP2 resulted in significantly upregulated FHL3 protein expression. To further determine the core recognition sequence in FHL3 mRNA 3′A, we inserted 4 clipped forms of 3′A (3′A-1, -2, -3, -4) into the pGEM-3zf vector (Figure 7A). The result of the biotin pull-down analysis showed that only FHL3-3′A-1 and the negative control FHL3-3′B failed to bind PCBP2 (Figure 7B). FHL3-3′A-2, FHL3-3′A-3, and FHL3-3′A-4, as well as unclipped FHL3-3′A, showed similar binding affinities to PCBP2 when the known core recognition sequences 5′-ACCC-3′ and 5′-CCCT/U-3′ at position +1212 to +1263 (34) were present (Figure 7C). We also tested the effect of PCBP2 knockdown on FHL3 3′ UTR activity and observed a 104.4% increase in luciferase reporter activity compared with T98G cells transfected with the control siRNA (Figure 6E And 7D). To understand how PCBP2 affects the translation of FHL3 mRNA, we designed an assay to assess the half-life of FHL3 mRNA. Our rationale for this experiment was that several examples exist in the literature in which PCBP2-binding sites occur in the 3′ UTR of specific mRNAs and regulate mRNA stability (35, 36). Two days after siRNA transfection, T98G cells were treated with the transcriptional inhibitor actinomycin D, and RNA was harvested at subsequent time points for the quantitative analysis of FHL3 mRNA with the RNase protection assay. This analysis revealed that the rate of FHL3 mRNA decay in PCBP2 siRNA–transfected cells was slower than in the control (Figure 7E). Linear regression analysis revealed an mRNA half-life of 3.67 hours in the control cells. In contrast, PCBP2 knockdown resulted in a prolonged half-life of 7.31 hours. This alteration in FHL3 mRNA stability in the PCBP2 knockdown cells is consistent with the observed increase in FHL3 protein levels.
Figure 6. Detection of FHL3 expression in glioma and downregulation of PCBP2 enhances FHL3 expression.

(A) Detection of FHL3 protein levels in 2 normal brain tissues (N1, N2) and 9 glioma tissues (T1–T3 grade II, T4–T6 grade III, and T7–T9 grade IV) by Western blotting. β-Actin was used as a loading control. (B) Immunohistochemical staining of FHL3 protein in glioma and pericarcinous tissues (Normal) using anti-human FHL3 antibodies. There was no staining with rabbit IgG. Original magnification, ×400. (C) Detection of FHL3 protein levels in 2 normal human astrocyte lines (NHA, HA) and the 5 indicated glioma cell lines by Western blotting. β-Actin was used as a loading control. (D) Representative Western blot showing FHL3 protein levels in glioma cell lines transiently transfected with the control siRNA or PCBP2 siRNA. β-Actin was used as a loading control. (E) (Top) Schematic diagram of an empty luciferase reporter construct and FHL3 3′UTR-A luciferase construct. T98G cells were transiently transfected with the control siRNA or PCBP2 siRNA, plus either an empty pLuc reporter plasmid or a pLuc-FHL3 3′ UTR reporter plasmid (bottom). Firefly luciferase activity was determined relative to a cotransfected Renilla luciferase internal control and expressed as a percentage of the matching pLuc empty vector control. Columns represent means and bars represent the SD of 3 independent experiments. *P < 0.05 compared with the control siRNA-transfected cells by a 2-tailed Student’s t test.
Figure 7. Identification of the FHL3 core recognition sequence; PCBP2 regulates FHL3 expression by stabilizing its mRNA.

(A) Schematic of FHL3 mRNA depicting the 3′ UTR-A in light gray rectangles. The templates for the synthesis of biotin-labeled 3′A-1 to 4 RNAs are indicated by dark lines and CU-rich patches are shown in red. (B) Detection of different binding abilities to PCBP2 by biotin pull-down analysis using the biotin-labeled 3′A-1 to 4 RNA. FHL3 mRNA 3′A and 3′B were included as positive and negative controls. (C) Nucleotide sequence of the identified FHL3 mRNA target binding site, with the core recognition sequence shown in red. (D) T98G cells were transiently transfected with the control siRNA or PCBP2 siRNA and either an empty pLuc reporter plasmid or a pLuc-FHL3 3′A-1,-2,-3,-4 reporter plasmid. *P < 0.001. (E) FHL3 mRNA stability assay in T98G cells transiently transfected with the control siRNA or PCBP2 siRNA. Transfectants were treated with the transcription inhibitor ActD for 0, 2, 4, and 6 hours; the whole-cell RNA was analyzed for FHL3 mRNA levels by an RNase protection assay. Representative blots are shown in the top panel. FHL3 mRNA levels were quantified by densitometric scanning. For each set of siRNA transfections, the band intensity at the 0 hour time point was set to 100%, and the percentage of mRNA remaining at the 2-, 4-, and 6-hour time points is plotted as shown in the bottom panel.
FHL2 has been reported to be overexpressed in gliomas, and suppression of FHL2 inhibits glioblastoma cell proliferation (37); however, there is no report on the role of FHL3 in gliomagenesis. To investigate the role of FHL3 in glioma development, we transfected a flag-FHL3 construct or an empty vector control into T98G cells (Figure 8A). After 48 hours of transfection, an MTT assay showed that the proliferation rate of glioma cells was reduced after overexpressing FHL3 (Figure 8B). Although flow cytometry did not reveal any significant changes in cell-cycle progression (Figure 8C), FHL3 induced P21 and P16 expression (Figure 8D). In T98G cells, FHL3 diminished the level of pRb phosphorylation at Ser780 and Ser795, but increased pRb phosphorylation at Ser608 and Ser807/811 (Figure 8E), which may be the reason for the absence of an effect on T98G cell-cycle progression. The results of the TUNEL assay showed an increased percentage of apoptotic cells from 7.8% to 50.4% in T98G cells, which is similar to the results in the PCBP2 knockdown cells (Figure 8F). Overexpression of FHL3 also induced cleavage of caspase-3 and its substrate PARP (Figure 8G). The pancaspase inhibitor zVAD-fmk blocked FHL-3–induced apoptosis in T98G cells (Supplemental Figure 2C), which suggests that the apoptotic response to FHL3 was caspase-3 mediated. To determine the role of FHL3 in cellular transformation, the ability of the FHL3-expressing U87MG and U251 cells to form colonies in soft agar was analyzed. An analogous recombinant adenovirus encoding FHL3 caused efficient FHL3 overexpression after infecting the glioma cells (Supplemental Figure 7B). The adenovirus vector cells formed many colonies in soft agar. In contrast, Ad FHL3 cells showed a significant 2- to 4-fold decrease in the number of colonies formed (Figure 8H). In an orthotopic tumor growth model, U87MG-Luc cells stably expressing FHL3 were injected into the brains of 6 mice, and 6 control mice were also generated (Supplemental Figure 7C). Tumor growth was quantified over time by imaging tumor-associated bioluminescence from days 6 to 22. The vector (control) U87MG tumors were enlarged by 18-fold on average; however, the FHL3-U87MG tumors only grew by approximately 5.2-fold (Figure 8I), suggesting that FHL3 significantly slowed the tumor growth rate. These results support the hypothesis that PCBP2 knockdown inhibits proliferation of glioma cells by upregulating FHL3 protein expression.
Figure 8. Overexpression of FHL3 inhibits glioma growth in vitro and in vivo.

(A) Western blot showing overexpression of Flag-FHL3 plasmid in T98G cells using anti-Flag or anti-FHL3 antibody. (B) MTT assay of T98G cells after transfection with the empty vector (—) or flag-FHL3 plasmid. (C) Approximately 48 hours after transfection, T98G cells were analyzed by flow cytometry. The proportions of cells in the G1, G2, and S phases are presented in the right histogram. (D and E) Representative Western blot showing P27, P21, and P16 protein levels (D) and expression levels of differentially phosphorylated pRb (E) in FHL3-overexpressing T98G cells. (F) Nuclear TUNEL staining for apoptotic T98G cells approximately 48 hours after transfection. The percentage of TUNEL-positive cells was calculated (n = 5) and plotted in the histogram. (G) Representative Western blot showing (cleaved) caspase-3 and its substrate (cleaved) PARP in FHL3-overexpressing T98G cells. (H) Anchorage-independent growth of U87MG and U251 cell lines after infection with the adenovirus vector or adenovirus FHL3 as evaluated by the soft agar assay. Scale bars: 50 μm. The number of colonies in each field was scored and the results are expressed as the means ± SD (n = 5; *P < 0.05 by a 2-tailed Student’s t test). (I) Vector U87MG and FHL3-U87MG stable transfectants (5 × 105) were each injected intracranially into 6 nude mice and then imaged after 6 days and 22 days. Tumor sizes were quantified by in vivo bioluminescence imaging. The graph illustrates the distribution of tumor growth factor (22nd day photon flux values versus sixth day photon flux value) between the 2 groups.
To further test the above hypothesis, we performed a co-knockdown of PCBP2 and FHL3 for a rescue study. FHL3 siRNA and FHL3 siRNA* were transfected individually into 3 glioma cell lines. Western blot analyses revealed that the 2 siRNAs inhibited their targeted protein (Figure 9A and Supplemental Figure 6A). PCBP2 siRNA and FHL3 siRNA were cotransfected into 3 glioma cells, resulting in the depletion of both PCBP2 and FHL3 (Figure 9B). The MTT assay demonstrated that the number of metabolically active cells after the siRNA cotransfection was closer to that of the control level than after PCBP2 siRNA transfection alone (Figure 9C). Similarly, the TUNEL assay showed that the co-knockdown of FHL3 and PCBP2 counteracted the majority of the apoptosis effect induced by PCBP2 knockdown alone in all glioma cell lines (Figure 9E). It should be noted that although the proliferation rate of glioma cells and the fraction of apoptotic cells after siRNA cotransfection were much closer to those of the control than the PCBP2 siRNA–transfected cells, there was still a significant difference between the co-knockdown and control cells. The levels of cleaved caspase-3 and cleaved PARP were reduced in the rescued cells (Figure 9F). No significant differences in cell-cycle progression were detected by flow cytometry between the cotransfected cells and the PCBP2 siRNA–transfected cells (Figure 9D), suggesting that PCBP2 likely has a stronger cell-cycle regulatory effect than FHL3. Cotransfection with PCBP2 siRNA* and FHL3 siRNA* showed glioma cell rescue effects that were similar to those of the cotransfection with PCBP2 siRNA and FHL3 siRNA (Supplemental Figure 6, B–F). To determine the effect of the PCBP2/FHL3 co-knockdown on cellular transformation, we coinfected cells with recombinant adenoviruses encoding shRNAs against PCBP2 (Ad shPCBP2) and FHL3 (Ad shFHL3). Both PCBP2 and FHL3 protein levels were reduced in the glioma cells coinfected with Ad shPCBP2 and Ad shFHL3 (Supplemental Figure 7D). In the U87MG line, coinfected cells showed a greater than 2-fold increase in the number of colonies formed in soft agar, and no significant difference in the number of colonies between the Ad shPCBP2 plus shFHL3 (rescue) and Ad control groups was observed. However, in the U251 line, although the rescued cells showed a stronger cellular transformation ability than the Ad shPCBP2 group, there was still a significant difference between the co-knockdown cells and the control cells (Figure 9G). To more directly test the possible in vivo effects of the rescue, U87MG cells were injected subcutaneously into 9 nude mice. After tumor formation, the mice were randomly divided into 3 groups: Ad control shRNA, Ad shPCBP2, and Ad rescue (Ad shPCBP2 + Ad shFHL3). Three mice in each group received intratumoral injection of adenoviruses at days 1, 2, 5, and 6, each at 109 PFUs per tumor. The tumors were measured with Vernier calipers weekly (Figure 9H). When mice began to die in the control or rescue group, the experiment was terminated. Using the Ad control shRNA group as the positive control, we found that the mice in the rescue group developed much larger tumors and had shorter survival times than those in the Ad shPCBP2 group (Figure 9H).
Figure 9. Co-knockdown of PCBP2 and FHL3 rescues the phenotype induced by PCBP2 knockdown alone.

(A) Western blot of FHL3 expression in 3 glioma cell lines transiently transfected with FHL3 siRNA. (B) Western blot showing PCBP2 and FHL3 protein levels in glioma cell lines transiently transfected with the PCBP2 siRNA alone, or PCBP siRNA plus FHL3 siRNA. (C) MTT assay of 3 glioma cell lines after transfection. *P < 0.05 compared with the control transfection and cotransfection by a 2-tailed Student’s t test). (D) The transfected cells above were analyzed by flow cytometry. The proportions of cells in the G1, G2, and S phases are depicted in the histograms. *P < 0.05 compared with the control. (E) Nuclear TUNEL staining for apoptosis in the glioma cell lines. The percentage of TUNEL-positive cells was calculated (n = 5) and plotted on the histogram. *P < 0.05 compared with the control transfection and cotransfection. (F) Western blot showing the levels of (cleaved) caspase-3 and (cleaved) PARP in the transfected glioma cell lines. (G) Anchorage-independent cell growth of U87MG and U251 cells after infection with Ad control shRNA, Ad shPCBP2, or Ad shPCBP2 plus Ad shFHL3 were evaluated by a soft-agar assay. Scale bars: 50 μm. The number of colonies in each field was expressed as the means ± SD (n = 5; *P < 0.05). (H) Subcutaneous tumors isolated from nude mice 4 weeks after adenovirus injection (top). Growth curves of tumors in the nude mice (n = 3 per group) are shown (bottom). Each intratumoral injection of adenovirus is indicated by an arrow.
Discussion
The results from this study indicate that in glioma tumors and cell lines, the mRNA and protein levels of PCBP2 are highly expressed compared with normal brain tissue and normal human astrocytes, as demonstrated by the real-time PCR, Western blotting, and IHC analyses. To our knowledge, this is the first report on PCBP2 protein expression in human gliomas. According to data presented by The Cancer Genome Atlas (TCGA), in 75% (316 of 424) of glioblastoma multiforme tissues, PCBP2 mRNA expression is at least 1.4-fold higher (log2 ≥0.5) compared with normal brain tissues. This result also supports our study. Cancer identification at an early stage may be possible by using biomarkers that are differentially expressed with the progression of the disease. The PCBP2 expression increased in grade III–IV glioma tissues compared with grade II tissues, suggesting that PCBP2 could potentially contribute to glioma malignancy. Although a large population study has not been performed, the results thus far indicate that PCBP2 may be a differentiation marker of human gliomas.
The functional significance of PCBP2 upregulation has been studied in vitro and in vivo. Our in vitro studies provide several insights into the mechanism of PCBP2 action. First, PCBP2 knockdown not only inhibited glioblastoma cell proliferation in the short-term MTT assays, but also significantly inhibited long-term colony formation in soft agar. Second, the expression levels of CDK inhibitors P27, P21, and P16 were increased, and the level of pRb phosphorylation was decreased in glioma cells transfected with PCBP2 siRNA. A G1 cell-cycle arrest was observed by flow cytometry. It was previously reported that PCBP1/PCBP2-codepleted cells exhibited a G1 arrest, which was not observed in cells depleted of either PCBP1 or PCBP2 alone (25). However, the cell line from that study, K562, was later revealed to be completely devoid of the p53 protein and therefore different from p53-dependent glioma cell lines. In addition, the relative proportions of G1 and G2 phases were also markedly different between K562 cells and glioma cells. Therefore, we believe that whether PCBP2 knockdown alone results in G1 cell-cycle arrest may depend on distinct cell characteristics. Third, PCBP2 siRNA transfection into the glioma cells induced caspase-3–mediated apoptosis. The relationship between PCBP2 and apoptosis has also been investigated in oral cancer (14). Together, these results may explain how PCBP2 could affect glioblastoma progression in vivo. By injecting adenovirus-mediated PCBP2 shRNA into nude mice glioma xenografts and observing tumor formation in the mice brain, we further confirmed that PCBP2 plays an important role in glioblastoma development. To the best of our knowledge, these findings suggest for the first time that PCBP2 is involved in gliomagenesis.
Our biochemical studies provide mechanistic insights into PCBP2 function in vitro and in vivo. We focused on molecules that: (a) directly or indirectly associate with the RNA-binding protein PCBP2 according to RIP-ChIP analysis, and (b) are known to affect cancer cell proliferation or apoptosis according to bioinformatics analysis. In this study, we focused on FHL3, which was verified as a PCBP2-binding target by biotin pull-down analysis. We also identified 6 other candidate binding targets of PCBP2 that might be regulated by other novel mechanisms and that might be involved in gliomagenesis. The detailed mechanisms must be further explored.
The identification of FHL3 mRNA as one of the novel PCBP2 target mRNAs is an interesting finding. The FHL family is composed of 5 members: FHL1, FHL2, FHL3, FHL4, and FHL5/ACT. By Northern analysis, FHL1, FHL2, FHL3, and FHL5 were found to be expressed at high levels in a variety of tumor cell lines, including squamous cell carcinoma, melanoma, and leukemia cell lines (38). Through interaction with other proteins, FHL proteins regulate various cellular processes, including proliferation, differentiation, apoptosis, adhesion, migration, transcription, and signal transduction (39). FHL3 has been reported to act as a coactivator or corepressor for transcription factors. Recently, it has been shown to be involved in carcinogenesis. FHL1-3 inhibited hepatocellular carcinoma (HCC) cell growth both in vitro and in nude mice. Furthermore, FHL protein expression was downregulated in patients with HCC and correlated with FHL-mediated TGF-β–like responses (40). FHL proteins also inhibit hypoxia-inducible factor 1 (HIF-1) transcriptional activity in HCC cells. FHL2 directly interacts with HIF-1 to repress transcription. FHL1 binds to the p300/CBP coactivators and disrupts their binding to HIF-1. However, FHL3 does not bind to HIF-1 or p300 and is likely to bind to another component of the coactivator complex that interacts directly or indirectly with the transactivation domains of HIF-1 (41). This previous study also demonstrated an interaction between FHL3 and CDC25B in mammalian cells by FRET analysis, but found that FHL3 had no effect on CDC25B phosphatase activity in vitro and that overexpression of FHL3 did not impair cell-cycle progression (42), which is a finding now supported by our data on T98G cells. FHL3 is also known to be downregulated in breast cancer. FHL3 inhibits both anchorage-dependent and anchorage-independent breast cancer cell growth. In addition, FHL3 induces both G1 and G2/M cell-cycle arrest and repression of the cell-cycle regulators cyclin D1 and cyclin B1, as well as activation of the cyclin-dependent kinase inhibitor P21 (43). In our study, FHL3 also upregulated P21 expression in glioma cells. In addition, we showed an FHL3-induced increase in P16 expression and a decrease in P27 expression in T98G cells. FHL3 has been previously identified as a novel angiogenin (Ang) binding partner. Furthermore, FHL3 was required for Ang-mediated HeLa cell proliferation and the nuclear translocation of Ang, a finding that highlights FHL3 as a potential candidate involved in the molecular mechanisms behind the antiapoptotic property of cancer cells (44). Our experiments show that FHL3 may induce caspase-3–mediated apoptosis in gliomas. Additional research is needed to determine whether FHL3 induces apoptosis through other mechanisms.
We believe our study is the first report on FHL3 downregulation in glioma tissues. FHL3 mainly displayed proapoptotic activity and inhibited glioma growth in vivo. We detected binding of PCBP2 to the FHL3 3′ UTR. In addition, PCBP2 knockdown stabilized the FHL3 mRNA and increased FHL3 protein levels. Our observations lead us to conclude that PCBP2 plays a significant role in the regulation of FHL3 expression, but it seems unlikely that the translational regulation of FHL3 by PCBP2 is the cause of the FHL3 underexpression in glioma cell lines. Knockdown of PCBP2 inhibited glioma cell proliferation in vitro and in vivo and induced caspase-3–mediated apoptosis. These functions appear to be mechanistically linked to the increase in FHL3 protein. Based on the previous and present experimental results, FHL3 appears to play a tumor suppressor–like role in cancers.
We also discovered that FHL3 induces G1 phase cell–cycle arrest in U87MG and U251 cells, along with an increased expression of P27 (data not shown). We speculated that reduced P27 expression might be the reason why the cell cycle is not subjected to FHL3 regulation in T98G cells. Our finding provides a better understanding of the molecular events of gliomagenesis. Furthermore, understanding the biological function of PCBP2 in glioma cell lines may help delineate its role in gliomas. Although the mechanism of how PCBP2 upregulation influences glioma is not known, our observation of the loss of glioma cell growth control after PCBP2 siRNA transfection suggests that PCBP2 exerts its function through the regulation of FHL3 expression. We believe that additional mRNAs, and even noncoding RNAs binding to PCBP2, will be sought in the future and that PCBP2 is potentially an important molecular target for anti-glioma therapy.
Methods
Cell lines and cell culture.
Human glioma cell lines T98G, U87MG, A172, and CCF-STTG1 were purchased from the ATCC and cultured according to the guidelines recommended by the ATCC. The U251 cell line (from the Cell Center of Peking Union Medical College) was cultured in minimum essential medium and Iscove’s modified Dulbecco’s medium supplemented with 10% FBS. All cells were maintained at 37°C with 5% CO2. The NHA cell line was purchased from the Lonza group and cultured with Clonetics medium and reagents. The other HA cell line was purchased from ScienCell Research Laboratories and cultured with astrocyte medium (catalog 1801).
Plasmids, siRNAs, and transfection.
The FHL3 expression construct was a gift from Qinong Ye of the Beijing Institute of Biotechnology (Beijing, China). The cDNA target sequences of siRNAs for PCBP2 have been described previously (24, 25), as well as the cDNA target sequences of siRNAs for FHL3 (40, 41). The cDNA target sequences of shRNAs for PCBP2 and FHL3 were described previously (27, 41). All of the plasmids were purified using the EndoFree Plasmid Maxi Kit (QIAGEN) and transfected into T98G cells using VigoFect (Vigorous Biotechnology). siRNAs were synthesized by the Shanghai GenePharma Company and transfected into glioma cell lines using Lipofectamine 2000 (Invitrogen) at a final concentration of 100 to 200 nM.
Western blotting.
Whole-cell extracts were obtained by lysing cells in TNTE buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 10 mM sodium pyrophosphate, 0.5% Triton X-100, 1 mM sodium vanadate, and 25 mM sodium fluoride) containing protease inhibitors (5 μg/ml PMSF, 0.5 μg/ml leupeptin, 0.7 μg/ml pepstatin, and 0.5 μg/ml aprotinin). The detailed Western blotting procedures have been described previously (45). The protein samples were analyzed using polyclonal rabbit anti-PCBP2 (1:2,000; Beijing Aviva Systems Biology), rabbit anti-FHL3 (1:500; Proteintech), mouse anti-survivin (1:1,000; Abcam), rabbit anti-P27, P21, P16, pRb, ppRb, caspase-3, and PARP (1:500; Cell Signaling Technology), rabbit anti-flag (1:3,000; Sigma-Aldrich), and mouse anti–β-actin (1:5,000; Sigma-Aldrich) antibodies.
Immunohistochemistry.
Immunohistochemical analyses of PCBP2 and FHL3 were conducted using paraffin section specimens of glioma and pericarcinous tissues from 20 patients. The sections were incubated overnight at 4°C with anti-PCBP2 or anti-FHL3 antibodies. Staining was performed using a diaminobenzidine staining kit (Zhongshan).
MTT assay, flow cytometric analysis, and TUNEL assay.
The MTT assays were performed after transfection for 0, 24, 48, and 96 hours as described previously (45).
Flow cytometric analysis was conducted at the Flow Cytometry facility of Peking Union Medical College. Briefly, after 48 to 72 hours of transfection, the cells were fixed and then incubated in PBS containing Rnase A (100 μg/ml; Sigma-Aldrich) and propidium iodide (50 μg/ml; Sigma-Aldrich) at room temperature for 30 minutes prior to analysis.
Glioma cells were infected with siRNAs or plasmids in 24-well plates for 48 to 72 hours. Then, the TUNEL assay was performed using a FragEL DNA fragmentation detection kit (Merck) according to the manufacturer’s instructions.
RNA immunoprecipitation (RIP).
RIP was performed following the method described in detail previously (46). Briefly, a tandem affinity-purified tag, biotin acceptor peptide (BAP) was fused to the 5′ terminal of the coding sequences of PCBP2 and GFP and then cotransfected with BirA (biotin ligase) into T98G cells. Biotin (1 mM) was added to the culture medium immediately after transfection. After 48 hours, the biotin-tagged proteins and RNA complexes were isolated from the cell lysate using high-affinity streptavidin-sepharose beads (GE Healthcare). RNP was eluted from the beads, and the RNA was purified from RNP using TRIzol (Invitrogen).
Microarray data analysis.
Microarray data have been deposited in the NCBI’s Gene Expression Omnibus public database (accession number GSE44048). The method for calculating the dye-normalized signal involves a linear normalization across the entire range of data, followed by the application of LOWESS normalization to the linearized dataset. We conservatively established the Log2 ratio of greater than or equal to 1 as the minimum average of fold enrichment, with an FDR-adjusted P value of less than 0.05 and a spot (gene) intensity equal to or above the average array signal as the cut-off values. The biological functions (gene ontology) of the preferential PCBP2-binding mRNAs identified by microarray analysis were determined with the web-based SBC analysis system. CSPN analysis was performed in the MOE Key Laboratory of Bioinformatics and Bioinformatics Division, TNLIST/Department of Automation at Tsinghua University (Beijing, China).
Biotin pull-down.
Fragments located in the 3′ UTR or 5′ UTR of candidate mRNAs were constructed into the pGEM-3zf vector containing a T7 promoter. The primers are shown in Supplemental Table 1. The biotin-labeled sense RNA probes were synthesized in vitro using T7 RNA polymerase (Takara Bio). Cytoplasmic cell extracts were isolated from T98G cells using nuclear protein extraction reagents (Thermo Scientific). RNA affinity capture was subsequently conducted with streptavidin-sepharose beads as described previously (47).
mRNA half-life and RNase protection assay.
Biotin-labeled antisense probes were synthesized in vitro with SP6 RNA polymerase (Takara Bio) from DNA templates constructed in a biotin pull-down assay. T98G cells were transfected with the control siRNA and PCBP2 siRNA. After 48 hours, actinomycin D (Sigma-Aldrich) (5 μg/ml) was added. Total RNA was collected at 0, 2, 4 and 6 hours. Ten micrograms of total RNA from each time point was cohybridized with the antisense probes at 45°C overnight. The excess probes and unhybridized RNA were eliminated by treatment with RNase A/T1 (Ambion, Invitrogen) at 37°C for 30 minutes. The protected RNA fragments in the precipitate were resolved on a 6% polyacrylamide gel containing 8 M urea and then electrophoretically transferred to a positively charged nylon membrane. After the membrane was cross-linked at 120 mJ/cm2, the protected fragments were immediately detected using Chemiluminescent Nucleic Acid Detection (Pierce Biotechnology) according to the manufacturer’s instructions. Quantitation was performed on the densitometric scan with Quantity One software, version 4.6.9 (Bio-Rad).
Soft agar colony formation assay.
U87MG and U251 cells infected by adenoviruses (1 × 104) were suspended in 1.5 ml complete medium supplemented with 0.45% low melting point agarose (Invitrogen). The cells were placed in 35-mm tissue culture plates containing 1.5 ml complete medium and agarose (0.75%) on the bottom layer. The plates were incubated at 37°C with 5% CO2 for 2 weeks. Cell colonies were stained with 0.005% crystal violet and analyzed using a microscope.
Xenograft model in nude mice.
T98G cells (5 × 106) in 100 μl of a 1:1 mixture of culture medium and growth factor–reduced Matrigel were implanted subcutaneously into the forelegs of 4- to 5-week-old male BALB/c athymic nu/nu mice (Vital River) according to the method described previously (48). When the tumors reached approximately 7 to 10 mm in diameter, they were prepared to form a brei and then injected subcutaneously into nude mice. The animals were separated into 3 groups (8 animals per group). Three weeks after injection, adenoviruses packaged at GeneChem were injected into the tumors 4 times with a 109 PFU per tumor each time, and the tumors were measured at each time point as indicated in Figure 3C. In Figure 9H, we used U87MG cells for subcutaneous implantation in mice. The data were analyzed by a 2-tailed Student’s t test.
Intracranial orthotopic xenografts were established by implanting 5 × 105 Luc-U87MG stable cells. The shNT-U87MG, shPCBP2-U87MG, vector-U87MG, and FHL3-U87MG stable transfectants were constructed by Shanghai Chempartner. Five- to 6-week-old BALB/c athymic nu/nu mice were anesthetized, and the implantation of stably constructed U87MG into the third ventricle was performed stereotactically (2 mm lateral and 0.5 mm anterior to the bregma; depth 1.5 mm from the dura). Tumor size was quantified by bioluminescence imaging.
Statistics.
Data are presented as the mean ± SD unless otherwise indicated. Statistical significance was determined by a paired or unpaired 2-tailed Student’s t test, and a P value of less than 0.05 was considered statistically significant.
Study approval.
All human tissue samples of normal brain and glioma were obtained from the Department of Neurosurgery, Beijing Tiantan Hospital. All samples were classified according to the third edition of the histological grades of tumors of the nervous system published by the WHO in 2000. Informed consent for the use of samples was obtained from all patients before surgery, and approval was obtained from the Medical Ethics Committee of the Beijing Tiantan Hospital (Beijing, China). All animal studies were approved by the IACUC of the Center for Experimental Animal Research (China), and all animal experiments were performed in accordance with institutional guidelines and abided by the declaration of ethical approval for experiments (animal experiment ethical investigation tab ACUC2010A02-123).
Supplementary Material
Acknowledgments
We thank members of the National Laboratory of Medical Molecular Biology (China) for their valuable input and support throughout this study. We especially thank Wenji Dong, Yongsheng Chang, and Ye Zhang for their helpful discussions. We are also grateful to Yang Chen of the MOE Key Laboratory of Bioinformatics and Bioinformatics Division, TNLIST/Department of Automation, Tsinghua University, for the CSPN analysis. This project was supported by grants from the National Basic Research Program of China (2011CBA01104, 2007CB946902, 2009CB825403, 2013CB531304), the National Natural Science Foundation of China (30825023, 31071203), the Program for New Century Excellent Talents in University (NCET-07-0505), and the 111 project (B08007).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(5):2103–2118. doi:10.1172/JCI61820.
Zhongshuai Xin’s present address is: Division of Biochemical and Gene Engineering Medicines, National Institute for Food and Drug Control, Beijing, China.
References
- 1.Makeyev AV, Liebhaber SA. The poly(C)-binding proteins: a multiplicity of functions and a search for mechanisms. RNA. 2002;8(3):265–278. doi: 10.1017/S1355838202024627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi HS, Hwang CK, Song KY, Law PY, Wei LN, Loh HH. Poly(C)-binding proteins as transcriptional regulators of gene expression. Biochem Biophys Res Commun. 2009;380(3):431–436. doi: 10.1016/j.bbrc.2009.01.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leffers H, Dejgaard K, Celis JE. Characterisation of two major cellular poly(rC)-binding human proteins, each containing three K-homologous (KH) domains. Eur J Biochem. 1995;230(2):447–453. doi: 10.1111/j.1432-1033.1995.tb20581.x. [DOI] [PubMed] [Google Scholar]
- 4.Makeyev AV, Liebhaber SA. Identification of two novel mammalian genes establishes a subfamily of KH-domain RNA-binding proteins. Genomics. 2000;67(3):301–316. doi: 10.1006/geno.2000.6244. [DOI] [PubMed] [Google Scholar]
- 5.Ritchie SA, et al. Identification of the SRC pyrimidine-binding protein (SPy) as hnRNP K: implications in the regulation of SRC1A transcription. Nucleic Acids Res. 2003;31(5):1502–1513. doi: 10.1093/nar/gkg246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans JR, et al. Members of the poly (rC) binding protein family stimulate the activity of the c-myc internal ribosome entry segment in vitro and in vivo. Oncogene. 2003;22(39):8012–2020. doi: 10.1038/sj.onc.1206645. [DOI] [PubMed] [Google Scholar]
- 7.Pillai MR, Chacko P, Kesari LA, Jayaprakash PG, Jayaram HN, Antony AC. Expression of folate receptors and heterogeneous nuclear ribonucleoprotein E1 in women with human papillomavirus mediated transformation of cervical tissue to cancer. J Clin Pathol. 2003;56(8):569–574. doi: 10.1136/jcp.56.8.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thakur S, et al. Regulation of BRCA1 transcription by specific single-stranded DNA binding factors. Mol Cell Biol. 2003;23(11):3774–3787. doi: 10.1128/MCB.23.11.3774-3787.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cloke B, et al. The poly(c)-binding protein-1 regulates expression of the androgen receptor. Endocrinology. 2010;151(8):3954–3964. doi: 10.1210//en.2009-1264. [DOI] [PubMed] [Google Scholar]
- 10.Chaudhury A, Hussey GS, Ray PS, Jin G, Fox PL, Howe PH. TGF-beta-mediated phosphorylation of hnRNP E1 induces EMT via transcript-selective translational induction of Dab2 and ILEI. Nat Cell Biol. . 2010;12(3):286–293. doi: 10.1038/ncb2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H, et al. PCBP1 suppresses the translation of metastasis-associated PRL-3 phosphatase. Cancer Cell. 2010;18(1):52–62. doi: 10.1016/j.ccr.2010.04.028. [DOI] [PubMed] [Google Scholar]
- 12.Tommerup N, Leffers H. Assignment of human KH-box-containing genes by in situ hybridization: HNRNPK maps to 9q21.32-q21.33, PCBP1 to 2p12-p13, and PCBP2 to 12q13.12-q13.13, distal to FRA12A. Genomics. 1996;32(2):297–298. doi: 10.1006/geno.1996.0121. [DOI] [PubMed] [Google Scholar]
- 13.Perrotti D, Calabretta B. Post-transcriptional mechanisms in BCR/ABL leukemogenesis: role of shuttling RNA-binding proteins. Oncogene. 2002;21(56):8577–8583. doi: 10.1038/sj.onc.1206085. [DOI] [PubMed] [Google Scholar]
- 14.Roychoudhury P, Paul RR, Chowdhury R, Chaudhuri K. HnRNP E2 is downregulated in human oral cancer cells and the overexpression of hnRNP E2 induces apoptosis. Mol Carcinog. 2007;46(3):198–207. doi: 10.1002/mc.20265. [DOI] [PubMed] [Google Scholar]
- 15.Sean P, Nguyen JH, Semler BL. The linker domain of poly(rC) binding protein 2 is a major determinant in poliovirus cap-independent translation. Virology. 2008;378(2):243–253. doi: 10.1016/j.virol.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sean P, Nguyen JH, Semler BL. Altered interactions between stem-loop IV within the 5′ noncoding region of coxsackievirus RNA and poly(rC) binding protein 2: effects on IRES-mediated translation and viral infectivity. Virology. 2009;389(1–2):45–58. doi: 10.1016/j.virol.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rieder E, Xiang W, Paul A, Wimmer E. Analysis of the cloverleaf element in a human rhinovirus type 14/poliovirus chimera: correlation of subdomain D structure, ternary protein complex formation and virus replication. J Gen Virol. 2003;84(pt 8):2203–2216. doi: 10.1099/vir.0.19013-0. [DOI] [PubMed] [Google Scholar]
- 18.Fukushi S, Okada M, Kageyama T, Hoshino FB, Nagai K, Katayama K. Interaction of poly(rC)-binding protein 2 with the 5′-terminal stem loop of the hepatitis C-virus genome. Virus Res. 2001;73(1):67–79. doi: 10.1016/S0168-1702(00)00228-8. [DOI] [PubMed] [Google Scholar]
- 19.Pan T, Fang C, Yi Z, Yang P, Yuan Z. Subproteomic analysis of the cellular proteins associated with the 3′ untranslated region of the hepatitis C virus genome in human liver cells. Biochem Biophys Res Commun. 2006;347(3):683–691. doi: 10.1016/j.bbrc.2006.06.144. [DOI] [PubMed] [Google Scholar]
- 20.You F, et al. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat Immunol. 2009;10(12):1300–1308. doi: 10.1038/ni.1815. [DOI] [PubMed] [Google Scholar]
- 21.Molinaro RJ, Jha BK, Malathi K, Varambally S, Chinnaiyan AM, Silverman RH. Selection and cloning of poly(rC)-binding protein 2 and Raf kinase inhibitor protein RNA activators of 2′,5′-oligoadenylate synthetase from prostate cancer cells. Nucleic Acids Res. 2006;34(22):6684–6695. doi: 10.1093/nar/gkl968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang JS, et al. High levels of the BCR/ABL oncoprotein are required for the MAPK-hnRNP-E2 dependent suppression of C/EBPalpha-driven myeloid differentiation. Blood. 2007;110(3):994–1003. doi: 10.1182/blood-2007-03-078303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eiring AM, et al. miR-328 functions as an RNA decoy to modulate hnRNP E2 regulation of mRNA translation in leukemic blasts. Cell. 2010. 140 5 652– 665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh D, Srivastava GP, Xu D, Schulz LC, Roberts RM. A link between SIN1 (MAPKAP1) and poly(rC) binding protein 2 (PCBP2) in counteracting environmental stress. Proc Natl Acad Sci U S A. 2008;105(33):11673–11678. doi: 10.1073/pnas.0803182105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waggoner SA, Johannes GJ, Liebhaber SA. Depletion of the poly(C)-binding proteins alphaCP1 and alphaCP2 from K562 cells leads to p53-independent induction of cyclin-dependent kinase inhibitor (CDKN1A) and G1 arrest. J Biol Chem. 2009;284(14):9039–9049. doi: 10.1074/jbc.M806986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uchida H, et al. Adenovirus-mediated transfer of siRNA against survivin induced apoptosis and attenuated tumor cell growth in vitro and in vivo. Mol Ther. 2004;10(1):162–171. doi: 10.1016/j.ymthe.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 27.Eiring AM, et al. Identification of novel posttranscriptional targets of the BCR/ABL oncoprotein by ribonomics: requirement of E2F3 for BCR/ABL leukemogenesis. Blood. 2008;111(2):816–828. doi: 10.1182/blood-2007-05-090472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waggoner SA, Liebhaber SA. Identification of mRNAs associated with alphaCP2-containing RNP complexes. Mol Cell Biol. 2003;23(19):7055–7067. doi: 10.1128/MCB.23.19.7055-7067.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Sousa Abreu R, et al. Genomic analyses of musashi1 downstream targets show a strong association with cancer-related processes. J Biol Chem. 2009;284(18):12125–12135. doi: 10.1074/jbc.M809605200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang Y, Li S. Detection of characteristic sub pathway network for angiogenesis based on the comprehensive pathway network. BMC Bioinformatics. 2010;11(suppl 1):1–9. doi: 10.1186/1471-2105-11-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chkheidze AN, Lyakhov DL, Makeyev AV, Morales J, Kong J, Liebhaber SA. Assembly of the alpha-globin mRNA stability complex reflects binary interaction between the pyrimidine-rich 3′ untranslated region determinant and poly(C) binding protein alphaCP. Mol Cell Biol. 1999;19(7):4572–4581. doi: 10.1128/mcb.19.7.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kong J, Sumaroka M, Eastmond DL, Liebhaber SA. Shared stabilization functions of pyrimidine-rich determinants in the erythroid 15-lipoxygenase and alpha-globin mRNAs. Mol Cell Biol. 2006;26(15):5603–5614. doi: 10.1128/MCB.01845-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ji X, Kong J, Liebhaber SA. In vivo association of the stability control protein alphaCP with actively translating mRNAs. Mol Cell Biol. 2003;23(3):899–907. doi: 10.1128/MCB.23.3.899-907.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Du Z, Lee JK, Fenn S, Tjhen R, Stroud RM, James TL. X-ray crystallographic and NMR studies of protein-protein and protein-nucleic acid interactions involving the KH domains from human poly(C)-binding protein-2. RNA. 2007;13(7):1043–1051. doi: 10.1261/rna.410107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stefanovic B, Hellerbrand C, Holcik M, Briendl M, Aliebhaber S, Brenner DA. Posttranscriptional regulation of collagen alpha1(I) mRNA in hepatic stellate cells. Mol Cell Biol. 1997;17(9):5201–5209. doi: 10.1128/mcb.17.9.5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paulding WR, Czyzyk-Krzeska MF. Regulation of tyrosine hydroxylase mRNA stability by protein-binding, pyrimidine-rich sequence in the 3′-untranslated region. J Biol Chem. 1999;274(4):2532–2538. doi: 10.1074/jbc.274.4.2532. [DOI] [PubMed] [Google Scholar]
- 37.Li M, et al. The four-and-a-half-LIM protein 2 (FHL2) is overexpressed in gliomas and associated with oncogenic activities. Glia. 2008;56(12):1328–1338. doi: 10.1002/glia.20701. [DOI] [PubMed] [Google Scholar]
- 38.Morgan MJ, Whawell SA. The structure of the human LIM protein ACT gene and its expression in tumor cell lines. Biochem Biophys Res Commun. 2000;273(2):776–783. doi: 10.1006/bbrc.2000.3006. [DOI] [PubMed] [Google Scholar]
- 39.Johannessen M, Møller S, Hansen T, Moens U, Van Ghelue M. The multifunctional roles of the four-and-a-half-LIM only protein FHL2. Cell Mol Life Sci. 2006;63(3):268–284. doi: 10.1007/s00018-005-5438-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ding L, et al. Human four-and-a-half LIM family members suppress tumor cell growth through a TGF-beta-like signaling pathway. J Clin Invest. 2009;119(2):349–361. doi: 10.1172/JCI35930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hubbi ME, Gilkes DM, Baek JH, Semenza GL. Four-and-a-half LIM domain proteins inhibit transactivation by hypoxia-inducible factor 1. J Biol Chem. 2012;287(9):6139–6149. doi: 10.1074/jbc.M111.278630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mils V, et al. LIM-only protein FHL3 interacts with CDC25B2 phosphatase. Exp Cell Res. 2003;285(1):99–106. doi: 10.1016/S0014-4827(03)00018-1. [DOI] [PubMed] [Google Scholar]
- 43.Niu C, et al. Downregulation and antiproliferative role of FHL3 in breast cancer. IUBMB Life. 2011;63(9):764–771. doi: 10.1002/iub.502. [DOI] [PubMed] [Google Scholar]
- 44.Xia W, et al. Identification and characterization of FHL3 as a novel angiogenin-binding partner. Gene. 2012;504(2):233–237. doi: 10.1016/j.gene.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, et al. MicroRNA-128 inhibits glioma cells proliferation by targeting transcription factor E2F3a. J Mol Med. 2009;87(1):43–51. doi: 10.1007/s00109-008-0403-6. [DOI] [PubMed] [Google Scholar]
- 46.Penalva LO, Keene JD. Biotinylated tags for recovery and characterization of ribonucleoprotein complexes. Biotechniques. 2004;37(4):604–610. doi: 10.2144/04374ST05. [DOI] [PubMed] [Google Scholar]
- 47.Lin JY, Li ML, Huang PN, Chien KY, Horng JT, Shih SR. Heterogeneous nuclear ribonuclear protein K interacts with the enterovirus 71 5’ untranslated region and participates in virus replication. J Gen Virol. 2008;89(pt 10):2540–2549. doi: 10.1099/vir.0.2008/003673-0. [DOI] [PubMed] [Google Scholar]
- 48.Teicher BA, Menon K, Alvarez E, Galbreath E, Shih C, Faul M. Antiangiogenic and antitumor effects of a protein kinase Cbeta inhibitor in human T98G glioblastoma multiforme xenografts. Clin Cancer Res. 2001;7(3):634–640. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.