

Abstract
Chronic pancreatitis is an inflammatory disease that causes progressive destruction of pancreatic acinar cells and, ultimately, loss of pancreatic function. We investigated the role of IκB kinase α (IKKα) in pancreatic homeostasis. Pancreas-specific ablation of IKKα (IkkαΔpan) caused spontaneous and progressive acinar cell vacuolization and death, interstitial fibrosis, inflammation, and circulatory release of pancreatic enzymes, clinical signs resembling those of human chronic pancreatitis. Loss of pancreatic IKKα causes defective autophagic protein degradation, leading to accumulation of p62-mediated protein aggregates and enhanced oxidative and ER stress in acinar cells, but none of these effects is related to NF-κB. Pancreas-specific p62 ablation prevented ER and oxidative stresses and attenuated pancreatitis in IkkαΔpan mice, suggesting that cellular stress induced by p62 aggregates promotes development of pancreatitis. Importantly, downregulation of IKKα and accumulation of p62 aggregates were also observed in chronic human pancreatitis. Our studies demonstrate that IKKα, which may control autophagic protein degradation through its interaction with ATG16L2, plays a critical role in maintaining pancreatic acinar cell homeostasis, whose dysregulation promotes pancreatitis through p62 aggregate accumulation.
Introduction
Chronic pancreatitis, a relatively common and potentially lethal disease, entails repetitive tissue destruction, which eventually leads to pancreatic failure (1). Chronic pancreatitis greatly increases the risk of pancreatic adenocarcinoma (PDAC), whose pathogenesis remains obscure and for which specific treatments and preventive approaches are still lacking (2). Usually, chronic pancreatitis is preceded by acute pancreatitis, whose hallmarks include inappropriate (intra-acinar) trypsinogen activation, vacuole formation, death of acinar cells through necrosis and apoptosis, and inflammation (3). Recurrent bouts of pancreatitis, which lead to progressive acinar destruction followed by healing and fibrosis, eventually result in chronic disease and loss of exocrine and, ultimately, endocrine pancreatic functions (1). Molecular mechanisms that initiate pancreatitis or drive its progression from acute to chronic disease are poorly understood. For a long time, the premature intra-acinar conversion of trypsinogen to active trypsin, which can trigger activation of other pancreatic zymogens, has been considered a key pathogenic mechanism (1). However, the acute and immediate cause of trypsinogen activation remains obscure. Furthermore, the critical role of premature trypsinogen activation has been challenged by generation of mice lacking trypsinogen isoform 7 (T7), which still develop experimental pancreatitis upon cerulein challenge, despite a large decrease in trypsin activity (4). Nonetheless, genetic alterations that increase or decrease pancreatitis risk were identified in genes that encode digestive proteases or their inhibitors, such as cationic trypsinogen (PRSS1), pancreatic secretory trypsin inhibitor (SPINK1), and chymotrypsinogen C (CTRC) (1). Yet intra-acinar trypsinogen activation does not lead to chronic pancreatitis accompanied by fibrosis and inflammation (5). Clearly, other pathways also play critical roles in acute and chronic pancreatitis. Such pathways may include NF-κB and Ras signaling and ER stress.
A pathogenic role for ER stress is supported by a mutation (p.R116C) that causes misfolding and intracellular retention of human PRSS1, but does not affect its activation (6). This mutation, which causes ER stress, results in chronic pancreatitis. ER stress, triggered by protein misfolding and accumulation of protein aggregates, is sensed by transmembrane transducers that signal the cell to lower rates of protein synthesis and translocation across membranes and upregulate expression of gene products that facilitate protein folding (7). In extreme cases, ER stress leads to cell death through induction of the proapoptotic transcription factor C/EBP homologous protein (CHOP) (7) and other mechanisms. Other cellular stresses, for instance, oxidative stress, can also trigger CHOP upregulation (8). As professional protein-secreting cells, pancreatic acinar cells are inherently susceptible to ER stress (7), and ethanol exposure was suggested to cause pancreatitis through an ER stress–related mechanism (9). Conversely, the protective unfolded protein response (UPR) attenuates pancreatitis and is necessary for maintenance of pancreatic homeostasis (7, 10, 11).
ER stress is also relieved by macroautophagy (hereafter referred to as autophagy), a major proteolytic pathway that eliminates unfolded proteins, inclusion bodies, and damaged organelles (12). Conversely, autophagy defects can cause ER stress (12, 13). During autophagy, protein aggregates, portions of cytoplasm, and damaged organelles are engulfed within a double-membrane vesicle, the autophagosome (14). Subsequently, autophagosomes fuse with late endosomes to form amphisomes that become single-membraned autolysosomes upon fusion with lysosomes, within which the sequestered macromolecules are degraded by lysosomal hydrolases (14). As a stress-preventing mechanism, autophagy suppresses degenerative and lysosomal storage diseases, inhibits tumor development, and slows down aging (14). Autophagic protein degradation is impaired during experimental pancreatitis (in particular upon cerulein administration), and this can cause acinar cell vacuolization and excessive trypsinogen activation (3). Autophagic flux is reduced during pancreatitis due to impaired cathepsin-mediated processing of lysosomal proteases (3). However, excessive autophagy can also promote lysosomal activation of trypsinogen and can thereby contribute to induction of pancreatitis in cerulein-treated mice (15), in which autophagic maturation and protein degradation are disrupted (3). Defective autophagic proteolysis is accompanied by marked accumulation of p62/sequestosome-1/SQSTM1, a selective autophagy substrate, a signaling scaffold, and a chaperone for polyubiquitinated proteins (16–19). Reduced p62 degradation leads to formation of ubiquitinated protein aggregates because p62 undergoes self oligomerization and at the same time binds polyubiquitinated proteins, whose degradation depends on normal autophagic flux (20). Accordingly, p62 is a major component of ubiquitin-containing inclusions, such as the “hepatocytic Mallory bodies” found in alcoholic hepatitis and steatohepatitis (21) and the protein aggregates that are typical of various degenerative and neurological diseases (22). Ablation of p62 in liver-specific Atg7-knockout mice, in which autophagy is blocked at the initiation stage, prevents liver damage and development of hepatic adenomas (22, 23).
IκB kinase α (IKKα) is a subunit of the IKK complex, which consists of 2 catalytic subunits, IKKα and IKKβ, and the regulatory subunit IKKγ/NEMO (24). A major function of IKK that mostly depends on IKKβ is phosphorylation of IκB proteins, which triggers their degradation and subsequent activation of NF-κB transcription factors (25). Although IKKα contributes to canonical NF-κB signaling in which RelA:p50 and cRel:p50 dimers are translocated to the nucleus, it is more important for alternative NF-κB signaling in which RelB:p52 dimers are activated (25). IKKα also has kinase-independent functions that are critical for epidermal development, keratinocyte differentiation, and limb and skeletal patterning. These functions are underscored by defects in skin and limb development in Ikkα-null mice (26–29) and in IKKα-deficient human fetuses (30). The role of NF-κB in pancreatic homeostasis was studied either through forced expression of a constitutively active IKKβ variant in pancreatic acinar cells (31) or pancreas-specific ablation of the RelA gene (32). Whereas chronic NF-κB activation in acinar cells can cause pancreatitis (31), insufficient NF-κB activity sensitizes acinar cells to cerulein-induced death (32). These seemingly opposing effects are due to the critical and distinct roles of NF-κB in induction of inflammation and maintenance of tissue integrity (33). To study the role of IKK subunits in pancreatic physiology, we ablated Ikkα and Ikkβ in pancreatic epithelial cells (PEC). Here, we report that PEC-specific Ikkα ablation (IkkαΔpan) results in acinar cell vacuolization and death, tissue damage, fibrosis, and inflammation, resembling chronic pancreatitis in humans. The role of IKKα in maintaining pancreatic homeostasis is independent of NF-κB or its protein kinase activity and is not shared with IKKβ. Loss of IKKα in acinar cells diminished autophagic protein degradation and caused accumulation of p62 aggregates and ER stress. PEC-specific p62 ablation ameliorated pancreatitis in IkkαΔpan mice. Downregulation of IKKα and accumulation of p62 aggregates were also observed in chronic human pancreatitis.
Results
Spontaneous and progressive acinar cell damage, fibrosis, and inflammation in IkkαΔpan mice.
“Floxed” IkkαF/F mice were crossed with Pdx1-Cre transgenic mice in which Cre-mediated recombination takes place in PEC, including acinar, ductal, and islet cells (34). Compound Pdx1-Cre;IkkαF/F (hereafter IkkαΔpan) mice were born at the expected Mendelian ratio and exhibited normal growth and development through weaning. Specific and efficient IKKα deletion in the pancreas was confirmed by immunoblotting (IB) (Figure 1A). Newborn IkkαΔpan pancreata appeared normal upon gross inspection, but by 3 weeks of age, patchy acinar cell vacuolization was observed (Figure 1, B and C, and Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI64498DS1). As the mice aged, the patchy abnormalities became more severe, including more extensive vacuolization, inter- and intralobular fibrosis, ductal metaplasia, acinar atrophy, adipose replacement of acini, immune cell infiltration, and ductal-complex formation with condensed luminal contents (Figure 1, D–J). Pancreatic fibrosis was confirmed by Sirius red staining (Figure 1K and Supplemental Figure 1B) and upregulation of α-SMA and collagen α1(I) and α1(III) mRNAs (Figure 1L). Expression of mRNAs encoding proinflammatory cytokines and chemokines, including TNF, IL-1β, IL-6, MCP-1, and RANTES, was also upregulated in IkkαΔpan pancreata, and this correlated with an increase in myeloid and lymphoid cell markers (Figure 1M). Immunofluorescence (IF) analysis confirmed infiltration of B and T cells, macrophages, and neutrophils into pancreata of IkkαΔpan mice that were 3 months or older (Supplemental Figure 1, C and D).
Figure 1. Loss of pancreatic IKKα results in spontaneous and progressive pancreatitis.

(A) IB analysis of pancreas (P) or liver (L) lysates of IkkαF/F and IkkαΔpan mice. (B–J) Histological analysis of pancreatic sections from IkkαF/F and IkkαΔpan mice. H&E staining of pancreatic sections (B, D, and E). Note acinar cell vacuolization (C, arrows), ductal metaplasia (F), BrdU incorporation in active ductal metaplasia (G), immune cell infiltration (H), fat tissue replacement (I), and condensed luminal contents in dilated ducts (J) in IkkαΔpan pancreata. Original magnification, ×200 (B, C, D, E, and I); ×400 (F, G, H, and J). (K) Sirius red staining showing pancreatic fibrosis in 3-month-old IkkαΔpan mice. Original magnification, ×200. (L and M) qRT-PCR analysis of pancreatic RNAs from 3-month-old mice for fibrogenic markers (L) and cytokine and chemokine genes and immune cell markers (M). Results are shown as mean ± SEM. n = 4–6 mice per condition. *P < 0.05; **P < 0.01. Scale bars: 50 μm; 20 μm (insets).
Increased acinar cell death and compensatory proliferation in IkkαΔpan pancreata.
Normally, amylase and lipase are secreted by acinar cells into pancreatic ducts and then reach the duodenum. However, when acinar cells are damaged, amylase and lipase leak into the circulation and their elevated serum concentrations are diagnostic markers of pancreatitis in humans. Serum amylase and lipase were significantly increased in IkkαΔpan mice relative to IkkαF/F controls, indicating acinar cell damage (Figure 2A). Intrapancreatic trypsin activity showed a progressively increasing trend in IkkαΔpan pancreata, which did not reach statistical significance (Figure 2B). Acinar cell vacuolization may lead to apoptosis and necrosis. Indeed, TUNEL and immunohistochemical (IHC) staining of cleaved caspase-3 revealed greatly enhanced acinar apoptosis in IkkαΔpan pancreata (Figure 2C and Supplemental Figure 2). H&E staining confirmed pyknotic nuclei in some IkkαΔpan acinar cells, as well as necrotic areas, which covered 1.3% ± 0.24% of the pancreatic parenchymal area (Figure 2, D and E). Acinar cell damage can trigger regenerative responses, including compensatory proliferation. Indeed, BrdU incorporation and Ki67 labeling of acinar cells were elevated in IkkαΔpan mice (Figure 2, F and G). Furthermore, acinar cell damage can trigger acinar-ductal metaplasia (ADM), a frequent feature of human chronic pancreatitis. Areas of ADM with BrdU incorporation were consistently observed in IkkαΔpan pancreata (Figure 1, F and G).
Figure 2. Loss of pancreatic IKKα causes acinar cell death and compensatory proliferation.

(A) Amylase and lipase activities in sera of 6- or 12-week-old IkkαF/F and IkkαΔpan mice. (B) Trypsin activity in pancreata of 2- or 5-month-old IkkαF/F and IkkαΔpan mice. (C) TUNEL assay and apoptotic cell index. Original magnification, ×200. (D and E) H&E staining of IkkαΔpan pancreatic sections showing acinar cell apoptosis (D) and necrosis (E). Original magnification, ×400. (F) BrdU incorporation and labeling index in pancreata of 3-month-old IkkαF/F and IkkαΔpan mice. Original magnification, ×400. (G) Ki67 staining and labeling index of pancreatic sections from 3-month-old mice. Original magnification, ×400. Results are shown as mean ± SEM. n = 4–6 mice per condition. **P < 0.01. Scale bars: 50 μm; 10 μm (insets).
Endocrine deficiency in IkkαΔpan mice.
Chronic pancreatitis can lead to diabetes mellitus (35). Most islets in IkkαΔpan mice appeared normal (Supplemental Figure 3A). Nonetheless, fasting serum insulin was reduced in IkkαΔpan mice over 3 months old compared with IkkαF/F littermates, and this was accompanied by increased fasting blood glucose (Supplemental Figure 3B). Furthermore, glucose tolerance tests (GTT) indicated that IkkαΔpan mice became more glucose intolerant as they aged (Supplemental Figure 3C). Glucose intolerance in IkkαΔpan mice can be explained by fibrosis and inflammation of the exocrine pancreas leading to islet damage. However, it cannot be ruled out that IKKα loss in β cells may impair insulin secretion more directly.
Pancreatic abnormalities in IkkαΔpan mice are independent of IKKα kinase activity or IKKβ.
In macrophages, IKKα kinase activity contributes to suppression of NF-κB activity by accelerating the turnover and promoter clearance of RelA and c-Rel, as seen in homozygous IkkαAA knockin mice in which IKKα cannot be activated due to Ser-to-Ala substitutions within its activation loop (36). Unlike IkkαΔpan mice, IkkαAA/AA mice did not exhibit any pancreatic abnormalities or changes in serum amylase or lipase (Supplemental Figure 4, A and B). We also did not detect elevated nuclear RelA in pancreata of 2-month-old IkkαΔpan mice (Supplemental Figure 4C). Consistent with previous reports (32), pancreas-specific Ikkβ ablation also did not result in any obvious histological or enzymatic abnormalities (Supplemental Figure 4, D and E). IkkβΔpan mice did not develop glucose intolerance either (Supplemental Figure 4F).
Although no increase in basal NF-κB activity was seen in IkkαΔpan pancreata, Ikkα ablation in keratinocytes results in elevated IKKβ activity (26, 27). We therefore determined whether additional Ikkβ ablation ameliorated pancreatitis in IkkαΔpan mice. However, IKKβ deletion did not attenuate any of the pancreatic pathologies in IkkαΔpan mice. At 3 weeks of age, IkkαΔpan;IkkβΔpan double mutants developed acinar cell vacuolization (Supplemental Figure 5A), and as they aged, these mice developed more acinar cell vacuolization, inter- or intralobular fibrosis, ductal metaplasia, and compensatory acinar cell proliferation and inflammation (Supplemental Figure 5, B–D). Serum amylase and lipase concentrations were also elevated in IkkαΔpan;IkkβΔpan mice compared with control littermates (Supplemental Figure 5E), and GTT indicated that IkkαΔpan;IkkβΔpan mice were glucose intolerant (Supplemental Figure 5F). EM revealed accumulation of enlarged autolysosomes encased by a single membrane in IkkαΔpan;IkkβΔpan acinar cells (Supplemental Figure 5G). Old (>1 yr) IkkαΔpan;IkkβΔpan mice exhibited marked acinar cell necrosis (Supplemental Figure 5H) and extensive adipose tissue replacement, with few remaining PanIN-like ductal lesions (Supplemental Figure 5I). Alcian blue staining indicated that the ductal lesions contained mucin, a potential marker of premalignancy and PDAC (Supplemental Figure 5J). However, no tumors were detected in IkkαΔpan;IkkβΔpan mice, although ductal hyperplasia (Supplemental Figure 5K), Ki67-positive ductal cells (Supplemental Figure 5L), periductal fibrosis (Supplemental Figure 5M), and periductal inflammation (Supplemental Figure 5, I and K) were prominent. IkkβΔpan single mutants did not exhibit any such defects. Therefore, IKKβ probably does not have a critical role in maintenance of pancreatic homeostasis in normal mice, although it appears to exert a protective function under pathological conditions such as those caused by the IKKα deficiency.
Loss of pancreatic IKKα impairs autophagy and causes p62 accumulation.
Acinar cell vacuolization was the first distinct feature in IkkαΔpan pancreata, appearing at 3 weeks of age (Figure 3A). EM analysis suggested that the vacuoles in IkkαΔpan acinar cells correspond to enlarged autolysosomes encased by a single membrane containing partially degraded organelles, mainly ER and ribosomes (Figure 3A and Supplemental Figure 6, A–D). Unlike starvation-induced autophagy, which results in appearance of multiple small vacuoles in pancreatic acinar cells (Supplemental Figure 6, E and F), in IkkαΔpan acinar cells, the vacuoles were fewer in number but larger in size (Supplemental Figure 6G). Fusion of lysosomes with autophagosomes was also observed and confirmed by IF analysis for the autophagosome and lysosome markers LC3 and LAMP1, respectively (Supplemental Figure 6H). These findings suggested that loss of IKKα may slow down autophagic protein degradation. Indeed, IkkαΔpan pancreata showed a marked increase in the autophagy substrates LC3 and p62 (Figure 3B). LC3, the mammalian homolog of yeast Atg8, undergoes site-specific proteolysis and lipidation, resulting in conversion of its LC3-I form to LC3-II, which specifically translocates to the autophagosomal membrane (37). IkkαΔpan pancreata exhibited marked accumulation of LC3-II and a more modest increase in LC3-I. p62 is a chaperone for polyubiquitinated proteins, responsible for their transport into autophagosomes and eventual degradation in the lysosome (19). p62 accumulation is a classical indicator of defective autophagy (20). IHC analysis confirmed accumulation of LC3, p62, and ubiquitin in IkkαΔpan pancreata, particularly in areas with acinar cell vacuolization (Figure 3C). Quantitative RT-PCR (qRT-PCR) analysis did not reveal significant changes in mRNA expression of multiple autophagy-related proteins, including p62, LC3, Beclin1, ATG12, and Vps34 (Figure 3D). Importantly, however, the turnover of p62 was much slower in IkkαΔpan acinar cells (Figure 3E). To confirm that p62 is degraded through autophagy and not via the proteasome, we examined p62 degradation in the presence of the proteasome inhibitor MG132 or the autophagy inhibitor 3-methyladenine (3-MA). MG132 treatment did not impair p62 degradation in normal acinar cells, whereas 3-MA treatment prevented degradation and caused p62 accumulation, confirming that acinar cell p62 is mainly degraded through autophagy (Supplemental Figure 7). We also measured autophagic flux in IkkαF/F and IkkαΔpan acinar cells and found it to be significantly reduced in the absence of IKKα (Figure 3F). LC3-II expression was also significantly higher (P < 0.05) in IkkαΔpan acinar cells than in IkkαF/F cells, and LC3-II turnover was slower (Supplemental Figure 8A). Treatment with bafilomycin A1 (Baf), a molecule that specifically inhibits lysosomal acidification, protein degradation, and autophagosome-lysosome fusion, increased the abundance of LC3-II in IkkαF/F cells more than in IkkαΔpan cells (Supplemental Figure 8B). In addition, autophagy induction with rapamycin increased LC3-II in IkkαF/F acinar cells, but not in IkkαΔpan cells, where LC3-II was already elevated (Supplemental Figure 8C). These results suggest that IKKα might be involved in late-stage autophagy (autophagosome maturation or protein degradation within autolysosomes) and that its absence in acinar cells impairs autophagic protein degradation.
Figure 3. Loss of pancreatic IKKα results in defective autophagic protein degradation and accumulation of p62 and ubiquitinated protein aggregates.

(A) H&E staining & EM analysis of pancreata from 3-week-old IkkαF/F and IkkαΔpan mice (note acinar cell vacuolization). EM shows enlarged autophagic vacuoles (autolysosomes) with partially digested organelles, including ER and ribosomes in IkkαΔpan pancreatic acinar cells (red arrows), but not in IkkαF/F mice (black arrows). Original magnification, ×400; scale bar: 50 μm (top panels); scale bar: 2 μm (bottom panels, EM). (B) IB analysis of pancreatic lysates from 3-month-old IkkαF/F and IkkαΔpan mice. (C) IF staining of LC3 and IHC staining of p62 and ubiquitin (Ub) in pancreatic sections from 3-month-old IkkαF/F and IkkαΔpan mice. Note p62 and Ub aggregates in IkkαΔpan acinar cells. Original magnification, ×400. Scale bars: 50 μm. (D) qRT-PCR of autophagy-related mRNAs. Results are shown as mean ± SEM. n = 3–5 mice per condition. (E) p62 turnover in acinar cells. Freshly isolated acinar cells of 2-month-old IkkαF/F and IkkαΔpan mice were incubated with cycloheximide (10 μg/ml) to block new protein synthesis. Protein lysates were prepared at the indicated times, followed by IB analysis. (F) ER stress–induced (10 μg/ml tunicamycin for 4 hours) autophagic flux in IkkαF/F and IkkαΔpan acinar cells in the absence or presence of the proteasome inhibitor MG132 (100 nM) or the autophagy inhibitor 3MA (10 mM). Results are shown as mean ± SEM. *P < 0.05.
IKKα deficiency enhances ER and oxidative stress.
Most autophagic vacuoles in IkkαΔpan acinar cells contained partially degraded ER, ribosomes, and occasional zymogen granules; proteinaceous aggregate accumulation was also observed (Figure 4, A–D, and Supplemental Figure 9A). IkkαΔpan acinar cells also contained distended and dilated ER instead of the long, thin, and densely packed cisterna in IkkαF/F cells (Figure 4, A and B). However, unlike the autophagic vacuoles, the ER lumen was devoid of any organelles or proteinaceous aggregates. ER stress markers, Bip, CHOP, and the spliced form of XBP1 (sXBP1), were upregulated in IkkαΔpan pancreata (Figure 4E). Pancreatic acinar cells have very high rates of protein synthesis and secretion and are therefore particularly susceptible to ER perturbations (7). Autophagy may relieve ER stress by degrading unfolded and abnormally folded proteins (12). Indeed, treatment with a low dose (10 μg/ml) of tunicamycin led to more CHOP accumulation in IkkαΔpan cells than in IkkαF/F cells (Supplemental Figure 9B). Correspondingly, tunicamycin-treated IkkαΔpan cells exhibited reduced viability relative to control acinar cells (Figure 4F). Because enhanced ER stress may interfere with enzyme secretion (7, 9), we measured amylase secretion in primary mouse pancreatic acinar cells. The characteristic, biphasic, cholecystokinin–induced (CCK-8–induced) amylase secretion was impaired in IkkαΔpan cells (Figure 4G).
Figure 4. Pancreas-specific IKKα deficiency results in ER and oxidative stress.

(A–D) EM images showing dilated ER and autophagic vacuoles containing damaged ER in IkkαΔpan acinar cells. Scale bars: 500 nm. (E) qRT-PCR analysis of ER stress markers in IkkαF/F and IkkαΔpan pancreata. (F) Viability of IkkαF/F and IkkαΔpan acinar cells incubated with the indicated concentrations of tunicamycin for 24 hours. (G) Amylase release from IkkαF/F and IkkαΔpan acinar cells incubated with the indicated concentrations of cholecystokinin. (H) EM images showing swollen and fragmented mitochondria in IkkαΔpan pancreata. Scale bars: 500 nm. (I) qRT-PCR analysis of oxidative stress genes in IkkαF/F and IkkαΔpan pancreata. (J) Cis-aconitase activity in pancreata of 3-month-old IkkαF/F and IkkαΔpan mice. Results are shown as mean ± SEM. n = 3–5 mice per condition. *P < 0.05; **P < 0.01; ***P < 0.001.
We utilized the AR42J rat pancreatic cell line as an additional in vitro system. IKKα-deficient AR42J cells were generated by infection with a lentivirus containing Ikkα shRNA. IKKα knockdown resulted in morphological changes; instead of spreading out, AR42J(ΔIKKα) cells tended to clump together (Supplemental Figure 10A). AR42J(ΔIKKα) cells exhibited enhanced ROS accumulation (Supplemental Figure 10B), reduced ATP content, elevated expression of ER stress markers (Supplemental Figure 10D), and enhanced eIF2A and JNK phosphorylation after incubation with tunicamycin (Supplemental Figure 10E). IKK-deficient cells were more likely to undergo ER stress–induced death compared with AR42J(ctrl) cells (Supplemental Figure 10F). The IKKα deficiency also augmented CCK-induced eIF2A and PERK phosphorylation (Supplemental Figure 10G). These data further suggest that the absence of IKKα augments ER stress.
Mitochondria in IkkαΔpan pancreatic acinar cells were fragmented and swollen and contained fewer cristae (Figure 4H). These morphological alterations correlated with upregulation of oxidative stress markers, including Alox15, Duox1, Duox2, and Cyp24α1, in IkkαΔpan pancreata (Figure 4I). Cis-aconitase activity in IkkαΔpan pancreata was reduced compared with that in age-matched IkkαF/F controls, suggesting ROS accumulation and oxidative damage to mitochondria (Figure 4J).
IKKα interacts with ATG16L2.
Accumulation of enlarged autolysosomes in IkkaΔpan acinar cells suggested that the defect could occur at the autophagosome-lysosome fusion step or the lysosome itself. We examined the size, number, and morphology of lysosomes in IkkαF/F and IkkαΔpan acinar cells by EM image analysis, but could not find any pronounced difference between the 2 groups (not shown). We analyzed lysosomal proteins involved in autophagy and lysosomal pH regulation, including LAMP1 and LAMP2, different V-ATPase subunits, and cathepsins. We did not observe significant differences in expression of these proteins or their mRNAs between IkkαF/F and IkkαΔpan pancreata (Supplemental Figure 11 and data not shown).
We performed a yeast 2-hybrid screen to identify IKKα interacting proteins. Using full-length IKKα as a bait to screen a mouse pancreas cDNA library, we identified the autophagy-related protein ATG16L2 as a potential IKKα interactor. We tested the interaction between IKKα and ATG16L2 in mammalian cells, and indeed, the 2 proteins interacted upon overexpression (Figure 5A). Importantly, the endogenous proteins also interacted: ATG16L2 was precipitated by an IKKα antibody from WT but not IkkαΔpan pancreatic lysates (Figure 5B). Induction of ER stress with thapsigargin or tunicamycin potentiated this interaction (Figure 5C). ATG16L2 is a mammalian ortholog of yeast Atg16; the other Atg16 ortholog is ATG16L1, which is involved in autophagosome formation, elongation, and closure (37). Gene ablation experiments suggest that ATG16L1 is an essential autophagy protein in Paneth cells and macrophages (38, 39). Notably, ATG16L2 is expressed at much higher amounts in the pancreas compared with the liver, in which the predominant form is ATG16L1 (Supplemental Figure 12). Conversely, the pancreas contains less ATG16L1 than the liver. The function of ATG16L2 in autophagy is not clear. We used Atg16l2 siRNA to downregulate endogenous Atg16l2 mRNA expression. Similar to IKKα-deficient cells, acinar cells with reduced ATG16L2 protein expression exhibited a modest LC3 and p62 accumulation (Figure 5D). Tunicamycin-treated Atg16l2-knockdown acinar cells exhibited reduced viability relative to control acinar cells (Figure 5E). Autophagic flux assay indicated reduced tunicamycin-induced autophagic proteolysis in Atg16l2 siRNA transfected acinar cells (Figure 5F). We also found that LC3-II turnover was slower in Atg16l2-knockdown acinar cells (Supplemental Figure 13A) and that rapamycin treatment did not cause much further LC3-II accumulation in these cells (Supplemental Figure 13B). These results suggest that ATG16L2 might function as an adaptor protein that is involved in autophagy completion.
Figure 5. IKKα interacts with ATG16L2.

(A) IKKα interacts with ATG16L2 in mammalian cells. HEK293 cells were transfected with HA-IKKα and Flag-ATG16L2 constructs as indicated. After 36 hours, the cells were lysed and immunoprecipitated with Flag antibody. The disrupted immunoprecipitates and original lysates were subjected to IB analysis with the indicated antibodies. (B) Endogenous IKKα interacts with ATG16L2 in pancreatic acinar cells. Lysates of IkkαF/F and IkkαΔpan acinar cells were immunoprecipitated with the indicated antibodies. The disrupted immunocomplexes and original lysates were IB with the indicated antibodies. (C) Mouse acinar cells were treated with 0.5 μM thapsigargin (Tg) or 10 μg/ml tunicamycin (Tm) for 4 hours. Cells were then collected and lysed, and the interaction between IKKα and ATG16L2 was analyzed as above. (D) Freshly isolated WT mouse pancreatic acinar cells were transfected with control or Atg16l2 siRNA. After 60 hours, cell lysates were collected and expression of the indicated proteins was analyzed by IB. (E) ATG16L2 knockdown increases susceptibility to ER stress–induced cell death. Freshly isolated mouse acinar cells were transfected with control or Atg16l2 siRNA. After 2 days, cells were incubated with or without tunicamycin (10 μg/ml) for 24 hours. An MTT assay was performed to analyze cell viability. (F) ER stress–induced (10 μg/ml tunicamycin for 4 hours) autophagic flux in control and Atg16l2 siRNA–transfected acinar cells in the absence or presence of the autophagy inhibitor 3MA (10 mM). Results are shown as mean ± SEM. *P < 0.05; ***P < 0.001.
p62 ablation attenuates pancreatitis in IkkαΔpan mice and reduces oxidative and ER stress.
Accumulation of p62 leads to formation of ubiquitinated protein aggregates, a common hallmark of degenerative and metabolic diseases (19, 20, 22). To determine whether p62 depletion can attenuate tissue injury and pancreatitis in Ikkαpan mice, we silenced p62 in IkkαΔpan mouse pancreatic acinar cells using a lentivirus containing p62 shRNA. Knockdown of p62 reduced expression of ER and oxidative stress markers (Figure 6A). We next generated p62Δpan and Ikkαpan;p62Δpan mice by crossing “floxed” p62F/F mice with Pdx1-Cre (Supplemental Figure 14) and IkkαΔpan mice. Pancreatic tissue damage, fibrosis, and inflammation were substantially reduced in IkkαΔpan;p62Δpan double mutants compared with age- and sex-matched IkkαΔpan mice, and the pancreata of p62Δpan mice appeared completely normal (Figure 6B). Serum amylase and lipase were also significantly reduced in IkkαΔpan;p62Δpan mice compared with IkkaΔpan single mutants (Figure 6C), and this was consistent with decreased acinar cell vacuolization in IkkαΔpan;p62Δpan mice (Figure 6B). Importantly, p62 ablation did not rescue the autophagy defect, as indicated by remaining LC3 accumulation in IkkαΔpan;p62Δpan pancreata (Figure 6D). p62 ablation, however, did reduce the susceptibility of IkkαΔpan acinar cells to ER stress–induced death (Figure 6E) and decreased the expression of ER stress markers (Figure 6F).
Figure 6. p62 ablation in IkkαΔpan mice attenuates pancreatitis and reduces oxidative and ER stress.

(A) Knockdown of p62 in IkkαΔpan pancreatic acinar cells reduces expression of ER stress and oxidative stress markers. Mouse acinar cells were transduced with control or p62 shRNA lentiviruses. After 60 hours, RNA was collected and expression of ER and oxidative stress markers was analyzed. (B) p62 ablation attenuates pancreatic damage in IkkαΔpan mice. H&E staining of pancreatic sections from 3-month-old mice of the indicated genotypes. Original magnification, ×400. Scale bars: 50 μm.(C) Amylase and lipase activities in sera of 3-month-old mice of the indicated genotypes. (D) IB analysis of autophagy markers and IKKα in pancreatic lysates of 3-month-old mice of indicated genotypes. (E) p62 ablation protects IKKα-deficient acinar cells from tunicamycin-induced death. Acinar cells were incubated with the indicated concentrations of tunicamycin and their viability was determined 24 hours later. Note that the IkkαΔpan data are the same as those shown in Figure 4F and all of the different cells were examined in parallel. (F) qRT-PCR analysis of ER stress markers in pancreatic RNA of 3-month-old mice of the indicated genotypes. Results are shown as mean ± SEM. n = 3–5 mice per condition. *P < 0.05; **P < 0.01; ***P < 0.001.
Downregulation of IKKα and p62 accumulation in human chronic pancreatitis.
We performed IHC staining of IKKα and p62 on human pancreatic tissue specimens (Figure 7). Interestingly, 20 out of 35 cases of human chronic pancreatitis showed accumulation of p62-containing aggregates, especially in areas of acinar cell vacuolization (Figure 7 and Supplemental Figure 15). Eight out of the 20 specimens with elevated p62 exhibited downregulation of IKKα in pancreatitis areas (Figure 7 and Table 1). Importantly, every chronic pancreatitis specimen with reduced IKKα expression exhibited p62 accumulation.
Figure 7. Reduced IKKα expression and accumulation of p62 aggregates in human chronic pancreatitis.
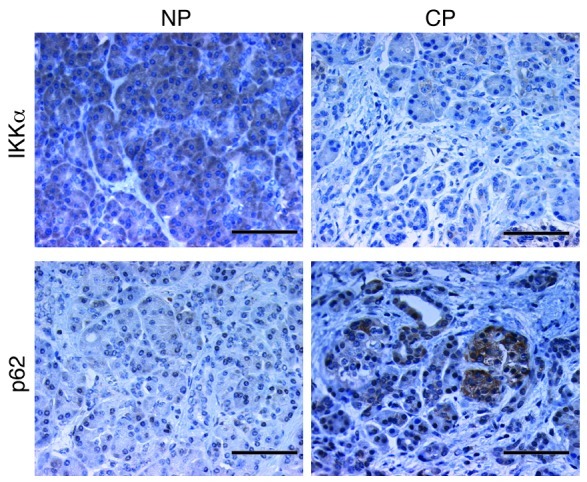
IHC staining for IKKα and p62 of human paraffin-embedded pancreas sections. The nonpancreatitis (NP) and chronic pancreatitis (CP) specimens are derived from different regions of the same pancreas. Original magnification, ×400. Scale bars: 50 μm.
Table 1.
Summary of IHC analysis of IKKα and p62 expression in human pancreas samples

Discussion
Pancreatitis is initiated by damage to the pancreatic acinar cell, and chronic pancreatitis is characterized by acinar cell injury and death, fibrosis, inflammation, fat replacement, ductal metaplasia, and dysregulated release of pancreatic enzymes (1). The same pathological features are exhibited by IkkαΔpan mice. Premature, intra-acinar trypsinogen activation was considered to be the major pathogenic event that leads to acute pancreatitis, but now it is known that the pathogenesis of chronic pancreatitis is more complex, and trypsinogen activation per se is just one of several factors that contribute to this poorly understood disease. Our results place reduced IKKα expression and accumulation of p62 aggregates among these factors. Acinar cell IKKα deficiency interferes with proper autophagic protein degradation and results in accumulation of p62 aggregates and subsequent upregulation of ER and oxidative stress markers. p62 is a key pathogenic mediator in this model of chronic pancreatitis, as its ablation in PEC in IkkαΔpan mice ameliorated tissue damage, pancreatitis, and fibrosis and reduced expression of ER and oxidative stress markers. Notably, p62 accumulation and, to a lesser extent, reduced IKKα expression were observed in human chronic pancreatitis, reinforcing the clinical relevance of the IkkαΔpan model described above.
The ER is the site for synthesis and maturation of cell surface and secretory proteins and is important for maintenance of Ca2+ homeostasis (7, 40). ER malfunction leads to accumulation of unfolded proteins and activation of the protective UPR, whose goal is to improve protein folding, reduce protein synthesis, and eliminate misfolded protein aggregates (40, 41). Possessing extremely high rates of protein secretion, pancreatic acinar cells are highly susceptible to ER perturbations, and ER stress occurs in pancreatitis (11, 42, 43). Reduced expression of Grp78/Bip, a critical ER chaperone and UPR component, blocks autophagosome formation and causes massive ER expansion and disorganization in acinar cells of cerulein-treated mice (11). Ethanol feeding of Xbp1+/– mice results in disorganized and dilated ER, loss of zymogen granules, accumulation of autophagic vacuoles, elevated CHOP expression, and acinar cell death (9). Similar pathological features occur spontaneously in IkkαΔpan mice, which exhibit activation of the 3 major UPR pathways, indicated by upregulation of Bip, Chop, and sXBP1, and dilated ER in pancreatic acinar cells. IkkαΔpan acinar cells are also more susceptible to ER stress–induced death, and, whereas acute UPR activation is an adaptive and protective response, sustained UPR upregulation is associated with cell death due to persistent expression of CHOP, a proapoptotic transcription factor (44). In the absence of CHOP-mediated death signaling, the UPR may continue to improve ER function and reduce accumulation of ROS and protein aggregates (9). Chop ablation improves ER function and protects against oxidative stress in β cells (45). Thus, it will be interesting to determine whether PEC-specific Chop ablation in IkkαΔpan mice will lead to amelioration of their exocrine and endocrine defects.
The key question, however, is how IKKα deficiency leads to ER stress and elevated CHOP expression. Our results suggest that defective autophagic protein degradation and the subsequent formation of p62-mediated protein aggregates are responsible for ER and oxidative stress in IkkαΔpan acinar cells. Ablation of the p62 gene ameliorated most of the clinical signs associated with pancreatitis in IkkαΔpan mice, with the exception of acinar vacuolization, which was reduced but not fully abolished. This is likely due to the fact that p62 ablation does not restore normal autophagic flux in IKKα-deficient cells, as indicated by persistent LC3 accumulation in IkkαΔpan;p62Δpan pancreata. Importantly, however, this genetic analysis clearly established that ER stress in the IKKα-deficient pancreas is fully dependent on p62 and not the other way around. Although p62 is a cytoplasmic protein, its defective autophagic clearance may lead to accumulation of misfolded proteins in the ER, thereby triggering ER stress. Notably, cerulein treatment, which interferes with the completion of autophagy in acinar cells and results in induction of pancreatitis, whose clinical and morphological signs are very similar to those observed in IkkαΔpan mice (3), also results in the buildup of p62 (A.S. Gukovskaya and I. Gukovsky, unpublished results). Although our results suggest that the defective completion of autophagy per se is not the most proximal cause of acinar cell death and pancreatitis in IkkαΔpan mice, they do support the importance of autophagy as a protective and antidegenerative mechanism that prevents the accumulation of p62-induced protein aggregates and ER stress. Notably, whole-body p62 ablation in liver-specific Atg7-deficient mice also prevented liver injury and development of hepatic adenomas without restoring autophagy (22, 23). In the pancreas, autophagy performs the standard functions of degrading damaged mitochondria and protein aggregates, and impairment of its completion using cerulein and other manipulations leads to acinar cell vacuolization and trypsinogen activation (3, 46, 47). However, other factors may be of greater importance in the development of pancreatitis than trypsinogen activation (4, 48). For instance, cathepsin L–deficient mice exhibit high levels of trypsinogen activation but reduced pancreatic injury (49), suggesting that the 2 parameters are not always linked. The recent generation of trypsin-deficient T7–/– mice, which still develop pancreatitis in response to cerulein administration (4), underscores the importance of trypsin-independent mechanisms, such as ER stress and protein aggregate formation, which may cause acinar cell death through CHOP induction. The 2 main routes for clearance of damaged proteins and inclusion bodies are the ubiquitin-proteasome system (UPS) and the autophagy-lysosome pathway (50). Accumulation of p62 may inhibit UPS-mediated protein degradation by interfering with delivery of ubiquitinated proteins to the proteasome (51). In addition, p62 accumulation can downregulate autophagy through activation of the TORC1 protein kinase complex (52). Thus, p62 accumulation, which we observed in approximately 50% of human chronic pancreatitis specimens, may be a cardinal event in the pathogenesis of this disease. Future research should be focused on pharmacological means to inhibit p62 accumulation or prevent formation of p62-mediated ubiquitinated protein aggregates.
It remains to be fully determined how reduced IKKα expression in PEC, observed in 25% of human chronic pancreatitis specimens, results in aberrant autophagic protein degradation and p62 accumulation. The IKK complex has been shown to contribute to starvation-induced autophagy, possibly by controlling autophagic gene expression via an NF-κB–independent mechanism (53, 54). However, in mice, pancreatic IKKα deficiency does not affect expression of essential autophagy proteins. Instead, IKKα interacts with the autophagy-related protein ATG16L2. Although ATG16L2 (55), unlike its close relative ATG16L1, does not seem to be an essential autophagy protein, in the pancreas, it is expressed in considerably higher amounts than ATG16L1. Thus, it is possible that in PEC, ATG16L2 may be required for autophagic maturation or protein degradation. Our preliminary results demonstrate that reduced ATG16L2 expression results in compromised acinar cell viability and upregulation of CHOP. Curiously, the ATG16-interacting protein Rab33B participates in autophagosome maturation (56), a function similar to that of IKKα. Further research is needed to determine whether IKKα, ATG16L2, and Rab33B form a functional network that controls autophagosome maturation and p62 turnover in acinar cells. Late-stage autophagy defects were previously proposed to be involved in acute alcoholic and nonalcoholic pancreatitis (3, 57). Such defects interfere with the autophagic clearance of abnormal organelles, and our results suggest that accumulation of dysfunctional mitochondria that leak electrons and lead to ROS formation can provide another impetus to CHOP induction and acinar cell death. Oxidative stress may further accentuate protein misfolding and thereby promote protein aggregation (58). Despite the remaining questions, the availability of the new IkkαΔpan model for spontaneous pancreatitis, whose features closely resemble those of human chronic pancreatitis, should facilitate our understanding of this rather common and potentially lethal disease and provide new avenues for its treatment or prevention.
Methods
Mice.
All mice were maintained in filter-topped cages on autoclaved food and water, and all experiments were performed in accordance to UCSD and NIH guidelines and regulations. Cre-negative mice were used as WT controls, and all experiments were conducted on age and sex-matched littermates. IkkαF/F, IkkβF/F, Pdx1Cre, and p62F/F mice have been described (59–62).
Reagents.
IB, IHC, and IF were conducted with antibodies to the following: IKKα (Imgenex), LC3B (Cell Signaling), p62 (Santa Cruz Biotechnology Inc. and Promgen), ERK (Cell Signaling), HA (Roche), Flag (Sigma-Aldrich), ATG16L2 (Abgent), Ub (Santa Cruz Biotechnology Inc.), LAMP1 and LAMP2 (Santa Cruz Biotechnology Inc.), Ki67 (Novocastra Laboratories), B220 (BD Biosciences — Pharmingen), CD3 (Abcam), F4/80 (AbD Serotec), PE-Gr1 (BD Biosciences — Pharmingen),insulin (Zymed), glucagon (Abcam), Sp1 (Santa Cruz Biotechnology Inc.), NF-κB/RelA (Santa Cruz Biotechnology Inc.), tubulin (Sigma-Aldrich), phospho-elF2α (Upstate), elF2α (Santa Cruz Biotechnology Inc.), phospho-ERK (Cell Signaling), JNK1/2 (Pharmingen), phospho-JNK (Cell Signaling), cathepsin B and cathepsin D (Santa Cruz Biotechnology Inc.), phospho-PERK (Cell Signaling), phospho-IKKα/β (Cell Signaling).
Histology and immunohistochemistry.
Pancreata were dissected and fixed in 4% paraformaldehyde in PBS and embedded in paraffin, and 5-μm sections were prepared and stained with H&E or Sirius red. Necrosis was quantified using H&E staining. Cells with swollen cytoplasm, loss of plasma membrane, and leakage of organelles into the interstitium were considered necrotic. IHC was performed as follows: after xylene deparaffinization and ethanol dehydration, sections were incubated with primary antibodies diluted in PBS with 5% goat serum for 1 hour at room temperature, followed by incubation with 1:200 biotinylated secondary antibodies conjugated with HRP for 30 minutes and 1:500 streptavidin-HRP for 30 minutes. Bound peroxidase was visualized by 1 to 10 minutes of incubation in a 3,3′-diaminobenzidine (DAB) solution. Sections were washed, counterstained with eosin, dehydrated, and mounted. In situ TUNEL staining was performed using the DeadEnd Colorimetric TUNEL System Kit (Promega). To measure in vivo proliferation, mice were i.p. injected with 100–200 μl 10 mg/ml BrdU solution in sterile PBS. After 4 hours, BrdU incorporation was detected with the BrdU In-Situ Detection Kit (BD Biosciences — Pharmingen).
IB and immunoprecipitation.
IB analysis was performed on tissue or cell lysates that were separated by SDS-PAGE and transferred to nitrocellulose membranes. Blots were incubated with 5% (w/v) nonfat dry milk in PBS with 0.05% (w/v) Tween 20 (PBST) at room temperature for 1 hour to block nonspecific binding, overnight at 4°C with primary antibodies in 3% BSA (w/v) in PBST, and finally with HRP-conjugated secondary antibody in blocking buffer. Blots were developed using an enhanced chemiluminescence detection kit (Plus-ECL Kit; PerkinElmer). Freshly isolated pancreata from age-matched WT and IkkaΔpan littermates were homogenized in lysis buffer (50 mM Tris HCl, pH 7.5, 150 mM NaCl, 3 mM EDTA, 3 mM EGTA, 1% Triton X-100, 0.1% SDS, 10% glycerol) supplemented with 1% proteinase inhibitor cocktail containing 1 mM pepstatin, 1.25 mg/ml leupeptin, 10 mg/ml aprotinin, and 10 mM bestatin. Protein lysates (1 mg) were incubated with either ATG16L2 or IKKα antibodies (1 μg each) overnight, and protein A/G beads were added for another 1 hour. The beads were then spun down and washed 3 times with lysis buffer. Bead-bound proteins were separated by SDS-PAGE and IB as above.
qRT-PCR.
Total tissue RNA was extracted using TRIzol (Invitrogen) or RNeasy Kit (QIAGEN). Total RNA was quantified by UV absorption and reverse transcribed into cDNA using SuperScript II reverse transcriptase and oligo-dT primers (Invitrogen). cDNA was further analyzed by real-time PCR on an iCycler iQ Real-Time PCR System (Bio-Rad). Data were normalized relative to cyclophilin or ribosomal protein L32 mRNAs.
Enzyme assays.
Serum amylase and lipase were measured by a Hitachi 707 analyzer (Antech). Trypsin activity was measured in pancreatic homogenates by a fluorimetric assay (3). Briefly, pancreata were homogenized on ice using a glass-Teflon homogenizer in a buffer containing 5 mM MES (pH 6.5), 1 mM MgSO4, and 250 mM sucrose. An aliquot of the homogenate was added to the assay buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM CaCl2, and 0.1 mg/ml BSA in a stirred cuvette at 37°C. The reaction was started by adding a specific substrate, Boc-Gln-Ala-Arg-MCA, which is converted to a fluorescent product that emits fluorescence at 440 nm upon excitation at 380 nm. The increase in fluorescence was linear during the 5-minute observation period. Trypsin activity in each sample was determined using a standard curve for purified trypsin (Worthington). Amylase secretion by isolated mouse pancreatic acinar cells was measured using Phadebas Amylase Kit (Magle AB) as described (3). Values for amylase secretion are the ratio between the amount of amylase released into the extracellular medium and total cellular amylase determined by permeabilizing cells with 1% Triton X-100. Cis-aconitase activity was measured using Aconitase Assay Kit (Abcam) according to the manufacturer’s instructions.
Isolation of pancreatic acinar cells, autophagic flux, and MTT assays.
Pancreata were subjected to collagenase digestion as described (3). Dispersed pancreatic acini were incubated at 37°C in F12/K medium (ATCC). Acinar cells were cultured for 6 hours on collagen IV–coated dishes in F12/K medium supplemented with [3H]-leucine and washed by centrifugation to remove unincorporated radioactivity and released amino acids. The cells were then replated and chased for 16 hours in fresh F12/K medium containing 2 mM cold leucine, during which time all short-lived proteins were degraded. After that, cells were washed 3 times by centrifugation in F12/K medium containing 2 mM cold leucine and further incubated for 4 hours with or without tunicamycin or other additives as indicated. At the end of the incubation, proteins were precipitated in ice-cold 10% trichloroacetic acid (TCA) (Sigma-Aldrich). Soluble radioactivity, which represents degradation products, was separated from intact proteins by centrifugation at 12,000 g for 10 minutes. The rate of degradation of long-lived proteins was calculated from the ratio of the acid-soluble radioactivity in the medium to that in the acid-precipitable cell fraction.
To determine cell viability, MTT solution (5 mg/ml in PBS) was added to each well of a 24-well plate containing cultured acinar cells that had been treated with or without tunicamycin or thapsigargin for 24 hours as indicated and incubated at 37°C for 2 hours to allow MTT to metabolize. The cells were spun down and resuspended in DMSO. The optical density of formazan (MTT metabolite) was read at 560 nm, and background at 670 nm was substracted.
Transfection and lentiviral infection.
For silencing of Atg16l2, islolated mouse pancreatic acinar cells were transfected with Atg16l2 siRNA (MSS232103; Invitrogen) and control siRNA (12935-113; Invitrogen). Atg16l2 ON-TARGET SMARTpool from Dharmacon and siCONTROL Non-Targeting Pool were also used. Transfections (100 pmoles of each siRNA) were performed using the Amaxa Nucleofactor Electroporation System. For silencing of p62, isolated mouse pancreatic acinar cells were infected with lentiviruses encoding p62 shRNA (generated in our laboratory) for 2 days with polybrene, and knock down efficiency was examined by RT-PCR or IB analysis. We used the following p62 (m) siRNA sequence: 5′-CCGCATCTACATTAAAGAGAA-3′.
Statistics.
Data are expressed as mean ± SEM. A 2-tailed Student’s t test was used for comparing 2 groups, and results were considered significant at P < 0.05.
Study approval.
This study was performed according to protocols approved by the Institutional Animal Care and Use Committee of UCSD. Pancreas tissue sections from the UCLA, UCSD, and Technical University of Munich pancreas tissue banks were provided by D. Dawson, S. VandenBerg, and M.C. Arkan, respectively. The study was approved by the respective Institutional Review Boards.
For further information, see Supplemental Methods.
Supplementary Material
Acknowledgments
This work was supported by NIH grants P50 AA011999 (Pilot Project to M. Karin), AI043477 (to M. Karin), CA163798 (to M. Karin), and CA167120 (to N. Li) and by Pancreatic Cancer Research Lustgarten Foundation grant RFP-B-007 (to M. Karin). X. Wu was supported by a postdoctoral fellowship from Susan G. Komen for the Cure (KG111506). J. Todoric was supported by the Austrian Science Fund’s Erwin Schroedinger Fellowship (Project# J 3233). We thank S.L. Gonias, D. Brenner, M. Erkan, J. Kleeff, and F.R. Greten for help in procuring human pancreatitis samples and for helpful advice; J. Xiao for technical assistance; and M.G. Farquhar and M. Niwa for helpful discussions regarding lysosomes and ER stress, respectively. We also thank Santa Cruz Biotechnology Inc. and Cell Signaling for antibody samples.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(5):2231–2243. doi:10.1172/JCI64498.
References
- 1.Chen JM, Férec C. Chronic pancreatitis: genetics and pathogenesis. Annu Rev Genomics Hum Genet. 2009;10:63–87. doi: 10.1146/annurev-genom-082908-150009. [DOI] [PubMed] [Google Scholar]
- 2.Zavoral M, Minarikova P, Zavada F, Salek C, Minarik M. Molecular biology of pancreatic cancer. World J Gastroenterol. 2011;17(24):2897–2908. doi: 10.3748/wjg.v17.i24.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mareninova OA, et al. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest. 2009;119(11):3340–3355. doi: 10.1172/JCI38674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dawra R, et al. Intra-acinar trypsinogen activation mediates early stages of pancreatic injury but not inflammation in mice with acute pancreatitis. Gastroenterology. 2011;141(6):2210–2217.e2. doi: 10.1053/j.gastro.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaiser S, et al. Intracellular activation of trypsinogen in transgenic mice induces acute but not chronic pancreatitis. Gut. 2011;60(10):1379–1388. doi: 10.1136/gut.2010.226175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kereszturi E, et al. Hereditary pancreatitis caused by mutation-induced misfolding of human cationic trypsinogen: a novel disease mechanism. Hum Mutat. 2009;30(4):575–582. doi: 10.1002/humu.20853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kubisch CH, Logsdon CD. Endoplasmic reticulum stress and the pancreatic acinar cell. Expert Rev Gastroenterol Hepatol. 2008;2(1):249–260. doi: 10.1586/17474124.2.2.249. [DOI] [PubMed] [Google Scholar]
- 8.Guyton KZ, Xu Q, Holbrook NJ. Induction of the mammalian stress response gene GADD153 by oxidative stress: role of AP-1 element. Biochem J. 1996;314(pt 2):547–554. doi: 10.1042/bj3140547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lugea A, et al. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology. 2011;140(3):987–997. doi: 10.1053/j.gastro.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hess DA, et al. Extensive pancreas regeneration following acinar-specific disruption of Xbp1 in mice. Gastroenterology. 2011;141(4):1463–1472. doi: 10.1053/j.gastro.2011.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ye R, et al. Grp78 heterozygosity regulates chaperone balance in exocrine pancreas with differential response to cerulein-induced acute pancreatitis. Am J Pathol. 2010;177(6):2827–2836. doi: 10.2353/ajpath.2010.100368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mathew R, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 15.Hashimoto D, et al. Involvement of autophagy in trypsinogen activation within the pancreatic acinar cells. J Cell Biol. 2008;181(7):1065–1072. doi: 10.1083/jcb.200712156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137(6):1001–1004. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moscat J, Diaz-Meco MT, Wooten MW. Signal integration and diversification through the p62 scaffold protein. Trends Biochem Sci. 2007;32(2):95–100. doi: 10.1016/j.tibs.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 18.Komatsu M, Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 2010;584(7):1374–1378. doi: 10.1016/j.febslet.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 19.Ichimura Y, Komatsu M. Selective degradation of p62 by autophagy. Semin Immunopathol. 2010;32(4):431–436. doi: 10.1007/s00281-010-0220-1. [DOI] [PubMed] [Google Scholar]
- 20.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7(3):279–296. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stumptner C, Fuchsbichler A, Heid H, Zatloukal K, Denk H. Mallory body--a disease-associated type of sequestosome. Hepatology. 2002;35(5):1053–1062. doi: 10.1053/jhep.2002.32674. [DOI] [PubMed] [Google Scholar]
- 22.Komatsu M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 23.Inami Y, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193(2):275–284. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rothwarf DM, Karin M. The NF-kappa B activation pathway: a paradigm in information transfer from membrane to nucleus. Sci STKE. 1999;1999(5):RE1. doi: 10.1126/stke.1999.5.re1. [DOI] [PubMed] [Google Scholar]
- 25.Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006(357):re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 26.Hu Y, et al. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science. 1999;284(5412):316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- 27.Hu Y, Baud V, Oga T, Kim KI, Yoshida K, Karin M. IKKalpha controls formation of the epidermis independently of NF-kappaB. Nature. 2001;410(6829):710–714. doi: 10.1038/35070605. [DOI] [PubMed] [Google Scholar]
- 28.Sil AK, Maeda S, Sano Y, Roop DR, Karin M. IkappaB kinase-alpha acts in the epidermis to control skeletal and craniofacial morphogenesis. Nature. 2004;428(6983):660–664. doi: 10.1038/nature02421. [DOI] [PubMed] [Google Scholar]
- 29.Takeda K, et al. Limb and skin abnormalities in mice lacking IKKalpha. Science. 1999;284(5412):313–316. doi: 10.1126/science.284.5412.313. [DOI] [PubMed] [Google Scholar]
- 30.Lahtela J, et al. Mutant CHUK and severe fetal encasement malformation. N Engl J Med. 2010;363(17):1631–1637. doi: 10.1056/NEJMoa0911698. [DOI] [PubMed] [Google Scholar]
- 31.Baumann B, et al. Constitutive IKK2 activation in acinar cells is sufficient to induce pancreatitis in vivo. J Clin Invest. 2007;117(6):1502–1513. doi: 10.1172/JCI30876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Algul H, et al. Pancreas-specific RelA/p65 truncation increases susceptibility of acini to inflammation-associated cell death following cerulein pancreatitis. J Clin Invest. 2007;117(6):1490–1501. doi: 10.1172/JCI29882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol. 2011;12(8):715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- 34.Hingorani SR, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4(6):437–450. doi: 10.1016/S1535-6108(03)00309-X. [DOI] [PubMed] [Google Scholar]
- 35.Chen N, Unnikrishnan IR, Anjana RM, Mohan V, Pitchumoni CS. The complex exocrine-endocrine relationship and secondary diabetes in exocrine pancreatic disorders. J Clin Gastroenterol. 2011;45(10):850–861. doi: 10.1097/MCG.0b013e31822a2ae5. [DOI] [PubMed] [Google Scholar]
- 36.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. 2005;434(7037):1138–1143. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- 37.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cadwell K, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456(7219):259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saitoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456(7219):264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 40.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 41.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 42.Pandol SJ, Gorelick FS, Gerloff A, Lugea A. Alcohol abuse, endoplasmic reticulum stress and pancreatitis. Dig Dis. 2010;28(6):776–782. doi: 10.1159/000327212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szmola R, Sahin-Toth M. Pancreatitis-associated chymotrypsinogen C (CTRC) mutant elicits endoplasmic reticulum stress in pancreatic acinar cells. Gut. 2010;59(3):365–372. doi: 10.1136/gut.2009.198903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13(3):184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest. 2008;118(10):3378–3389. doi: 10.1172/JCI34587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Czaja MJ. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology. 2011;140(7):1895–1908. doi: 10.1053/j.gastro.2011.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohmuraya M, Yamamura K. Autophagy and acute pancreatitis: a novel autophagy theory for trypsinogen activation. Autophagy. 2008;4(8):1060–1062. doi: 10.4161/auto.6825. [DOI] [PubMed] [Google Scholar]
- 48.Ji B, Logsdon CD. Digesting new information about the role of trypsin in pancreatitis. Gastroenterology. 2011;141(6):1972–1975. doi: 10.1053/j.gastro.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wartmann T, et al. Cathepsin L inactivates human trypsinogen, whereas cathepsin L-deletion reduces the severity of pancreatitis in mice. Gastroenterology. 2010;138(2):726–737. doi: 10.1053/j.gastro.2009.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010;584(7):1393–1398. doi: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 51.Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33(4):517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duran A, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44(1):134–146. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Criollo A, et al. The IKK complex contributes to the induction of autophagy. EMBO J. 2010;29(3):619–631. doi: 10.1038/emboj.2009.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Comb WC, Cogswell P, Sitcheran R, Baldwin AS. IKK-dependent, NF-kappaB-independent control of autophagic gene expression. Oncogene. 2011;30(14):1727–1732. doi: 10.1038/onc.2010.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mizushima N, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116(pt 9):1679–1688. doi: 10.1242/jcs.00381. [DOI] [PubMed] [Google Scholar]
- 56.Itoh T, Kanno E, Uemura T, Waguri S, Fukuda M. OATL1, a novel autophagosome-resident Rab33B-GAP, regulates autophagosomal maturation. J Cell Biol. 2011;192(5):839–853. doi: 10.1083/jcb.201008107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fortunato F, et al. Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology. 2009;137(1):350–360, 360.e1–360.e5. doi: 10.1053/j.gastro.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 58.Gregersen N, Bross P. Protein misfolding and cellular stress: an overview. Meth Mol Biol. 2010;648:3–23. doi: 10.1007/978-1-60761-756-3_1. [DOI] [PubMed] [Google Scholar]
- 59.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121(7):977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 60.Liu B, et al. IKKalpha is required to maintain skin homeostasis and prevent skin cancer. Cancer Cell. 2008;14(3):212–225. doi: 10.1016/j.ccr.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dessimoz J, Bonnard C, Huelsken J, Grapin-Botton A. Pancreas-specific deletion of beta-catenin reveals Wnt-dependent and Wnt-independent functions during development. Curr Biol. 2005;15(18):1677–1683. doi: 10.1016/j.cub.2005.08.037. [DOI] [PubMed] [Google Scholar]
- 62.Muller TD, et al. p62 Links beta-adrenergic input to mitochondrial function and thermogenesis. J Clin Invest. 2013;123(1):469–478. doi: 10.1172/JCI64209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.