

Abstract
Platinum compounds display clinical activity against a wide variety of solid tumors; however, resistance to these agents is a major limitation in cancer therapy. Reduced platinum uptake and increased platinum export are examples of resistance mechanisms that limit the extent of DNA damage. Here, we report the discovery and characterization of the role of ATP11B, a P-type ATPase membrane protein, in cisplatin resistance. We found that ATP11B expression was correlated with higher tumor grade in human ovarian cancer samples and with cisplatin resistance in human ovarian cancer cell lines. ATP11B gene silencing restored the sensitivity of ovarian cancer cell lines to cisplatin in vitro. Combined therapy of cisplatin and ATP11B-targeted siRNA significantly decreased cancer growth in mice bearing ovarian tumors derived from cisplatin-sensitive and -resistant cells. In vitro mechanistic studies on cellular platinum content and cisplatin efflux kinetics indicated that ATP11B enhances the export of cisplatin from cells. The colocalization of ATP11B with fluorescent cisplatin and with vesicular trafficking proteins, such as syntaxin-6 (STX6) and vesicular-associated membrane protein 4 (VAMP4), strongly suggests that ATP11B contributes to secretory vesicular transport of cisplatin from Golgi to plasma membrane. In conclusion, inhibition of ATP11B expression could serve as a therapeutic strategy to overcome cisplatin resistance.
Introduction
Platinum-based drugs such as cisplatin, carboplatin, and oxaliplatin have a broad range of activity in malignant diseases and are used to treat many types of cancer (1). Since the introduction of cisplatin into clinical trials almost 40 years ago, this drug has had a major impact on the therapeutic management of several tumors, such as those of the ovary, testes, and head and neck (2). Its cytotoxic mode of action is mediated by its interaction with DNA to form adducts, mostly intrastrand crosslink adducts, which activate several signal transduction pathways, including those involving ATR, p53, p73, and MAPK, culminating in the activation of apoptosis (3). Unfortunately, emergence of primary or acquired resistance to platinum agents eventually limits their efficacy (1).
The molecular mechanisms of resistance to cisplatin can be broadly divided into 2 main groups: (a) mechanisms that reduce cisplatin accumulation and, thereby, attenuate DNA damage and (b) mechanisms that inhibit propagation of cisplatin-mediated DNA damage signaling to the apoptotic machinery (3). Earlier studies have documented the development of resistance as a result of increased cisplatin efflux (4, 5). Recently, interest in this resistance mechanism has reemerged as new exporter proteins have been identified. For example, the multidrug resistance-associated gene family includes at least 7 (MRP1–MRP7) ABC-membrane proteins that are involved in the cellular efflux of a variety of drugs (6). However, only MRP2 (also known as cMOAT) seems to be important in cisplatin efflux (7). A second important group of proteins associated with cisplatin resistance comprises ATP7A and ATP7B, 2 copper-membrane transporting P-type ATPases that are overexpressed in cisplatin-resistant tumor cells (8, 9). These copper transporters have been shown to be involved in intracellular cisplatin trafficking and export (10, 11).
Here, we undertook a search for candidate genes that could confer cisplatin resistance in cancer cells. Using genomic analyses, we found that ATP11B gene expression was substantially increased in cisplatin-resistant cells. Moreover, ATP11B enhanced cisplatin efflux and ATP11B silencing restored sensitivity of ovarian cancer cells to cisplatin. These findings identify ATP11B as a potential target for overcoming cisplatin resistance.
Results
Gene expression analyses in cisplatin-sensitive and cisplatin-resistant cells.
To identify new potential gene candidates that could affect platinum resistance, we performed genomic analyses in the progressively resistant ovarian cancer cell lines A2780-CP20, CP40, CP55, and CP70, compared with the cisplatin-sensitive A2780-PAR cells. Among the various classes of genes with increased expression, many of the ATPase membrane transporters had increased expression in the cisplatin-resistant cells (Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI65425DS1). ATP11B, a P-type class VI ATPase transport protein, was highly upregulated in all cell lines tested. A2780-CP20 cells had the highest ATP11B expression (Supplemental Figure 1B). Next, we examined ATP11B expression in a panel of additional ovarian cancer cell lines. We found that ATP11B gene expression was significantly elevated in the cisplatin-resistant cells (A2780-CP20, IGROV1-CP20, RMG2, and ES2) compared with that in the highly sensitive A2780-PAR cells (Supplemental Figure 1B). Protein expression was also confirmed to be increased in A2780-CP20 cells compared with that in A2780-PAR cells (Supplemental Figure 1D). In addition, tumor samples from patients with ovarian cancer (cisplatin responsive and nonresponsive) were analyzed to determine ATP11B mRNA and protein expression. Supplemental Figure 1E shows significant correlation between ATP11B mRNA and protein expression.
Clinical significance of ATP11B expression in human ovarian carcinoma.
To determine whether ATP11B expression was related to patient outcome, we examined 111 ovarian cancer specimens and 10 normal ovarian surface epithelial cell specimens by using quantitative RT-PCR for ATP11B mRNA expression. Among these patients, 92.8% had high-stage disease and 75.7% had undergone optimal surgical cytoreduction. ATP11B expression was significantly related to higher tumor stage (P = 0.04; Table 1). High ATP11B expression was associated with worse overall survival (mean 2.75 vs. 9.25 years for high vs. low ATP11B expression, respectively; P < 0.001) and progression-free survival (mean 1.2 vs. 2.8 years for high vs. low ATP11B expression, respectively; P < 0.001; Figure 1). To examine for concordance between protein and mRNA expression, we examined a subset of human ovarian cancer samples. There was a significant relationship between mRNA and protein expression (Supplemental Figure 1E). Next, we examined ATP11B expression in cancer cells from patients with ovarian cancer treated with platinum-based chemotherapy. The PE01 (chemotherapy-sensitive) and PE04 (chemotherapy-resistant) cells were tested for ATP11B expression. There was a 2.4-fold increase in ATP11B expression in the PE04 cells compared with that in the PE01 cells (Supplemental Figure 2).
Table 1.
Patient and tumor characteristics
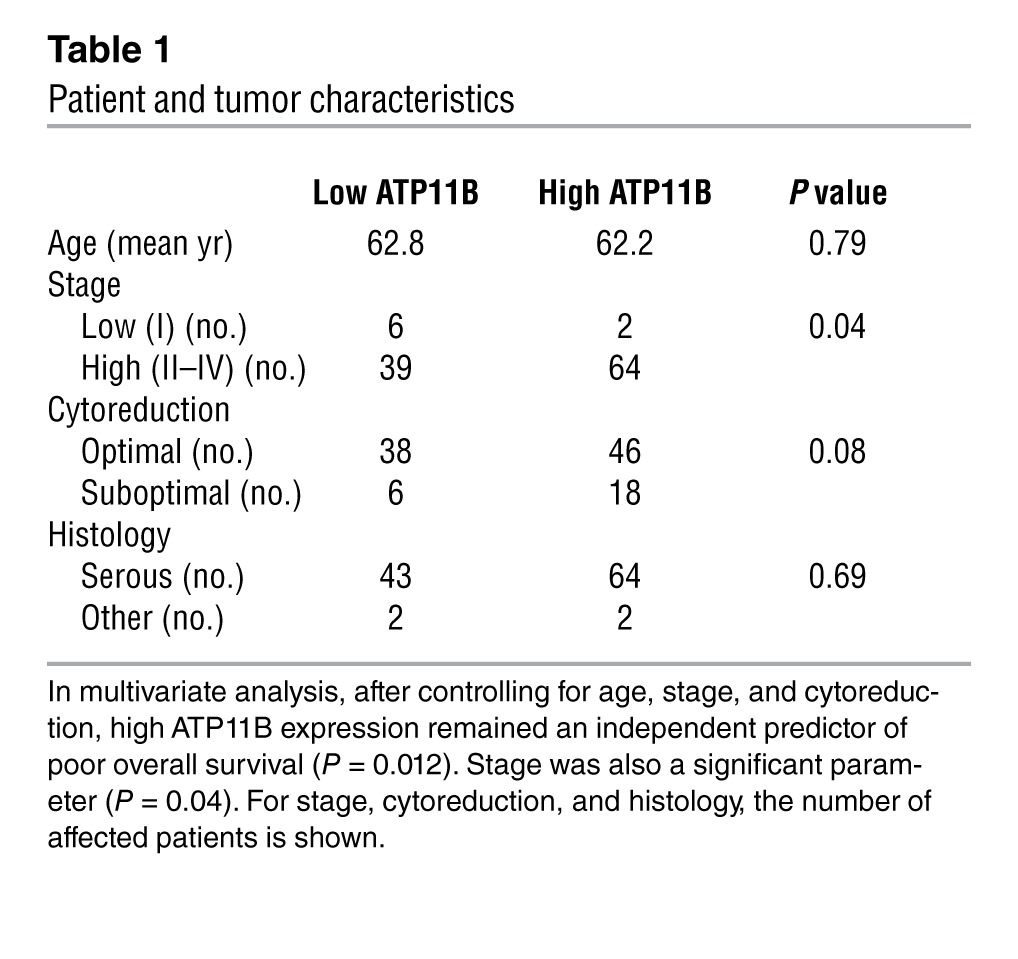
Figure 1. Clinical significance of ATP11B in human ovarian carcinoma.

Kaplan-Meier curves for patients with epithelial ovarian carcinoma based on ATP11B expression. (A) Overall and (B) progressive-free survival (PFS) curves showed that high ATP11B expression remained an independent predictor of poor overall survival (n = 111; P = 0.012).
Effect of increased ATP11B expression in A2780-PAR cells.
We next asked whether increased ATP11B expression in the A2780-PAR cells would result in resistance to cisplatin. To determine the sensitivity of ATP11B-transfected A2780-PAR cells to cisplatin, these cells were grown in the presence of cisplatin. Among the various clones, clone 6 had the highest survival rate, comparable to that for A2780-CP20 cells. In the presence of 5 μM cisplatin, A2780-ATP11B-6 cells yielded 12 times more colonies, compared with A2780-PAR cells and the other clones (Figure 2A). Half-maximal inhibitory concentration (IC50) was determined by means of the 2-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay using a wide range of cisplatin concentrations. IC50 for A2780–empty vector–transfected (A2780-EV–transfected) cells and A2780-ATP11B-6 cells was 0.52 and 2.0 μM, respectively (Supplemental Figure 3A). In contrast, the IC50 of A2780 cells transfected with C-terminal partial ATP11B sequence was very similar to the IC50 for A2780-PAR cells (Supplemental Figure 3B), suggesting that the full-length sequence is required for its function.
Figure 2. ATP11B affects cisplatin sensitivity of ovarian cancer cells.

(A) A2780-PAR cells transfected with ATP11B construct were plated onto 6-well culture plates and treated with 5 and 7.5 μM cisplatin for 24 hours. Cisplatin-resistant clones were selected by clonogenic assay. Colonies formed were stained with 0.25% Crystal Violet in 20% ethanol for 5 minutes and counted under the light microscope. Among the resistant clones, clone no. 6 showed the highest survival rate. In the presence of 5 μM cisplatin, clone no. 6 yielded 12 times more colonies, compared with A2780-PAR cells and the other clones. ATP11B downregulation increased the sensitivity to cisplatin of (B) A2780-PAR and (C) A2780-CP20 cells. ATP11B silencing decreased the IC50 7 fold and 9 fold in A2780-PAR and A2780-CP20 cells, respectively, compared with A2780-PAR cells transfected with control siRNA (ctr siRNA). Data represent the mean ± SEM. *P < 0.05, **P < 0.01, §P < 0.001, †P < 0.0001, ††P < 0.00001.
Downregulation of the ATP11B gene sensitizes ovarian cancer cells to cisplatin in vitro.
On the basis of our findings with clinical samples and the increased expression observed in cisplatin-resistant cell lines, we next examined the effect of ATP11B silencing in vitro. Compared with control siRNA–transfected cells, ATP11B mRNA and protein expression levels were decreased by >50% in the ATP11B siRNA–transfected cells (Supplemental Figure 3C). After ATP11B silencing, the IC50 was 0.07 μM, a 7-fold decrease of IC50 (P < 0.01) compared with control siRNA–treated cells (Figure 2B). The effect of ATP11B silencing was even stronger (P < 0.001; 9-fold increase in cisplatin sensitivity) in the cisplatin-resistant A2780-CP20 cells (Figure 2C). Control siRNA did not affect cisplatin sensitivity in either ovarian cancer cell line. In addition, ATP11B downregulation increased cisplatin sensitivity in the PE04 ovarian (chemoresistant) cancer cells (Supplemental Figure 4A). Next, we examined whether ATP11B gene silencing affects sensitivity to other chemotherapy drugs. In multiple ovarian cancer cell lines (ES2, A2780-PAR, and A2780-CP20), ATP11B silencing did not significantly affect sensitivity to paclitaxel or topotecan (Supplemental Figure 4B). Survival of these cell lines was not significantly affected after ATP11B silencing (Supplemental Figure 4C).
RNA expression of ATP11B and ATP7B is not coordinately regulated.
To test whether ATP7B, another member of the family of the P-type ATP-membrane transporters, could be involved in the ATP11B-mediated effect on cisplatin resistance, we examined ATP7B and ATP11B mRNA expression in ATP7B siRNA–silenced as well as in ATP11B-silenced cisplatin-resistant (A2780-CP20) ovarian cancer cells. There was no significant change in ATP7B expression levels following ATP11B gene silencing; similarly, no significant changes in ATP11B levels were noted following ATP7B gene silencing. These data have now been included in Supplemental Figure 5.
ATP11B gene silencing restores sensitivity to cisplatin in vivo.
On the basis of our in vitro findings, we next tested the ability of ATP11B siRNA, incorporated in dioleoylphosphatidylcholine (DOPC) nanoliposomes, to downregulate ATP11B expression in vivo. Female nude mice bearing A2780-CP20 tumors (approximately 3 weeks after i.p. injection) were treated with a single dose of ATP11B siRNA–DOPC (150 μg/kg i.p.). Tumors from mice treated with ATP11B siRNA demonstrated 90% reduction in ATP11B mRNA and protein expression levels 96 hours after the single ATP11B siRNA injection (Supplemental Figure 3D). Therefore, we used twice-weekly administration of siRNA-DOPC for subsequent therapy experiments. ATP11B expression was not affected by control siRNA–DOPC. Next, we examined whether ATP11B silencing could sensitize resistant tumors to cisplatin therapy. Nude mice were injected with A2780-CP20 (cisplatin-resistant) cells, and, a week later, siRNA therapy was started using the following groups (n = 10 mice per group): (a) empty liposomes, (b) control siRNA–DOPC, (c) ATP11B siRNA–DOPC, (d) control siRNA–DOPC plus cisplatin, and (e) ATP11B siRNA–DOPC plus cisplatin. All siRNA treatments were given at 150 μg/kg i.p. twice weekly, and cisplatin was given at 160 μg i.p. once per week. None of the single treatments had a significant effect on tumor growth (Figure 3A). In contrast, treatment with ATP11B siRNA–DOPC plus cisplatin resulted in a 73% reduction (P < 0.01) in tumor growth compared with that in the control siRNA–DOPC plus cisplatin group and in a 85% reduction (P < 0.01) compared with that in the empty liposome group (Figure 3A). ATP11B siRNA plus cisplatin treatment also resulted in the greatest efficacy in the SKOV3ip1, RMG2, and IGROV1-CP20 models (Figure 3A). We examined the biological effects of ATP11B-targeted therapy on tumor cell proliferation. Both the mice treated with ATP11B siRNA and those treated with ATP11B siRNA plus cisplatin showed significant decreases in cell proliferation (42%, P < 0.001, and 70%, P < 0.001, respectively) compared with their corresponding controls (Figure 3B). The apoptotic rates (percentages of TUNEL-positive cells) in tumor cells from mice treated with ATP11B siRNA–DOPC, control siRNA–DOPC plus cisplatin, or ATP11B siRNA plus cisplatin were considerably higher than rates for the empty liposome and control siRNA–DOPC groups. Mice treated with ATP11B siRNA and those treated with ATP11B siRNA plus cisplatin had significantly higher apoptotic rates compared with their corresponding controls (P < 0.05; Figure 3B). To test for consistency of effect, we also used a second independent ATP11B siRNA sequence (sequence no. 2) for an in vivo experiment with the A2780-CP20 cancer model. Treatment with ATP11B siRNA–DOPC (sequence no. 1) or ATP11B siRNA–DOPC (sequence no. 2) with cisplatin resulted in similar antitumor effects (P < 0.01; Supplemental Figure 6).
Figure 3. Therapeutic effectiveness of ATP11B siRNA plus cisplatin in ovarian cancer models.

(A) Tumor growth in the A2780-CP20, SKOV3ip1, RMG2, and IGROV-CP20 ovarian cancer models. Each bar represents mean tumor weight + SEM (n = 10). Significantly decreased tumor growth was observed only in mice treated with ATP11B siRNA plus cisplatin. *P < 0.05 (B) Silencing ATP11B reduced tumor proliferation and increased tumor cell apoptosis in ovarian cancer tumors. Tissue sections from A2780-CP20 tumors were immunostained for the detection of proliferating cell nuclear antigen (PCNA) and TUNEL-positive cells to assess tumor proliferation and apoptosis, respectively. Tumor tissues from mice treated with ATP11B siRNA and those treated with ATP11B siRNA plus cisplatin showed significantly lower (*P < 0.001) proliferation and significantly higher (**P < 0.05) apoptotic rates compared with their corresponding controls. Percentages of proliferating and apoptotic cells were calculated in 5 fields for each tissue sample (n = 5). Data represent the mean ± SEM. Original magnification, ×200.
ATP11B regulates platinum transport in ovarian cancer cells.
To explore mechanisms by which ATP11B could play a functional role in cisplatin resistance, we assessed cellular uptake of platinum and DNA adduct formation in A2780-EV and A2780-ATP11B-6 cells as well as in the A2780-CP20 (cisplatin-resistant) cells. Cellular platinum uptake by A2780-ATP11B-6 and A2780-CP20 cells was significantly reduced (28%; P < 0.01; and 80% P < 0.001; respectively), compared with that by A2780-EV cells (Figure 4A). The same cell lines exhibited decreased DNA adduct formation (43%, P < 0.01, and, 82%, P < 0.01, respectively; Figure 4B) compared with A2780-EV cells. Next, we determined the DNA adduct formation after ATP11B downregulation (Figure 4C) in both cisplatin-sensitive and -resistant cells. Silencing ATP11B expression with specific ATP11B siRNA enhanced DNA adduct formation in the A2780-CP20 cells to values close to those of cisplatin-sensitive cells (P < 0.01). ATP11B silencing in A2780-PAR cells led to higher adduct formation than in control siRNA–transfected A2780-PAR cells (P < 0.001); control siRNA treatment had no effect on these cell lines.
Figure 4. ATP11B regulates platinum transport in ovarian cancer cells.

(A) Platinum (Pt) uptake was significantly decreased in A2780-ATP11B and A2780-CP20 cells compared with that in A2780-EV–transfected cells, whereas (B) DNA adduct formation was significantly increased in the same cell lines. (C) DNA adduct formation after ATP11B silencing resulted in significantly increased DNA adduct formation in both cell lines compared with that in A2780-EV cells. Time kinetics of cisplatin efflux showing the percentage of intracellular remaining platinum content in (D) A2780-PAR, A2780-EV, A2780-ATP11B, and A2780-CP20 cells as well as (E) A2780 control siRNA, A2780 ATP11B siRNA, A2780-CP20 control siRNA, and A2780-CP20 ATP11B siRNA cells after 10 to 30 minutes of incubation without cisplatin (see Methods). Remaining platinum amounts were significantly decreased in A2780-ATP11B–upregulated and A2780-CP20 cells at all time points compared with that in their corresponding control siRNA controls. In contrast, compared with their controls and at all time points, significantly higher platinum contents were determined in ATP11B-silenced A2780-PAR or A2780-CP20 cells. Platinum determinations (duplicates) were performed by flame atomic absorption spectroscopy. Data represent the mean ± SEM from 3 independent experiments. *P < 0.05; **P < 0.01; †P < 0.001.
Next, we performed kinetic studies of cisplatin efflux to examine the effect of increased expression of ATP11B on the cellular pharmacology of cisplatin in A2780-PAR cells. The time course of cisplatin efflux was determined over a 30-minute period, after 10 minutes of cisplatin exposure. There was a significant decrease (P < 0.05) in platinum accumulation in A2780 cells transfected with ATP11B construct (A2780-ATP11B cells) and A2780-CP20 cells compared with that in A2780-EV cells (Figure 4D). The percentage of platinum remaining in A2780-ATP11B and A2780-CP20 cells was significantly decreased at all time points compared with that in A2780-PAR cells (P < 0.05 for all cell lines). We also examined cisplatin efflux kinetics in ATP11B-silenced A2780-PAR and A2780-CP20 cells. The platinum content was significantly (P < 0.01) increased in both cell lines after ATP11B silencing compared with that in their respective nontransfected parental cell lines (Figure 4E).
ATP11B is involved in platinum trafficking.
To further examine a potential role for ATP11B in the intracellular trafficking and/or vesicular export of cisplatin, we performed a series of immunofluorescence studies in the presence or absence of fluorescent cisplatin (FDDP). A2780-PAR and A2780-CP20 cells were exposed to 2 μM FDDP for different times (5 minutes to up to 1 hour) and immunostained for ATP11B. Supplemental Figure 7 shows representative confocal images from these cells. After 30 minutes and 1 hour of FDDP exposure, FDDP was frequently detected in the nuclei of A2780-PAR cells. In contrast, FDDP was seldom found in the nuclei of A2780-CP20 cells (Supplemental Figure 7). To examine the subcellular localization of ATP11B by immunofluorescence staining, we used specific cell organelle markers. In both cell lines, ATP11B partially colocalized with early endosome and lysosome-positive structures (data not shown). ATP11B was mostly detected in the trans-Golgi network (TGN) (Figure 5A), colocalizing with syntaxin-6 (STX6, a TGN marker). ATP11B strongly colocalized with STX6 in the absence and presence of FDDP (2 μM) at all time points examined (15, 30, and 60 minutes). Figure 5 shows representative images from A2780-PAR (Figure 5B) and A2780-CP20 (Figure 5C) cells in the absence or presence of FDDP. FDDP accumulation was detected in the TGN, colocalizing with STX6 and also with ATP11B at all exposure times in both cell lines. This suggests that FDDP is first sequestered into the Golgi and eventually enters into TGN vesicles. To determine whether the presence of different concentrations of nonfluorescent cisplatin could trigger relocalization of ATP11B from the TGN to other cell compartments, we performed immunofluorescence experiments in both cell lines after exposure to cisplatin for 2 hours. Figure 5D includes confocal images of A2780-PAR and A2780-CP20 cells, which were exposed to cisplatin concentrations ranging from 0 to 2 μM. In the presence of cisplatin, ATP11B and STX6 were detected in the TGN and in the nuclei of A2780-PAR cells. In A2780-CP20 cells, both proteins colocalized mostly in the TGN. However, in the presence of 2 μM cisplatin, ATP11B was found in the nuclei of a limited number of cells. To further analyze the subcellular localization of ATP11B in the absence or presence of cisplatin, membrane lysates (post-nuclear and mitochondrial) from A2780-PAR and A2780-CP20 cells, treated or untreated with 2 μM cisplatin, were subjected to sucrose gradient fractionation. In A2780-CP20 cells, ATP11B protein was more abundant than in A2780-PAR cells and distributed in gradient fractions 6–12 (Supplemental Figure 8A). ATP11B was mainly detected in fraction 11 in both cell lines treated with 2 μM cisplatin. Soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) proteins (STX6 and vesicular-associated membrane protein 4 [VAMP4]) and adaptor protein 1 (AP1, a TGN marker) were also found in this gradient fraction. ATP11B localization was also analyzed in cytosolic and nuclear fractions from these cells. Cisplatin treatment induced the redistribution of ATP11B from cytoplasm to nucleus in both cell lines (Supplemental Figure 8B), which is consistent with data in Figure 5 related to cytoplasm-to-nuclear translocation.
Figure 5. Subcellular localization of ATP11B in cisplatin-sensitive and cisplatin-resistant cells.

(A) Immunofluorescence staining using antibodies against ATP11B and specific cell markers showed the ATP11B signal mostly in the TGN colocalizing with STX6 (a TGN marker) in A2780-PAR and A2680-CP20 cells (green, STX6; red, ATP11B; blue, nuclei). Higher-magnification images (yellow boxes) from merged images show colocalization of ATP11B and STX6. Color histograms from these areas are included. (B) In both cell lines, FDDP, STX6, and ATP11B colocalized at the TGN at different times of FDDP exposure (15–60 minutes) (green, FDDP; red, ATP11B; blue, STX6). (C) Colocalization of FDDP, VAMP4 (vesicular transport marker), and ATP11B during FDDP exposure (green, FDDP; red, ATP11B; blue, VAMP4) in A2780-PAR and A2780-CP20 cells. Representative confocal images after 0, 15, 30, and 60 minutes of FDDP exposure. (D) Confocal images from A2780-PAR and (E) A2780-CP20 cells in the absence and presence of nonfluorescent cisplatin (0–2 μM), immunostained for the detection of ATP11B/ STX6 and ATP11B/lamin B1 (nuclear marker). In both cell lines colocalization between ATP11B and STX6 was stronger in the presence of cisplatin (asterisks). ATP11B was frequently observed in nuclei from A2780-PAR cells colocalizing with lamin B1 (green, STX6, lamin B1; red, ATP11B; blue, nuclei). Original magnification, ×200; scale bars: 5 μM (A); 10 μM (B–E).
Gene array analysis of ATP11B-silenced cells.
To identify potential effects of ATP11B on downstream pathways, we examined gene expression in cisplatin-sensitive and cisplatin-resistant cells following ATP11B gene silencing. In A2780-CP20 ovarian cancer cells, the ErbB, p53, glycolysis, and SNARE signaling pathways were significantly deregulated (Supplemental Table 1). Based on the putative transport function of ATP11B and our results from platinum efflux kinetics, we next focused on the SNARE vesicular transport pathway. Supplemental Table 2 shows the fold change and P values of 5 deregulated genes from the SNARE pathway. SNARE proteins make up a superfamily of small proteins that are involved in membrane fusion events, intracellular protein trafficking, and secretory processes. By means of immunoprecipitation analyses, we next examined the potential interaction between ATP11B and the 5 deregulated SNARE proteins: STX4, STX6, VAMP1, VAMP4, and Golgi synaptosomal associated protein receptor 1 in cell lysates from A2780-PAR and A2780-CP20 cells. Among these, ATP11B coimmunoprecipitated with STX6 and VAMP4. In the presence of cisplatin (0.5 μM), the interaction was stronger in cell lysates from both cell lines (Figure 6A).
Figure 6. ATP11B, STX6, and VAMP4 contribute to cisplatin export.

(A) Coimmunoprecipitation of ATP11B with STX6 or VAMP4 in A2780-PAR and A2780-CP20 cell lysates. All coimmunoprecipitation lanes were run on the same gel but were noncontiguous. Input lanes (1:10 of original cell lysate) were run on a second gel simultaneously and detected with anti-ATP11B. Association of ATP11B with STX6 or VAMP4 was stronger in the presence of 0.5 μM cisplatin. Densitometry analysis of ATP11B protein was performed with ImageJ software. Data were normalized to a loading control (β-actin from original lysate) and are representative of 2 independent experiments. (B) Time course of cisplatin efflux in STX6- or VAMP4-silenced A2780-PAR cells and (C) STX6- or VAMP4-downregulated A2780-CP20 cells. Silencing of STX6 in A2780-CP20 cells resulted in significantly increased cellular platinum contents after 10 to 30 minutes of incubation without cisplatin (*P < 0.05). (D) Time kinetics of cisplatin efflux in ATP11B-upregulated STX6- or VAMP4-downregulated A2780-PAR cells and (E) in ATP11B-upregulated STX6- or VAMP4-silenced A2780-CP20 cells. Percentages of remaining cellular platinum were significantly increased in ATP11B-upregulated VAMP4-silenced A2780-PAR cells at all time points compared with those in A2780-PAR ATP11B-upregulated cells. In contrast, in ATP11B-upregulated A2780-CP20 cells, downregulation of STX6 led to increased platinum contents at all time points compared with those of ATP11B-upregulated A2780-CP20 cells. Data represent the mean ± SEM. *P < 0.05.
ATP11B, STX6, and VAMP4 are involved in platinum cell transport.
To determine the role of STX6 and/or VAMP4 along with ATP11B in cisplatin export, we performed kinetic studies of cisplatin efflux, as described above, in STX6- or VAMP4-silenced cisplatin-sensitive and -resistant cells. In A2780-CP20 cells, but not in A2780-PAR cells, silencing of STX6 caused a significant increase in percentage of platinum content after 10, 20, and 30 minutes (P < 0.05) compared with that in A2780-CP20 control siRNA–transfected cells (Figure 6, B and C). VAMP4 silencing did not affect platinum export in either cell line. These data suggest that STX6 might be essential for cisplatin export in the cisplatin-resistant cells. We next addressed the question of whether STX6 and/or VAMP4 could play a role in the ATP11B-mediated effect on cisplatin export. For this purpose, we measured the kinetics of cisplatin efflux in ATP11B-upregulated or STX6- VAMP4-downregulated cells. Figure 6, D and E, show the results of these analyses. The percentage of remaining platinum was higher in both ATP11B-upregulated STX6- or VAMP4-silenced A2780-PAR cells after 10, 20, and 30 minutes compared with that in A2780-PAR ATP11B-upregulated cells. However, these changes were significant only in ATP11B-upregulated VAMP-silenced cells (P < 0.05). Furthermore, significantly higher percentages of platinum content were determined in ATP11B-upregulated STX6-downregulated A2780-CP20 cells at all 3 time points compared with that in A2780-CP20 ATP11B-transfected cells (Figure 6, D and E). These findings indicate that downregulation of these proteins retards cisplatin efflux and suggest a role of VAMP4 and STX6 in the ATP11B-mediated effect on cisplatin export in A2780-PAR and A2780-CP20 cells, respectively.
Discussion
The results reported here provide evidence that ATP11B regulates sensitivity to platinum in ovarian cancer cells. ATP11B is a member of the family of P-type ATPases and encodes for a transmembrane protein of structure similar to that encoded by the other family members (12). ATP11B downregulation through specific siRNA sequences in vitro led to significantly increased sensitivity to cisplatin in cisplatin-sensitive and -resistant ovarian cancer cells. This finding was further confirmed by our in vivo therapy experiments.
On the basis of their substrates and specific sequence motifs, members of the superfamily of P-type ATPases have been categorized into 5 subfamilies (13–16). The type 1 ATPases transport heavy metals, such as copper, cadmium, and zinc. The copper transporters ATP7A and ATP7B belong to this subfamily of type 1 P-type ATPases. ATP11B, however, is included in the subfamily of type 4 P-type ATPases. These proteins are thought to translocate phospholipids (rather than cations) from the outer and inner leaflet of membrane bilayers (13, 17–23). Although the putative role of ATP11B, based on its protein structure, would not have predicted effects on platinum transport, the unexpected results, with regard to its role in cisplatin efflux from cells, provide a new conceptual model (Supplemental Figure 9) for this protein.
Studies in yeast have recently demonstrated an association between the translocase activity of type 4 P-type ATPases and vesicular (protein) trafficking (13, 20, 24–26). These findings are supportive of our findings in that ATP11B is involved in the vesicular transport of cisplatin. STX6 and VAMP4 are known to contribute to cellular vesicular transport at the level of the TGN (27, 28). Our pharmacological studies demonstrated that both of these SNARE proteins were essential for ATP11B-mediated cisplatin export. Furthermore, the interaction of ATP11B with STX6 and VAMP4 and also with FDDP points to engagement of vesicular trafficking and export of cisplatin.
Reduced platinum uptake, which leads to decreased DNA adduct formation, is a characteristic feature of cisplatin-resistant cells (29). In this study, we demonstrated that upregulation of ATP11B in cisplatin-sensitive and -resistant cells leads to increased platinum export and to decreased cellular platinum levels and DNA adduct formation. On the other hand, silencing ATP11B expression had opposite effects: decreased cisplatin export and increased platinum levels, with subsequent enhancement of cisplatin-DNA adduct formation. These findings indicate a role for ATP11B in pharmacokinetics and transport of cellular platinum. A growing list of membrane transporters has been shown to be involved in the intracellular trafficking, import, and/or export of cisplatin (10). Among these transporters are ATP7A and ATP7B, 2 copper transporters that are members of the P-type family of ATPases (8, 30, 31). Recently, we described the functional and biological effects of targeting ATP7B to reverse cisplatin resistance in ovarian cancer cells (31). In these studies, we demonstrated that ATP7B interacts directly with cisplatin through its metal-binding methionine-rich sequences at its amino terminus (31). ATP11B shares a high homology in protein structure with ATP7A and ATP7B; however, the ATP11B protein structure lacks metal-binding motifs at the amino terminus that are essential to bind platinum.
A recent study demonstrated that class 5 (ATP10A, ATP10B, and ATP10D) and class 6 (ATP11A, ATP11B, and ATP11C) P4-ATPases require CDC50 proteins, primarily CDC50A, for their exit from the ER and final subcellular localization (32). These proteins were mainly found in the ER. Our studies provide evidence that ATP11B can also reside in other cell compartments (e.g., TGN, transport vesicles, endosomes, lysosomes) in addition to ER. This suggests that ATP11B can exit the ER more efficiently to reach its final localization and exert its cellular (biological) function.
Use of RNA interference as a therapeutic modality is highly attractive due to sequence specificity and the ability to target traditionally “undruggable” genes (33). While systemic delivery has been a challenge, highly efficient methods, such as the one described here, hold potential for clinical development (34, 35). The use of ATP11B siRNA in DOPC nanoliposomes could be an attractive therapeutic strategy to enhance the effectiveness of cisplatin in patients with cancer. However, formal safety testing would be essential prior to such clinical development.
Collectively, our data demonstrate that overexpression of ATP11B confers cisplatin resistance in ovarian cancer cells by enhancing the efflux of cisplatin via the vesicular secretory pathway. On the basis of these findings, ATP11B appears to be an important target for overcoming cisplatin resistance.
Methods
Reagents.
Cisplatin (cis-diamminedichloroplatinum, CDDP) was purchased from Roche; paclitaxel and topotecan were obtained from Tecoland. FDDP was synthesized according to a procedure already described (36). Restriction enzymes were obtained from New England BioLabs.
Cell culture.
The human epithelial ovarian cancer cell lines, A2780 parental (PAR) and SKOV31p1 (cisplatin-sensitive) as well as A2780-CP20, IGROV1CP20, RMG2, and ES2 (cisplatin-resistant), were maintained as previously described (37). The A2780-CP20 cell line was developed by sequential exposure of the A2780 cell line to increasing concentrations of cisplatin. The A2780-ATP11B–transfected cell line was grown in the same medium containing neomycin (0.6 mg/ml). All in vitro experiments were conducted at 60% to 80% cell confluence. For in vivo injections, cells were trypsinized and centrifuged at 600 g for 7 minutes at 4°C, washed twice with PBS, and reconstituted in serum-free Hank’s balanced salt solution (Life Technologies) at a concentration of 1 × 106 cells/ml. Only single-cell suspensions with more than 95% viability (determined by trypan blue exclusion) were used for in vivo i.p. injections. The human ovarian cancer cell lines PE01 and PE04 were a gift from Michael White (UT Southwestern Medical Center at Dallas, Dallas, Texas, USA). These cell lines were derived from the ascites of a single patient, who was diagnosed as having stage III ovarian adenocarcinoma. PE01 and PE04 cell lines were isolated before and after the onset of resistance to chemotherapy involving cis-platinum, chlorambucil, and 5-fluorouracil, respectively (38, 39). For our in vitro studies, PE01 and PE04 cells were cultured at 37°C, 5% CO2 in RPMI-1640, 4 mM l-glutamine, 25 mM HEPES (Gibco) with 10% FBS, streptomycin (100 μg/ml), and penicillin (100 IU/ml). Confluent cultures were passaged by 1:5 or 1:10 splits and cell passages below 25 were used for the experiments.
ATP11B construct.
A 3.8-kb nucleotide sequence containing the coding region of the ATP11B gene was cloned into the pcDNA-h(HM)1 vector (Invitrogen) between the XhoI/HindIII restriction sites. The Xho1/HindIII fragment was then subcloned into the expression vector pcDNA 3.1 (Invitrogen) with cytomegalovirus promoter. To confirm the correct cloning of the ATP11B insert, restriction enzyme analysis, PCR amplification, and bidirectional sequence analysis were performed. Plasmid DNA preparations were performed with the Qiagen Mini and Maxi Prep Kits (Qiagen). The concentration of plasmid DNA was determined spectrophotometrically at 260 nm.
Transfection of A2780-PAR cells with ATP11B construct.
A2780-PAR cells were seeded at a density of 3 × 105 cells in 10-cm plates. After 24 hours of incubation, cells were transfected with empty vector p-cDNA 3.1 or ATP11B construct using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Briefly, cDNA and Lipofectamine solution (1:2.5 ratio) were diluted with serum-free medium, and the two solutions were mixed and incubated for 20 minutes at room temperature. This mixture was added to cells, and serum-free medium was replaced with regular serum-containing medium after 6 to 8 hours of incubation. After 48 hours, cells were trypsinized and processed for determination of RNA and protein expression.
Clonogenic assay.
A2780-PAR cells transfected with ATP11B construct were plated onto 6-well culture plates and treated with 5 and 7.5 μM cisplatin for 24 hours. Colonies formed were stained with 0.25% Crystal Violet (Sigma-Aldrich) in 20% ethanol for 5 minutes and counted under the light microscope.
ATP711B gene silencing by siRNA.
ATP11B-targeted siRNA sequences were designed and purchased from Sigma-Aldrich. From them, two pairs of target sequences were selected based on their ability to downregulate ATP11B expression. Target sequences for sequence no. 1 were sense, 5′-GAAACAGCAUUAUUACAAA, and antisense, 5′-UUUGUAAUAAUGCUGUUUC. Target sequences for sequence no. 2 were sense, 5′-GAUGGUUCCUGUCACGUUA and, antisense 5′-UAACGUGACAGGAACCAUC. Transient transfections were done as described previously (40). Briefly, cells were transfected with ATP11B-specific or scrambled (control) siRNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. At selected time intervals, cells were harvested to measure mRNA levels of ATP11B. An oligonucleotide sequence that did not have homology to any human mRNA (scrambled siRNA as determined by a National Center for Biotechnology Information BLAST search) served as a control.
Analysis of ATP11B expression.
A quantitative reverse-transcriptase–PCR (RT-PCR) assay was used to measure ATP11B mRNA in RNA extracted from ovarian cancer cell lines and ovarian tumors. Quantitative and conventional RT-PCR was performed using the primers designed from the C terminus of the ATP11B gene: sense, 5′-GATCAACCATCTCGTTACCTGGGGA-3′; antisense, 5′-GATCAACCATCTCGTTACCTGGGGA-3′. The expected 437-bp PCR product was obtained under these experimental conditions. Total RNA was isolated using a Qiagen RNeasy Kit. Reverse transcription of 5 μg total RNA was performed using SuperScript II and random hexamer primers (Invitrogen). 18S was using as a housekeeping gene to normalize ATP11B mRNA levels. Protein expression was examined by means of Western blotting (see below).
Gene array analyses.
Gene array data from A2780-PAR and A2780-CP20 ovarian cancer cells were deposited into the GEO database (accession no. GSE44022; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=ttcpnwcayimckpc&acc=GSE44022). Total RNA was extracted and labeled by oligo-dT primed reverse transcription with either Cy5-dUTP or Cy3-dUTP as previously described (41). Each cisplatin-resistant cell line cDNA (Cy5) was pooled with the parental cell line cDNA (Cy3) and hybridized to a spotted DNA microarray comprising 15,000 probes derived from IMAGE consortium clones (41). Each hybridization was conducted in duplicate. Slides were scanned in an Agilent scanner, and the signal was extracted with DeArray software (42). Gene array data from ATP11B-silenced A2780-PAR and A2780-CP20 cells were deposited into the GEO database (accession no. GSE43694; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE43694). Array data processing were performed on Illumina BeadStudio software. We normalized gene expression data using Quantile normalization and log2 transformation. To export to a data matrix, Sample Gene Profile option of this software was used. BRB-ArrayTools were primarily used to identify genes differentially expressed between the 2 subgroups (43), and all other statistical analyses were performed in the R language environment. Gene expression differences were considered significant if P values were less than 0.001. Integrated function and pathway analyses were performed using DAVID bioinformatics resources ( http://david.abcc.ncifcrf.gov/) (44) and significant features were clustered.
Western blot and immunoprecipitation analyses.
Western blot analysis was performed as previously described (45). Briefly, lysates from cultured cells were prepared using modified RIPA buffer, and protein concentrations were determined using the BCA Protein Assay Reagent Kit (Pierce Biotechnology). Protein lysates were subjected to 10% SDS-PAGE separation and transferred onto a nitrocellulose membrane. Blots were probed with a customized antibody (Sigma-Aldrich) raised against a peptide sequence (CKFTSKEYEEIDKRI; Supplemental Figure 10) of ATP11B protein and a HRP-conjugated secondary antibody (Amersham) and subsequently developed with the ECL Detection Kit (Pierce Biotechnology). Equal protein loading was confirmed by reprobing membranes with an anti–β-actin antibody (Sigma-Aldrich). To examine the association between ATP11B protein and the SNARE proteins, A2780-PAR and A2780-CP20 cells were incubated in the absence or presence of cisplatin (0–2.5 μM) at 37°C for 2 hours. Cell lysates were then prepared and immunoprecipitated with STX6 (BD Biosciences) or VAMP4 (Santa Cruz Biotechnology Inc.) at 4°C for 16 hours. Immunocomplexes were captured with 2% protein A-agarose beads (Upstate Biotechnology). Protein was eluted in reducing sample Laemmli buffer, subjected to 10% SDS-PAGE separation, and transferred to a nitrocellulose membrane. Anti-ATP11B was used as primary antibody. Immunodetection of ATP11B protein was carried out as described earlier.
Cytotoxicity assay.
The sensitivity to cisplatin of A2780-PAR, A2780-CP20, A2780-ATP11B–transfected, and A2780-CP20 ATP11B siRNA–transfected cells was determined using the MTT assay as previously described (45). Briefly, 2 × 103 cells were seeded onto 96-well plates and incubated at 37°C overnight. Cells were then transfected with control or ATP11B siRNA for 48 hours, and subsequently the medium was exchanged with fresh medium containing different cisplatin concentrations (0–32 μM). After incubation for 72 hours, 50 μl of the MTT solution (0.15%) was added to each well, and the plates were further incubated for 2 hours. One hundred microliters of DMSO was added to solubilize the MTT formazan product. Absorbance at 540 nm was measured with a Falcon microplate reader (BD — Labware). Dose-response curves were plotted on a semilog scale as the percentage of the control cell number, which was obtained from the sample with no drug exposure. IC50 was determined by the intersection of the cisplatin concentration and the midpoint of the 570-nm reading.
Animal care and therapeutic studies.
Female athymic nude mice (NCr-nu) were purchased from the National Cancer Institute-Frederick Cancer Research and Development Center and housed under specific pathogen–free conditions. Tumors were established by i.p. injection of cells prepared as described above. This model reflects the i.p. growth pattern of advanced ovarian cancer, as intra-abdominal spread is the main mechanism of ovarian cancer metastasis. Mice (10 mice per group) were monitored for adverse effects of therapy and killed when any of the mice in a group began to appear moribund. Mouse weight, tumor weight, and tumor distribution were recorded. Tissue specimens were snap frozen for lysate preparation, fixed in formalin for paraffin embedding, or frozen in OCT medium for frozen-slide preparation. To evaluate the therapeutic effectiveness of ATP11B siRNA in combination with cisplatin, we first determined the optimal dose of ATP11B siRNA. A2780-CP20 cells were prepared as described above and injected i.p. Treatment was initiated 21 days after the cell suspension injection, when tumors could be detected by palpation. Mice were divided into 3 groups (10 mice per group) and treated with a single dose of PBS, control siRNA–DOPC, or ATP11B siRNA–DOPC at a dose of 150 or 300 μg/kg i.p. For siRNA delivery, the i.p. route was chosen based on comparable uptake and therapy effectiveness as described in the literature (46). Three mice in each group were killed after 24 hours, 48 hours, and 6 days of treatment. Downregulation of the ATP11B gene was observed at both siRNA doses. Therefore, a dose of 150 μg/kg was used for subsequent experiments. The optimal cisplatin dose was also determined in nude mice bearing cisplatin-sensitive A2780-PAR tumors. These mice were treated i.p. with 80, 160, and 200 μg cisplatin per week. The lowest effective dose (160 μg/week) was chosen for all experiments performed. On the basis of the data from our preliminary dose-response experiments, therapy experiments with ATP11B siRNA were conducted as follows: tumor cells were injected i.p., and 7 days later, mice were divided into 5 treatment groups: empty liposome (150 μg/kg), control siRNA–DOPC (150 μg/kg), ATP11B siRNA–DOPC (150 μg/kg), control siRNA–DOPC (150 μg/kg) in combination with 160 μg cisplatin per week, and ATP11B siRNA–DOPC (150 μg/kg) in combination with 160 μg cisplatin per week. All treatments were given twice weekly.
Immunohistochemistry.
Analyses of microvessel density, assessment of tumor cell proliferation, and tumor cell apoptosis were conducted following procedures described previously (45, 47, 48).
Immunofluorescence and confocal microscopy.
Immunofluorescence cell staining for the detection of ATP11B was performed as follows: A2780-PAR and A2780-CP20 cells were plated onto cover slips in 6-well plates at a density of 3 × 105 cells per cover slip. The following day, cells were fixed in acetone at –20°C for 10 minutes and washed with PBS. Cells were then permeabilized with PBS/0.25% Triton for 15 minutes and blocked with 1% normal goat serum (NGS) in PBS for 1 to 2 hours. Afterward, cells were incubated with rabbit anti-ATP11B (1:100; Sigma customized) for 2 hours at room temperature. The cover slips were subsequently washed 3 times with PBS and then incubated with the appropriate fluorescent-conjugated secondary antibody (1:1,000; Invitrogen) for 1 hour at room temperature. Nuclei were stained with Hoechst (Invitrogen) or SYTOX orange (Invitrogen). Double immunofluorescence staining for ATP11B/STX6 or ATP11B/VAMP4 was performed following a procedure previously described (47) using the tyramide/streptavidin system (Perkin Elmer Inc.) to enhance the signal of ATP11B protein. A2780-PAR and A2780-CP20 cells were first stained for ATP11B as described above, and then cells were blocked in 5% NHS/1% NGS for 10 minutes and incubated with mouse anti-STX6 (BD Biosciences; 1:100) or mouse anti-VAMP4 (Santa Cruz Biotechnology Inc.; 1:100) for 16 hours at 4°C. The cover slips were subsequently washed 3 times with PBS and then incubated with the appropriate fluorescent-conjugated secondary antibody (1:1,000; Invitrogen) for 1 hour at room temperature. Nuclei were stained as described earlier. Immunofluorescence microscopy was carried out using a Zeiss Axioplan fluorescence microscope (Carl Zeiss Inc.) equipped with a 100-W Hg lamp and narrow band-pass excitation filters. Representative images were obtained using a cooled charge–coupled device camera (model C5810, Hamamatsu Photonics) and Optimas software (Media Cybernetics). Confocal fluorescence images from Z sections were collected under a ×20 objective lens on a Zeiss LSM 510 laser scanning microscopy system (Carl Zeiss Inc.) equipped with a motorized Axioplan microscope, argon laser (458/477/488/514 nm, 30 mW), HeNe laser (413 nm, 1 mW, and 633 nm, 5 mW), LSM 510 control and image acquisition software, and appropriate filters (Chroma Technology Corp.). Composite images were constructed with Photoshop software (Adobe Systems Inc.).
Subcellular fractionation by discontinuous sucrose density gradient centrifugation.
Endomembrane fractions from A2780-PAR and A2780-CP20 cells were prepared as described previously (49). These sonicated fractions were transferred to a 12-ml polycarbonate ultracentrifuge tube (Beckman) and adjusted to 40% (w/v) sucrose by the addition of an equal volume of 80% (w/v) sucrose in 10 mM Tris-HCl (pH 7.4), 1 mM EDTA, and 1 mM EGTA to give a final volume of 4 ml. The sample was overlaid with 4 ml of 35% (w/v) sucrose, followed by 4 ml of 5% (w/v) sucrose, and subjected to discontinuous equilibrium sucrose density gradient centrifugation overnight at 4°C at 180,000 g using a swing-out SW41 Beckman rotor. Then, 1-ml gradient fractions were harvested, beginning from the top of the tube, and stored at –20°C.
Determination of platinum cell uptake and platinum efflux kinetics.
Cells were incubated at 37°C in the presence of cisplatin (20 μM) for 4 hours. Cells were then centrifuged at 400 g for 10 minutes. Pellets were washed twice with ice-cold PBS and digested overnight at 60°C with 1 M benzethonium hydroxide (Sigma-Aldrich). A small aliquot of cell pellet was taken for protein determination before digestion. After digestion, samples were acidified with 1 N HCl, and the platinum amount was determined by flame atomic absorption spectroscopy (Varian). Protein concentrations were determined using a BCA Protein Assay Reagent Kit (Pierce Biotechnology). To examine cisplatin efflux kinetics, cells were exposed to cisplatin (500 μM) for 10 minutes (0 time); cisplatin was then removed, and the cells were further incubated in regular medium for 10, 20, and 30 minutes. At each time point, cells were processed as described above for platinum and protein determination.
Determination of platinum bound to DNA in cells.
Cells incubated with cisplatin (20 μM) were centrifuged (400 g; 10 minutes), and the pellet was washed 2 times with cold PBS. High-molecular-weight DNA was isolated from cell pellets according to standard procedures. Briefly, the pellets were lysed using the extraction buffer (10 mM Tris,100 mM EDTA, 20 μg/ml RNase, 0.5% SDS, pH 8.0) and treated with proteinase K (1 mg/mI) at 37°C for 1 hour and then at 50°C for 2 hours. DNA was precipitated by phenol chloroform extraction and then washed twice with 70% ethanol. The DNA pellet was partially dried and dissolved in 50 μl of buffer (10 mM Tris, 1 mM EDTA, pH 8.0). The DNA content and purity were assessed by spectrophotometry at 260 and 280 nm, and the amount of platinum in the sample was determined directly by flame atomic absorption spectroscopy.
Statistics.
To determine distribution of ATP11B levels around cutoff points, histograms were created on a log2 scale of the expression ratio and tested for normality with the Kolmogorov-Smirnov test. The Wilcoxon test was used to compare rank distributions of continuous variables not conforming to the assumptions of normality. Kaplan-Meier plots were constructed and a log-rank test was used to determine differences among survival curves according to ATP11B expression levels. Multivariate analyses were performed with the use of a Cox proportional hazards model to examine the effects ATP11B expression on death from disease while adjusting for other covariates. The relationship between the expression of the ATP11B gene and survival was explored in qRT-PCR data sets by separating the cases from each cohort into a group with a high median level of expression and a group with a low median level of expression. A kappa statistic was calculated to assess the agreement between qRT-PCR results and histochemical scores. All P values were 2 sided, and a P value of less than 0.05 was considered to indicate statistical significance. Continuous variables were compared with the 2-tailed Student’s t test (between 2 groups) or analysis of variance (for all groups) if normally distributed and the Mann-Whitney rank-sum test if distributions were nonparametric. For in vivo therapy experiments, 10 mice in each group were used, and a P value of less than 0.05 with 2-tailed testing was considered significant. To control for the effects of multiple comparisons, a Bonferroni adjustment was made; for the analysis, a P value of less than 0.017 was considered statistically significant.
Study approval.
Approval for studies in patient samples was obtained from the Institutional Review Board of The University of Texas MD Anderson Cancer Center. The study was exempt from informed consent because it used previously collected residual tissue samples. Animals were treated humanely and cared for in accordance with guidelines set forth by the American Association for Accreditation of Laboratory Animal Care and the US Public Health Service Policy on Humane Care and Use of Laboratory Animals, and all studies were approved and supervised by the Institutional Animal Care and Use Committee at The University of Texas MD Anderson Cancer Center.
Supplementary Material
Acknowledgments
The authors thank Svetlana Lutsenko for helpful discussions and guidance. We also thank Michael White (UT Southwestern Medical Center) for providing the patient cell lines and Donna Reynolds for assistance with immunohistochemistry. This work was supported in part by grants from the CPRIT (RP110595-P2), Gynecologic Cancer Foundation; grants (CA109298, P50 CA083639, P50 CA098258, CA128797, RC2GM092599, U54 CA151668) from the National Institutes of Health; a Program Project Development Grant from the Ovarian Cancer Research Fund; grants (OC073399, W81XWH-10-1-0158, and BC085265) from the Department of Defense; the Baylor College of Medicine and MD Anderson Cancer Center Multidisciplinary Research Program; the Zarrow Foundation; the Marcus Foundation; the Estate of C.G. Johnson Jr.; the Blanton-Davis Ovarian Cancer Research Program; the Gilder Foundation; the RGK Foundation; the Bettyann Asche Murray Distinguished Professorship; and a training grant (T32 CA101642 supported J. Bottsford-Miller) from the National Cancer Institute.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(5):2119–2130. doi:10.1172/JCI65425.
References
- 1.Martin LP, Hamilton TC, Schilder RJ. Platinum resistance: role of DNA repair pathways. Clin Cancer Res. 2007;14(5):1291–1295. doi: 10.1158/1078-0432.CCR-07-2238. [DOI] [PubMed] [Google Scholar]
- 2.Prestayko AW, D’Aoust JC, Issell BF, Crooke ST. Cisplatin (cis-diamminedichloroplatinum II). Cancer Treat Rev. 1979;6(1):17–39. doi: 10.1016/S0305-7372(79)80057-2. [DOI] [PubMed] [Google Scholar]
- 3.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22(47):7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 4.Teicher BA, et al. Characterization of a human squamous carcinoma cell line resistant to cis-diamminedichloroplatinum(II). Cancer Res. 1987;47(2):388–393. [PubMed] [Google Scholar]
- 5.Andrews PA, Velury S, Mann SC, Howell SB. cis-Diamminedichloroplatinum(II) accumulation in sensitive and resistant human ovarian carcinoma cells. Cancer Res. 1988;48(1):68–73. [PubMed] [Google Scholar]
- 6.Borst P, Evers R, Kool M, Wijnholds J. A family of drug transporters: the multidrug resistance-associated proteins. J Natl Cancer Inst. 2000;92(16):1295–1302. doi: 10.1093/jnci/92.16.1295. [DOI] [PubMed] [Google Scholar]
- 7.Kool M, et al. Analysis of expression of cMOAT (MRP2), MRP3, MRP4, and MRP5, homologues of the multidrug resistance-associated protein gene (MRP1), in human cancer cell lines. Cancer Res. 1997;57(16):3537–3547. [PubMed] [Google Scholar]
- 8.Komatsu M, et al. Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res. 2000;60(5):1312–1316. [PubMed] [Google Scholar]
- 9.Katano K, Safaei R, Samimi G, Holzer A, Rochdi M, Howell SB. The copper export pump ATP7B modulates the cellular pharmacology of carboplatin in ovarian carcinoma cells. Mol Pharmacol. 2003;64(2):466–473. doi: 10.1124/mol.64.2.466. [DOI] [PubMed] [Google Scholar]
- 10.Kuo MT, Chen HH, Song IS, Savaraj N, Ishikawa T. The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 2007;26(1):71–83. doi: 10.1007/s10555-007-9045-3. [DOI] [PubMed] [Google Scholar]
- 11.Safaei R, Otani S, ChengLarson BJ, Rasmussen ML, Howell SB. Transport of cisplatin by the copper efflux transporter ATP7B. Mol Pharmacol. 2008;73(2):461–468. doi: 10.1124/mol.107.040980. [DOI] [PubMed] [Google Scholar]
- 12.Halleck MS, Schlegel RA, Williamson PL. Reanalysis of ATP11B, a type IV P-type ATPase. J Biol Chem. 2002;277(12):9736–9740. doi: 10.1074/jbc.M200240200. [DOI] [PubMed] [Google Scholar]
- 13.Paulusma CC, Oude Elferink RP. The type 4 subfamily of P-type ATPases, putative aminophospholipid translocases with a role in human disease. Biochim Biophys Acta. 2005;1741(1–2):11–24. doi: 10.1016/j.bbadis.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 14.Axelsen KB, Palmgren MG. Evolution of substrate specificities in the P-type ATPase superfamily. J Mol Evol. 1998;46(1):84–101. doi: 10.1007/PL00006286. [DOI] [PubMed] [Google Scholar]
- 15.Axelsen KB, Palmgren MG. Inventory of the superfamily of P-type ion pumps in Arabidopsis. Plant Physiol. 2001;126(2):696–706. doi: 10.1104/pp.126.2.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palmgren MG, Axelsen KB. Evolution of P-type ATPases. Biochim Biophys Acta. 1998;1365(1–2):37–45. doi: 10.1016/s0005-2728(98)00041-3. [DOI] [PubMed] [Google Scholar]
- 17.Auland ME, Roufogalis BD, Devaux PF, Zachowski A. Reconstitution of ATP-dependent aminophospholipid translocation in proteoliposomes. Proc Natl Acad Sci U S A. 1994;91(23):10938–10942. doi: 10.1073/pnas.91.23.10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pomorski T, Lombardi R, Riezman H, Devaux PF, van Meer G, Holthuis JC. Drs2p-related P-type ATPases Dnf1p and Dnf2p are required for phospholipid translocation across the yeast plasma membrane and serve a role in endocytosis. Mol Biol Cell. 2003;14(3):1240–1254. doi: 10.1091/mbc.E02-08-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daleke DL, Lyles JV. Identification and purification of aminophospholipid flippases. Biochim Biophys Acta. 2000;1486(1):108–127. doi: 10.1016/s1388-1981(00)00052-4. [DOI] [PubMed] [Google Scholar]
- 20.Graham TR. Flippases and vesicle-mediated protein transport. Trends Cell Biol. 2004;14(12):670–677. doi: 10.1016/j.tcb.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 21.Halleck MS, et al. Differential expression of putative transbilayer amphipath transporters. Physiol Genomics. 1999;1(3):139–150. doi: 10.1152/physiolgenomics.1999.1.3.139. [DOI] [PubMed] [Google Scholar]
- 22.Williamson P, Schlegel RA. Transbilayer phospholipid movement and the clearance of apoptotic cells. Biochim Biophys Acta. 2002;1585(2–3):53–63. doi: 10.1016/s1388-1981(02)00324-4. [DOI] [PubMed] [Google Scholar]
- 23.Dolis D, Moreau C, Zachowski A, Devaux PF. Aminophospholipid translocase and proteins involved in transmembrane phospholipid traffic. Biophys Chem. 1997;68(1–3):221–231. doi: 10.1016/S0301-4622(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 24.Tang X, Halleck MS, Schlegel RA, Williamson P. A subfamily of P-type ATPases with aminophospholipid transporting activity. Science. 1996;272(5267):1495–1497. doi: 10.1126/science.272.5267.1495. [DOI] [PubMed] [Google Scholar]
- 25.Hoekstra D, van IJzendoorn SC. In search of lipid translocases and their biological functions. Dev Cell. 2003;4(1):8–9. doi: 10.1016/S1534-5807(02)00405-7. [DOI] [PubMed] [Google Scholar]
- 26.Pomorski T, Holthuis JC, Herrmann A, van Meer G. Tracking down lipid flippases and their biological functions. J Cell Sci. 2004;117(pt 6):805–813. doi: 10.1242/jcs.01055. [DOI] [PubMed] [Google Scholar]
- 27.Bock JB, Klumperman J, Davanger S, Scheller RH. Syntaxin 6 functions in trans-Golgi network vesicle trafficking. Mol Biol Cell. 1997;8(7):1261–1271. doi: 10.1091/mbc.8.7.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steegmaier M, Klumperman J, Foletti DL, Yoo JS, Scheller RH. Vesicle-associated membrane protein 4 is implicated in trans-Golgi network vesicle trafficking. Mol Biol Cell. 1999;10(6):1957–1972. doi: 10.1091/mbc.10.6.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohmichi M, Hayakawa J, Tasaka K, Kurachi H, Murata Y. Mechanisms of platinum drug resistance. Trends Pharmacol Sci. 2005;26(3):113–116. doi: 10.1016/j.tips.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 30.Holzer AK, et al. The copper influx transporter human copper transport protein 1 regulates the uptake of cisplatin in human ovarian carcinoma cells. Mol Pharmacol. 2004;66(4):817–823. doi: 10.1124/mol.104.001198. [DOI] [PubMed] [Google Scholar]
- 31.Mangala LS, et al. Therapeutic targeting of ATP7B in ovarian carcinoma. Clin Cancer Res. 2009;15(11):3770–3780. doi: 10.1158/1078-0432.CCR-08-2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takatsu H, et al. ATP9B, a P4-ATPase (a putative aminophospholipid translocase), localizes to the trans-Golgi network in a CDC50 protein-independent manner. J Biol Chem. 2011;286(44):38159–38167. doi: 10.1074/jbc.M111.281006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pecot CV, Calin GA, Coleman RL, Lopez-Berestein G, Sood AK. RNA interference in the clinic: challenges and future directions. Nat Rev Cancer. 2011;11(1):59–67. doi: 10.1038/nrc2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 35.Tabernero J, et al. Cancer Discov. First-in-man trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement [published online ahead of print January 28, 2013]. doi: 10.1158/2159-8290.CD-12-0429 . [DOI] [PubMed] [Google Scholar]
- 36.Katano K, et al. Confocal microscopic analysis of the interaction between cisplatin and the copper transporter ATP7B in human ovarian carcinoma cells. Clin Cancer Res. 2004;10(13):4578–4588. doi: 10.1158/1078-0432.CCR-03-0689. [DOI] [PubMed] [Google Scholar]
- 37.Sood AK, et al. Molecular determinants of ovarian cancer plasticity. Am J Pathol. 2001;158(4):1279–1288. doi: 10.1016/S0002-9440(10)64079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolf CR, et al. Cellular heterogeneity and drug resistance in two ovarian adenocarcinoma cell lines derived from a single patient. Int J Cancer. 1987;39(6):695–702. doi: 10.1002/ijc.2910390607. [DOI] [PubMed] [Google Scholar]
- 39.Langdon SP, et al. Characterization and properties of nine human ovarian adenocarcinoma cell lines. Cancer Res. 1988;48(21):6166–6172. [PubMed] [Google Scholar]
- 40.Landen CN, et al. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 2005;65(15):6910–6918. doi: 10.1158/0008-5472.CAN-05-0530. [DOI] [PubMed] [Google Scholar]
- 41.DeRisi J, et al. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat Genet. 1996;14(4):457–460. doi: 10.1038/ng1296-457. [DOI] [PubMed] [Google Scholar]
- 42.Chen Y, Dougherty ER, Bittner ML. Ratio-based decisions and the quantitative analysis of cDNA microarray images. J Biomed Opt. 1997;2(4):364–374. doi: 10.1117/12.281504. [DOI] [PubMed] [Google Scholar]
- 43.Hu W, et al. Biological roles of the Delta family Notch ligand Dll4 in tumor and endothelial cells in ovarian cancer. Cancer Res. 2011;71(18):6030–6039. doi: 10.1158/0008-5472.CAN-10-2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lahat G, et al. Vimentin is a novel anti-cancer therapeutic target; insights from in vitro and in vivo mice xenograft studies. PLoS One. 2010;5(4):e10105. doi: 10.1371/journal.pone.0010105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Landen CN, et al. Efficacy and antivascular effects of EphA2 reduction with an agonistic antibody in ovarian cancer. J Natl Cancer Inst. 2006;98(21):1558–1570. doi: 10.1093/jnci/djj414. [DOI] [PubMed] [Google Scholar]
- 46.Vasey PA. Resistance to chemotherapy in advanced ovarian cancer: mechanisms and current strategies. Br J Cancer. 2003;89(suppl 3):S23–S28. doi: 10.1038/sj.bjc.6601497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langley RR, Ramirez KM, Tsan RZ, Van Arsdall M, Nilsson MB, Fidler IJ. Tissue-specific microvascular endothelial cell lines from H-2K(b)-tsA58 mice for studies of angiogenesis and metastasis. Cancer Res. 2003;63(11):2971–2976. [PubMed] [Google Scholar]
- 48.Lu C, et al. Regulation of tumor angiogenesis by EZH2. Cancer Cell. 2010;18(2):185–197. doi: 10.1016/j.ccr.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Waugh MG, Chu KM, Clayton EL, Minogue S, Hsuan JJ. Detergent-free isolation and characterization of cholesterol-rich membrane domains from trans-Golgi network vesicles. J Lipid Res. 2011;52(3):582–589. doi: 10.1194/jlr.D012807. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.