

Abstract
Many neurodegenerative disorders, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and frontotemporal dementia, are proteinopathies that are associated with the aggregation and accumulation of misfolded proteins. While remarkable progress has been made in understanding the triggers of these conditions, several challenges have hampered the translation of preclinical therapies targeting pathways downstream of the initiating proteinopathies. Clinical trials in symptomatic patients using therapies directed toward initiating trigger events have met with little success, prompting concerns that such therapeutics may be of limited efficacy when used in advanced stages of the disease rather than as prophylactics. Herein, we discuss gaps in our understanding of the pathological processes downstream of the trigger and potential strategies to identify common features of the downstream degenerative cascade in multiple CNS proteinopathies, which could potentially lead to the development of common therapeutic targets for multiple disorders.
Introduction
Multiple lines of evidence show that many degenerative CNS disorders are proteinopathies (1–3). Pivotal support for this assertion has come from two and half decades of genetic and biochemical studies, including the identification of gene mutations that cause various neurodegenerative diseases as well as pathological and experimental studies demonstrating that these mutations enhance the propensity of the encoded protein to aggregate and accumulate inside various CNS cells, outside of cells, or both (Table 1). In other instances, genetic alterations, such as duplication or triplication of a gene (e.g., the amyloid β [Aβ] precursor protein [APP] gene in Alzheimer’s disease [AD] and the α-synuclein [SNCA] gene in Parkinson’s disease [PD]), do not produce an altered protein but simply result in increased levels that lead to abnormal protein aggregation and accumulation (4, 5). In a third scenario, as observed with progranulin (GRN) mutations that cause frontotemporal dementia, haploinsufficiency at the GRN locus results in aggregation and aberrant localization of the TAR DNA binding protein 43 (TDP-43) but not the mutant GRN-encoded protein (6, 7). Accumulated proteins are often deposited as amyloid or amyloid-like structures, but in recent years there has been an increasing focus on the smaller more soluble aggregates, generically referred to as oligomers, whose formation can also be enhanced by mutation or overexpression (8, 9). The identification of mutant genes linked to neurodegenerative diseases not only provided concrete evidence for causality, but also enabled the generation of transgenic rodent models of these diseases (Figure 1). As there are few naturally occurring animal models of human neurodegenerative diseases, the transgenic models have proven to be essential tools for understanding pathogenesis and also developing novel therapies (10). Though many neurodegenerative disorders are solely heritable disorders, several of the more common disorders, such as AD and PD, have more complex etiologies, with both familial and sporadic forms. However, the overall similarities in pathology and clinical presentations between sporadic and familial forms indicate that the pathological cascades are likely to be more conserved than disparate.
Table 1.
CNS proteinopathies: proteins, genes, and pathologies
Figure 1. Modeling neurodegenerative proteinopathies in transgenic rodents has provided preclinical disease models that support therapeutic discovery.
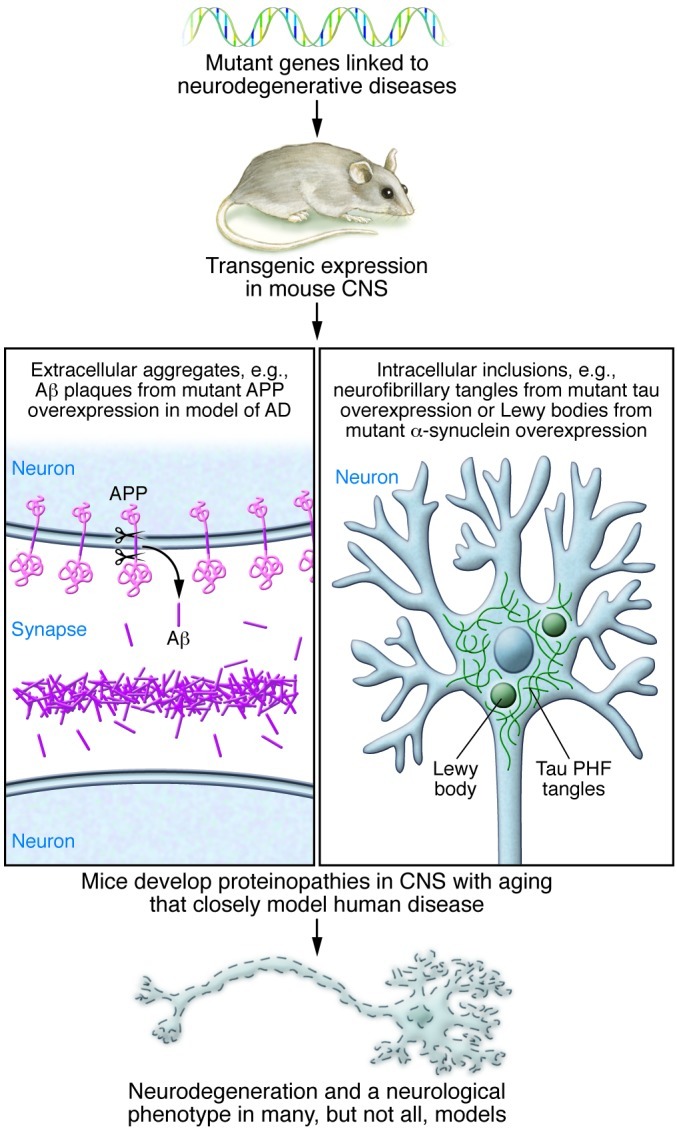
For most human neurodegenerative diseases there are not naturally occurring animal models. By expressing mutant genes associated with various human neurodegenerative diseases, it has been possible to develop models that often are excellent phenocopies of the CNS proteinopathy associated with the human disease. In many cases, but not all, these models also show neurodegenerative phenotypes. The development of transgenic rodent models has been critical for both mechanistic understanding of neurodegenerative diseases and also for the preclinical testing of novel therapeutics. PHF, paired helical filaments.
Most neurodegenerative diseases are characterized by a long prodromal phase, during which neuropathological and neurodegenerative changes precede the presentation of overt neurological symptoms. For example, before classic motor symptoms arise in PD, typically 75% or more of the dopaminergic input to the striatum has been lost (11, 12). Progressive loss of this input, reflecting dopaminergic cell death in the substantia nigra pars compacta, is thought to occur over decades. In AD, the triggering Aβ proteinopathy begins 15 or more years before the onset of dementia and plateaus 5–10 years before dementia is diagnosed (reviewed in ref. 13). The long delay between disease onset (defined as the time when the triggering proteinopathy is detectable) and clinical symptoms presents a dilemma in developing and testing treatments that target the proteinopathy. In the current paradigm for clinical development, novel, trigger-targeting therapeutics are typically tested in symptomatic patients. While such trigger-targeting therapies might be extremely effective if tested as prophylactics or even in the early prodromal phases, they can be predicted to have little or no efficacy in symptomatic patients (13). There is a growing recognition that this therapeutic development paradigm may limit our ability to demonstrate efficacy of novel disease-modifying therapies, which has resulted in some shift toward trials in prodromal disease states in AD (14). However, the current road map for developing disease-modifying therapies for most neurodegenerative disease would still require an initial therapeutic trial in symptomatic individuals and then a series of trials progressively targeting earlier stages of disease. In this scenario, the time lines from initial human trials to definitive testing of a therapy in an optimal early intervention or primary prevention trial could span multiple decades.
Though we all hope that the barriers to conducting true primary prevention studies with trigger-targeting therapies will, over time, fall and that such therapies will be efficacious, the time lines, along with the general challenge of developing and testing these therapies, raise serious questions about whether we can wage a successful campaign to treat or prevent these devastating CNS disorders. This is especially true if we only focus on trigger-targeting therapies. Because of the extended time lines of current therapeutic development paradigms, a generation or more will develop these devastating disorders. We believe that it is imperative for the field to bolster efforts to identify therapies that might work at later disease stages. Although the clinical and pathological phenotypes and the initiating proteinopathies that trigger various neurodegenerative diseases are distinct, it is our premise that (a) there are more similarities than differences in the mechanisms downstream of various triggering proteinopathies that drive neurodegeneration and (b) an understanding of these similarities and key differences might lead to novel therapeutic strategies that could lead to more effective symptomatic therapies. Herein, we discuss how an integrated approach to understanding CNS proteinopathies and therapeutic target identification, with a focus on mechanisms of disease spread within the brain, can be achieved by looking laterally across a spectrum of diseases to understand common pathological mechanisms downstream of the triggering proteinopathy.
How do proteinopathies spread?
Prionoid mechanisms?
Amyloid formation is a concentration-dependent, seeded, polymerization reaction (15, 16). In this reaction, formation of an initial assembly, referred to as a nucleation event or seed, catalyzes the conversion of the normal protein into the pathological amyloid state. Once seeded, growth of amyloid and amyloid-like structures is typically exponential, resulting in rapid formation of macromolecular structures that appear as intracellular inclusions or extracellular deposits used to pathologically define the disease. Building on seminal studies in prion diseases, in which the data unequivocally demonstrate that conversion of the normal cellular prion protein (PrPc) into the abnormal disease-causing prion (PrPsc) results in pathology spread within the infected CNS and enables disease transmissions (17, 18), there is now abundant evidence that many CNS proteinopathies can spread via a prion-like mechanism (reviewed in refs. 19–21). Indeed, this evidence has been used to support human pathological studies that suggest that neurofibrillary tau pathology spreads into the neocortex in AD (22) and α-synuclein pathology spreads into the brain from enteric nerves in PD (23, 24). However, unlike prion disease, there is no evidence that these and other human CNS proteinopathies are transmissible, and, thus, they have been termed by some as prionoids (19).
Despite the emergence of the prionoid hypothesis to explain proteinopathy spread within the brain, at present, there are many unanswered questions regarding the importance and relevance of prionoid mechanism in most human CNS proteinopathies. Answering these questions will enhance our understanding of disease progression and provide insight into the therapeutic tractability of targeting this pathway. One of the more elusive challenges for the field has been to define a seed. Though it is hypothesized that seeds contain structurally altered forms of the normal proteins, a precise structural understanding is lacking (25). Even for prions and Aβ, the presence of seeds is empirically defined by showing that some extract or preaggregated solution of the purified protein can catalyze aggregate formation, without a nucleation or lag phase. Given the likelihood that seeds are present in trace quantities in initial disease states, the seeds theoretically represent attractive, albeit elusive, targets.
A second perplexing question for intracellular proteinopathies relates to how cell-to-cell transmission occurs. Proteins implicated in intracellular CNS proteinopathies are largely localized to the cytoplasm, nucleus, or part of the scaffolding network of the cell, but there is growing evidence that these proteins can be secreted and are also present at low levels in interstitial and cerebrospinal fluid (19, 20, 26, 27). Proteins such as tau and α-synuclein may be secreted in exosomes and containment within exosomes might enhance cellular uptake, but the contribution of this pathway in vivo remains unclear (28–30). Furthermore, there are little data addressing (a) whether aggregated proteins are preferentially released, (b) whether secretion of these proteins has a normal physiologic role or is a disposal mechanism, or (c) whether the secreted proteins present in interstitial fluid or cerebrospinal fluid contain seeds capable of inducing pathology. Notably, the recognition that tau and α-synuclein aggregates may be secreted already provides one explanation for the apparent preclinical efficacy of immunotherapy targeting these proteins, and further elucidation of the mechanism of cell-to-cell transmission may provide a basis for novel therapies (31, 32).
Another unknown relates to the difference between prionoid and prion mechanisms. By definition, prions are transmissible; prionoids are not (33). Though some experiments raise the specter that certain proteinopathies could be transmitted from one genetically manipulated mouse to another, there is no human data to support human-to-human transmission of any CNS proteinopathy other than prions. So why are PrPsc prions uniquely transmissible? Indeed, many of the normal proteins involved in CNS proteinopathies are quite easily converted into aggregates that can behave as seeds, in contrast to PrPc (17). Speculatively, this difference could be explained by the kinetics of the templating reaction that follows seeding rather than the initial kinetic barriers to conversion. De novo conversion of PrPc into PrPsc is extremely challenging, but there is evidence that PrPsc may rapidly convert PrPc — in the order of minutes or seconds (34). In contrast, it is quite easy to nucleate Aβ, but the subsequent templating still takes hours to days to reach completion (15). Differences in peripheral expression levels might also play a role; if the normal protein is not present at sufficient levels to support templating, then the disease will not be transmitted peripherally. A final issue may relate to immunogenicity. Perhaps, in the periphery, PrPsc evades the immune system, whereas prionoids do not. If a seed or aggregate is easily recognized by the immune systems, a formidable barrier to transmission would exist.
A final question that serves as an excellent reminder that other mechanisms may account for the apparent spreading of pathology is whether spreading and cell-to-cell transmission of pathology can occur under physiologic levels of protein expression. The vast majority of data generated to date supporting prionoid mechanisms for tau, Aβ, superoxide dismutase-1 (SOD1), TDP-43, and α-synuclein use cell or animal models that have substantial overexpression of the protein and exogenous administration of the seed (21, 35–43). Whether pathology spread can occur under physiological expression levels is not as clear. Except for a recent study of α-synuclein seeding in nontransgenic mice (44), in most other instances induction of pathology is quite limited in the absence of overexpression (45–47). Furthermore, human studies, such as those regarding pathology in transplanted neurons in human patients with PD, cannot exclude other mechanisms such as those discussed below (48, 49). Though the physiologic relevance remains to be definitively established, the ability to rapidly induce seeding of pathological inclusions that phenocopy human proteinopathies is a major advance for the field. Indeed, armed with such models, it should be feasible to not only dissect out the consequences of inclusion formation in various cell types, but also to screen for both genetic and pharmacologic modifiers of pathology. Collectively, such studies may reveal novel strategies for therapy.
Prionoids as danger-associated molecular patterns that propagate via a toxic environment.
One of the hallmarks of a proteinopathy is that the proteins that accumulate are either alternatively folded or misfolded and are found in a substantial, ordered assembly. Thus, there is significant potential for recognition of repetitive, pathological, conformational epitopes in the aggregate as non-self antigens (50, 51). Prionoid self-protein aggregates represent what are referred to immunologically as danger-associated molecular patterns (DAMPs) that are capable of inducing a robust immune response (52). Notably, a large number of studies show that prionoids associated with CNS proteinopathies, when applied exogenously to glial cells, activate innate immunity through pattern recognition receptors and induce a proinflammatory response (50, 53). Furthermore, there is some evidence that the resulting response to the DAMPs could modify the protein aggregate via various posttranslational modifications, such nitration, oxidation, or proteolysis, enhancing toxicity or promoting additional aggregation (54–56). Though studies of Aβ aggregates acting as DAMPs have until recently dominated this area of investigation, the finding that intracellular protein aggregates can be secreted provides a mechanism whereby even tau, α-synuclein, and other intracellular aggregates could activate the innate immune system upon secretion (57–59). Indeed, recent identification of AD risk alleles within the triggering receptor expressed on monocytes 2 (TREM2) gene highlight the potential relevance of this pathway to neurodegenerative disease (60, 61).
It is likely that innate immune activation can have positive or negative effects on proteostasis, behavior, and neurodegeneration, with the balance between positive and negative effects dependent on the nature, timing, duration, and strength of the specific signals. For example, there are conflicting data regarding the relationship between alterations in innate immune activation states and effects on extracellular Aβ accumulation and behavior (53, 62–64). There are more consistent data that indicate that proinflammatory stimuli may promote tau and α-synuclein pathology but also many fewer studies in this area (53). For example, LPS and various other proinflammatory stimuli have been shown to induce tau and α-synuclein pathology (65–67). Thus, at least in tau- and α-synuclein-opathies, there is evidence that a proinflammatory neurotoxic environment could induce or promote spread of pathology. Extracellular aggregates acting as both DAMPs and “seeds” may set off a vicious cycle of aggregate secretion, inflammation, and reseeding that propagates pathology. Inflammatory factors may enhance cell permeability, resulting in enhanced remodeling or destruction of synapses and other cellular processes. Further, such factors may lead to increased levels of extracellular aggregates, seeds, and other types of DAMPs (cell debris, nucleic acids, etc.) that in turn trigger more inflammation and reinforce this positive feedback loop (Figure 2).
Figure 2. Schematic of mechanism of possible spread of neurodegenerative proteinopathies and contribution to cellular demise.

In this scheme, initiation of a proteinopathy can trigger a series of events that illicit feedback that contributes to spread of pathology and cellular demise. Danger signals might include DAMPs but also other signals indicative of cellular stress (ATP release, expression of MHC, etc.). Although there is strong evidence that microglia (and possibly astrocytes) may secrete neurotoxic factors, these factors have not been definitively identified in human neurodegenerative proteinopathies. Although most in the field have focused on mechanisms of neuronal decline, it is also important to consider the possibility that proteinopathies might result in functional decline and death of other CNS cells, including microglia, astrocytes, and oligodendrocytes (not shown). Indeed, in multiple system atrophy, oligodendrocytes are the primary cell affected by α-synuclein inclusions (97), and there is evidence for microglial dystrophy in AD (98).
An area that has received much less attention is whether cytoplasmic or organelle-bound intracellular aggregates can also act as DAMPs and activate intracellular immune pathways. Both fibrillar and oligomeric assemblies of proteins resemble intracellular pathogens. As there are established links among intracellular innate immunity, autophagic pathways, and the induction of other stress responses, further investigation of the action of intracellular protein aggregates as DAMPs might provide novel insights into how these protein aggregates alter cellular function (68–70). Extending this concept further, many of the responses of neurons to intracellular proteinopathies are highly similar to responses observed following sublytic viral infection of a neuron — including both induction of various elimination signals and general suppression of protein synthesis pathways (71). Furthermore, evidence from ALS models suggests that expression of mutant SOD1 in astrocytes and microglia plays a key role in mediating disease progression, implicating some role for these aggregates in activating intrinsic pathways that may contribute to neurotoxicity (72).
Gliosis and altered CNS immune activation states have long been recognized as an invariant accompanying feature of CNS proteinopathies, but the role of altered innate immune activation remains poorly understood (53). Though epidemiological studies have suggested that antiinflammatory strategies might be useful in AD, PD, and ALS, testing of antiinflammatory therapies in various disease states has not shown much promise to date (73, 74). However, as with therapies targeting the triggering proteinopathy, there is some concern that these therapies may have been tested in disease states that are too far advanced (13). Despite this lack of success to date, the notion that the immune system could be therapeutically harnessed to clear the underlying proteinopathy is attractive. In this light, we would simply point out that coordinated innate immune activation can clear a virus from the brain (71, 75). Given the evidence for prionoid- and DAMP-like properties of these protein aggregates, one might hypothesize that appropriate activation of the innate immune system could be used to restore normal proteostasis.
As with prionoid-like mechanisms, more insight is needed in a number of areas to fully comprehend the role of innate immunity in CNS proteinopathies. First, we need a better understanding of the temporal sequence of innate immune activation in the brain, and even the periphery, during disease progression. Such immunophenotyping might not only identify novel therapeutic targets but also novel biomarkers for disease. Indeed, a recent study in SOD1 mutant mice and human patients with ALS illustrates how detailed temporal immunophenotyping can inform both biomarker and therapeutic development (76). Second, there needs to be a more thorough understanding of the pattern recognition receptors activated by prionoids as well as the signaling cascades induced (77). Such studies should not only focus on glia as the resident immune cells of the brain, but also explore whether neurons activate innate immune pathways in response to prionoid aggregates. Third, there needs to be a much more intensive study of various protein aggregates acting as DAMPs. At this time, it is not clear whether all protein aggregates as well as the various types of assemblies formed act as DAMPs and, if they do, whether they behave in similar or distinct fashions. Finally, it will be important, both conceptually and from a therapeutic development perspective, to establish the relative contribution of prionoid spread compared with that of induction of the toxic environment for all of the CNS proteinopathies.
Spread via intrinsic disruptions of proteostatic mechanisms?
The proteostasis network is a concept used to define the myriad of activities and functions that work in concert to maintain the proteome (78). A major conceptual premise of the proteostasis network is that the system is tightly balanced and regulated to optimize efficient use of cellular resources (79). Components of the proteostasis network (e.g., protein synthesis, protein chaperones, and protein degradation activities) are proposed to be present in sufficient, but not excess, levels; insults that diminish or burden the function of one or more elements of the network could create a condition of insufficiency and an environment that is unable to prevent the accumulation of misfolded proteins. Nonneuronal cells may counteract this insult by an upregulation of chaperones (for example, heat-shock response). However, aspects of this response are largely absent in the CNS of neurodegenerative disease models (80, 81). The proteostasis network is often conceptualized only as the protein machinery and intracellular compartments that assist in the folding of intracellular proteins, recognize inappropriately folded or modified proteins, and target them for degradation via the proteosome or autophagic pathways (82, 83). Given the role of extracellular clearance pathways and innate immunity in mediating detection and removal of protein DAMPs, we believe that it may be appropriate to more critically consider the role of the immune system in proteostasis. Nevertheless, proteostasis provides a conceptual framework to explain not only the origin of some of the pathologic features in neurodegenerative disease, but possibly both spread of pathology and cellular demise.
Age-related disruptions of the proteostasis network have often been invoked to explain why many CNS proteinopathies are late-onset diseases and some show dramatic, continuous increases in prevalence with age (80, 83). A number of elegant studies in model organisms link various pathways that perturb the proteostasis network to both life span and predisposition to develop a proteinopathy (84). But, it has been challenging to definitively identify any aging-related changes within the proteostasis networks that clearly trigger a CNS proteinopathy in mammalian model, let alone in humans (84). Furthermore, genetic alterations, such as triplet expansion in Huntington’s disease and various spinocerebellar ataxias, more aggressive presenilin mutations associated with high levels of Aβ42 production, and increased copy number of normal α-synuclein, all demonstrate that intrinsic factors that promote self-aggregation of proteins can trump any aging effects, causing disease in early life. Thus, triggering events in most proteinopathies may reflect aging-related disturbances in proteostasis but may simply be stochastic in nature.
Ultimately, the presence of the proteinopathy demonstrates a failure of the proteostasis network to handle the altered protein. This intrinsic failure can the set off the cycle illustrated in Figure 2 that promotes spread and induces a toxic environment. Indeed, even for prion disease, the vast majority of the cases are not caused by exogenously transmitted prions but are sporadic cases or arise from genetic mutations that predispose PrPc to intrinsically convert to PrpPsc. In either case, the accumulation of one protein may then enhance the propensity of other aggregation-prone, metastable proteins to aggregate by depleting chaperones required for proteostasis. If such disruptions occur, then one might predict the consequence would be induction of multiple proteinopathies. Indeed, there is a great deal of pathological evidence to suggest that many CNS proteinopathies are, at least at end stage, mixed. AD, for example, is characterized not only by accumulation of Aβ and tau pathology but, in a subset of cases, also α-synuclein and TDP-43 pathology (see Table 1 and Figure 3). In addition, studies in model organisms support the idea that an initiating proteinopathy can trigger induction of other proteinopathies (80, 85). However, it remains an open question as to why various secondary proteinopathies are induced to varying extents in AD and other neurodegenerative diseases? An alternative explanation for these observations is that cross-seeding via a prionoid-like mechanisms could result in one proteinopathy triggering another. For example, “amyloidogenic” intermediates or oligomers formed from different proteins may share sufficient structural homology to cross-seed each other into the amyloid pathway (39, 86, 87). Curiously, the intracellular proteins predominantly do not make heterogeneous inclusions; individual proteins accumulate in distinct inclusions (88). Further, some in vitro studies show that many amyloidogenic proteins prefer to form homopolymers (39, 88). On the surface, such an observation argues against a cross-seeding mechanism. However, there is ample evidence for cross-seeding in experimental systems, and it appears that once cross-seeding occurs, subsequent templating preferentially results in aggregation of each distinct protein. Therefore, structurally homologous intermediates may be involved in cross-seeding, but most proteins are kinetically inclined to assemble as homopolymers, which reflects the unique primary sequence characteristic of each protein.
Figure 3. Schematic of the interrelated neurodegenerative proteinopathies.

Diseases are organized in color blocks that indicate their primary proteinaceous aggregate. AD has primary proteinaceous aggregates of both Aβ (yellow) and tau (red) and is therefore designated orange. Diseases are connected to proteinaceous aggregates that can be observed in at least some cases of the disease with lines. AGD, argyrophilic grain disease; CBD, corticobasal degeneration; DLB, dementia with Lewy bodies; FTD, frontotemporal dementia; HD, Huntington’s disease; MSA, multiple system atrophy; Perry synd., Perry syndrome; PDC, parkinsonism-dementia complex; PiD, Pick’s disease; PSP, progressive supranuclear palsy; αSyn, α-synuclein.
Irrespective of the role of disruptions in the proteostasis networks in disease induction and spread, the notion that one can treat CNS proteinopathies by augmenting the proteostasis network is highly intriguing. One might envision that augmenting chaperone systems or clearance pathways might be applicable to multiple CNS proteinopathies, especially those involving intracellular proteins. Again, several seminal studies suggest the feasibility of such an approach, but the long-term consequences of perturbing such networks in humans are largely unknown (89–93).
Thinking laterally to understanding functional demise in CNS proteinopathies
A curious observation that arises from looking across the spectrum of research conducted in various neurodegenerative diseases is how the investigators with different primary disease foci have prioritized investigation in different ways. For example, a major focus within the AD field has been an attempt to identify “the” toxic species of Aβ and now more recently “the” toxic species of tau (8, 94). In contrast, the ALS field has been less focused on the aggregate states of mutant SOD1 and more focused on defining various perturbations in cellular and animal models that link mutant SOD1 to cellular and functional demise as well the contribution of various cell types to disease (72). Despite the availability of what are perhaps the best animal models, in terms of an authentically phenocopied human prion disease, prion researchers have focused on issues such as attempting to define the still elusive structure of the seed and more esoteric concept of prion strains (19). Until recently, the PD field has focused on the selective vulnerability of dopaminergic neurons, despite the evidence that these neurons are neither the most vulnerable nor the only vulnerable cells within the brain (95, 96). Although there are good reasons for the perpetuation of these long-standing research foci, the unmet medical need and the lack of proven disease-modifying therapies for any neurodegenerative proteinopathy indicate that innovative strategies are needed. Exploring commonalties among these disorders may provide the critical insights needed to accelerate therapeutic discovery. As highlighted herein, the recent efforts to understand how various proteinopathies spread in the brain reveal a number of potential and unexpected commonalties as well as important differences, thus, illuminating the existing gaps in our knowledge. Perhaps, by thinking laterally, we may be able to answer other critical question about degeneration and functional demise triggered by CNS proteinopathies, including addressing why various populations of neurons succumb to the underlying proteinopathy and which signaling cascades that are triggered by proteinopathies in neurons and other cells of the CNS result in cellular dystrophy and death. Indeed, more definitive answers to such questions will likely lead to the development of novel therapeutic strategies that might work in later disease stage and possibly in multiple disorders.
Acknowledgments
T.E. Golde, D.R. Borchelt, and J. Lewis are supported by grants from the NIH. T.E. Golde is also supported by the Ellison Medical Foundation, Thome Medical Foundation, and ALSA.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(5):1847–1855. doi:10.1172/JCI66029.
References
- 1.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(suppl):S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 2.Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med. 2004;10(10):1055–1063. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- 3.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 4.Singleton AB, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 5.Rovelet-Lecrux A, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38(1):24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 6.Baker M, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 7.Cruts M, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442(7105):920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 8.Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15(3):349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 9.Bitan G, Fradinger EA, Spring SM, Teplow DB. Neurotoxic protein oligomers — what you see is not always what you get. Amyloid. 2005;12(2):88–95. doi: 10.1080/13506120500106958. [DOI] [PubMed] [Google Scholar]
- 10.Gama Sosa MA, De Gasperi R, Elder GA. Modeling human neurodegenerative diseases in transgenic systems. Hum Genet. 2012;131(4):535–563. doi: 10.1007/s00439-011-1119-1. [DOI] [PubMed] [Google Scholar]
- 11.Pakkenberg B, Moller A, Gundersen HJ, Mouritzen Dam A, Pakkenberg H. The absolute number of nerve cells in substantia nigra in normal subjects and in patients with Parkinson’s disease estimated with an unbiased stereological method. J Neurol Neurosurg Psychiatry. 1991;54(1):30–33. doi: 10.1136/jnnp.54.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damier P, Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain. 1999;122(pt 8):1437–1448. doi: 10.1093/brain/122.8.1437. [DOI] [PubMed] [Google Scholar]
- 13.Golde TE, Schneider LS, Koo EH. Anti-abeta therapeutics in Alzheimer’s disease: the need for a paradigm shift. Neuron. 2011;69(2):203–213. doi: 10.1016/j.neuron.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Callaway E. Alzheimer’s drugs take a new tack. Nature. 2012;489(7414):13–14. doi: 10.1038/489013a. [DOI] [PubMed] [Google Scholar]
- 15.Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73(6):1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 16.Glenner GG. Amyloid deposits and amyloidosis. The beta-fibrilloses (first of two parts). N Engl J Med. 1980;302(23):1283–1292. doi: 10.1056/NEJM198006053022305. [DOI] [PubMed] [Google Scholar]
- 17.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95(23):13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prusiner SB, Scott MR. Genetics of prions. Annu Rev Genet. 1997;31:139–175. doi: 10.1146/annurev.genet.31.1.139. [DOI] [PubMed] [Google Scholar]
- 19.Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron. 2009;64(6):783–790. doi: 10.1016/j.neuron.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 20.Polymenidou M, Cleveland DW. Prion-like spread of protein aggregates in neurodegeneration. J Exp Med. 2012;209(5):889–893. doi: 10.1084/jem.20120741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol. 2011;70(4):532–540. doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18(4):351–357. doi: 10.1016/S0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- 23.Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett. 2006;396(1):67–72. doi: 10.1016/j.neulet.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 24.Braak H, et al. Pathology associated with sporadic Parkinson’s disease — where does it end? J Neural Transm Suppl. 2006;(70):89–97. doi: 10.1007/978-3-211-45295-0_15. [DOI] [PubMed] [Google Scholar]
- 25.Auer S, Dobson CM, Vendruscolo M, Maritan A. Self-templated nucleation in peptide and protein aggregation. Phys Rev Lett. 2008;101(25):258101. doi: 10.1103/PhysRevLett.101.258101. [DOI] [PubMed] [Google Scholar]
- 26.Yamada K, et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. 2011;31(37):13110–13117. doi: 10.1523/JNEUROSCI.2569-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee SJ. Origins and effects of extracellular alpha-synuclein: implications in Parkinson’s disease. J Mol Neurosci. 2008;34(1):17–22. doi: 10.1007/s12031-007-0012-9. [DOI] [PubMed] [Google Scholar]
- 28.Saman S, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. 2012;287(6):3842–3849. doi: 10.1074/jbc.M111.277061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Danzer KM, et al. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener. 2012;7:42. doi: 10.1186/1750-1326-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emmanouilidou E, et al. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30(20):6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sigurdsson EM. Tau-focused immunotherapy for Alzheimer’s disease and related tauopathies. Curr Alzheimer Res. 2009;6(5):446–450. doi: 10.2174/156720509789207930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bae EJ, et al. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J Neurosci. 2012;32(39):13454–13469. doi: 10.1523/JNEUROSCI.1292-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aguzzi A, O’Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 2010;9(3):237–248. doi: 10.1038/nrd3050. [DOI] [PubMed] [Google Scholar]
- 34.Goold R, et al. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat Commun. 2011;2:281. doi: 10.1038/ncomms1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012;209(5):975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clavaguera F, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11(7):909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284(19):12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286(17):15317–15331. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giasson BI, et al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300(5619):636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- 40.Desplats P, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106(31):13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hansen C, et al. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest. 2011;121(2):715–725. doi: 10.1172/JCI43366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chia R, Tattum MH, Jones S, Collinge J, Fisher EM, Jackson GS. Superoxide dismutase 1 and tgSOD1 mouse spinal cord seed fibrils, suggesting a propagative cell death mechanism in amyotrophic lateral sclerosis. PLoS One. 2010;5(5):e10627. doi: 10.1371/journal.pone.0010627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Munch C, O’Brien J, Bertolotti A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc Natl Acad Sci U S A. 2011;108(9):3548–3553. doi: 10.1073/pnas.1017275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luk KC, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338(6109):949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ridley RM, Baker HF, Windle CP, Cummings RM. Very long term studies of the seeding of beta-amyloidosis in primates. J Neural Transm. 2006;113(9):1243–1251. doi: 10.1007/s00702-005-0385-2. [DOI] [PubMed] [Google Scholar]
- 46.Baker HF, Ridley RM, Duchen LW, Crow TJ, Bruton CJ. Induction of beta (A4)-amyloid in primates by injection of Alzheimer’s disease brain homogenate. Comparison with transmission of spongiform encephalopathy. Mol Neurobiol. 1994;8(1):25–39. doi: 10.1007/BF02778005. [DOI] [PubMed] [Google Scholar]
- 47.Baker HF, Ridley RM, Duchen LW, Crow TJ, Bruton CJ. Evidence for the experimental transmission of cerebral beta-amyloidosis to primates. Int J Exp Pathol. 1993;74(5):441–454. doi: 10.1007/BF02778005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li JY, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14(5):501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 49.Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14(5):504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 50.Golde TE, Miller VM. Proteinopathy-induced neuronal senescence: a hypothesis for brain failure in Alzheimer’s and other neurodegenerative diseases. Alzheimers Res Ther. 2009;1(2):5. doi: 10.1186/alzrt5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T. Inflammation in Alzheimer’s disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol. 2009;87(3):181–194. doi: 10.1016/j.pneurobio.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 52.Rubartelli A, Lotze MT. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007;28(10):429–436. doi: 10.1016/j.it.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 53.Czirr E, Wyss-Coray T. The immunology of neurodegeneration. J Clin Invest. 2012;122(4):1156–1163. doi: 10.1172/JCI58656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kummer MP, et al. Nitration of tyrosine 10 critically enhances amyloid beta aggregation and plaque formation. Neuron. 2011;71(5):833–844. doi: 10.1016/j.neuron.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 55.Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S. Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3(pE), in senile plaques. Neuron. 1995;14(2):457–466. doi: 10.1016/0896-6273(95)90301-1. [DOI] [PubMed] [Google Scholar]
- 56.Wang Y, Garg S, Mandelkow EM, Mandelkow E. Proteolytic processing of tau. Biochem Soc Trans. 2010;38(4):955–961. doi: 10.1042/BST0380955. [DOI] [PubMed] [Google Scholar]
- 57.Tukel C, Wilson RP, Nishimori JH, Pezeshki M, Chromy BA, Baumler AJ. Responses to amyloids of microbial and host origin are mediated through toll-like receptor 2. Cell Host Microbe. 2009;6(1):45–53. doi: 10.1016/j.chom.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Béraud D, Maguire-Zeiss KA. Misfolded alpha-synuclein and Toll-like receptors: therapeutic targets for Parkinson’s disease. Parkinsonism Relat Disord. 2012;18(suppl 1):S17–S20. doi: 10.1016/S1353-8020(11)70008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fellner L, et al. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia. 2013;61(3):349–360. doi: 10.1002/glia.22437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guerreiro R, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jones B. Alzheimer disease: TREM2 linked to late-onset AD. Nat Rev Neurol. 2012;9(1):5. doi: 10.1038/nrneurol.2012.254. [DOI] [PubMed] [Google Scholar]
- 62.Chakrabarty P, et al. Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010;24(2):548–559. doi: 10.1096/fj.09-141754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chakrabarty P, Tianbai L, Herring A, Ceballos-Diaz C, Das P, Golde TE. Hippocampal expression of murine IL-4 results in exacerbation of amyloid deposition. Mol Neurodegener. 2012;7:36. doi: 10.1186/1750-1326-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee CY, Landreth GE. The role of microglia in amyloid clearance from the AD brain. J Neural Transm. 2010;117(8):949–960. doi: 10.1007/s00702-010-0433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gao HM, Zhang F, Zhou H, Kam W, Wilson B, Hong JS. Neuroinflammation and alpha-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson’s disease. Environ Health Perspect. 2011;119(6):807–814. doi: 10.1289/ehp.1003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee DC, et al. LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflammation. 2010;7:56. doi: 10.1186/1742-2094-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68(1):19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Martinelli C, Reichhart JM. Evolution and integration of innate immune systems from fruit flies to man: lessons and questions. J Endotoxin Res. 2005;11(4):243–248. doi: 10.1179/096805105X37411. [DOI] [PubMed] [Google Scholar]
- 69.Nallagatla SR, Toroney R, Bevilacqua PC. Regulation of innate immunity through RNA structure and the protein kinase PKR. Curr Opin Struct Biol. 2011;21(1):119–127. doi: 10.1016/j.sbi.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deretic V. Autophagy: an emerging immunological paradigm. J Immunol. 2012;189(1):15–20. doi: 10.4049/jimmunol.1102108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Griffin DE, Metcalf T. Clearance of virus infection from the CNS. Curr Opin Virol. 2011;1(3):216–221. doi: 10.1016/j.coviro.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187(6):761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rees K, et al. Non-steroidal anti-inflammatory drugs as disease-modifying agents for Parkinson’s disease: evidence from observational studies. Cochrane Database Syst Rev. 2011;(11):CD008454. doi: 10.1002/14651858.CD008454.pub2. [DOI] [PubMed] [Google Scholar]
- 74.Lleo A, Galea E, Sastre M. Molecular targets of non-steroidal anti-inflammatory drugs in neurodegenerative diseases. Cell Mol Life Sci. 2007;64(11):1403–1418. doi: 10.1007/s00018-007-6516-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ousman SS, Kubes P. Immune surveillance in the central nervous system. Nat Neurosci. 2012;15(8):1096–1101. doi: 10.1038/nn.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Butovsky O, et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest. 2012;122(9):3063–3087. doi: 10.1172/JCI62636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 78.Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475(7356):324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 79. Gidalevitz T, Prahlad V, Morimoto RI. The stress of protein misfolding: from single cells to multicellular organisms. Cold Spring Harb Perspect Biol. 2011. 3 6 ). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22(11):1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Voisine C, Pedersen JS, Morimoto RI. Chaperone networks: tipping the balance in protein folding diseases. Neurobiol Dis. 2010;40(1):12–20. doi: 10.1016/j.nbd.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6(4):352–361. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- 83.Kikis EA, Gidalevitz T, Morimoto RI. Protein homeostasis in models of aging and age-related conformational disease. Adv Exp Med Biol. 2010;694:138–159. doi: 10.1007/978-1-4419-7002-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dillin A, and Cohen E. Ageing and protein aggregation-mediated disorders: from invertebrates to mammals. Philos Trans R Soc Lond B Biol Sci. 2011;366(1561):94–98. doi: 10.1098/rstb.2010.0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311(5766):1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- 86.Lundmark K, Westermark GT, Olsen A, Westermark P. Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. Proc Natl Acad Sci U S A. 2005;102(17):6098–6102. doi: 10.1073/pnas.0501814102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.O’Nuallain B, Williams AD, Westermark P, Wetzel R. Seeding specificity in amyloid growth induced by heterologous fibrils. J Biol Chem. 2004;279(17):17490–17499. doi: 10.1074/jbc.M311300200. [DOI] [PubMed] [Google Scholar]
- 88.Lee VM, Giasson BI, Trojanowski JQ. More than just two peas in a pod: common amyloidogenic properties of tau and alpha-synuclein in neurodegenerative diseases. Trends Neurosci. 2004;27(3):129–134. doi: 10.1016/j.tins.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 89.Calamini B, et al. Small-molecule proteostasis regulators for protein conformational diseases. Nat Chem Biol. 2012;8(2):185–196. doi: 10.1038/nchembio.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee BH, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467(7312):179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cattaneo M, Dominici R, Cardano M, Diaferia G, Rovida E, Biunno I. Molecular chaperones as therapeutic targets to counteract proteostasis defects. J Cell Physiol. 2012;227(3):1226–1234. doi: 10.1002/jcp.22856. [DOI] [PubMed] [Google Scholar]
- 92.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319(5865):916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 93.Bulawa CE, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109(24):9629–9634. doi: 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ashe KH, Zahs KR. Probing the biology of Alzheimer’s disease in mice. Neuron. 2010;66(5):631–645. doi: 10.1016/j.neuron.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Thomas B, Beal MF. Parkinson’s disease. Hum Mol Genet. 2007. 16 Spec No. 2: R183 R194 10.1093/hmg/ddm159 [DOI] [PubMed] [Google Scholar]
- 96.Henchcliffe C, Severt WL. Disease modification in Parkinson’s disease. Drugs Aging. 2011;28(8):605–615. doi: 10.2165/11591320-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 97.Tu PH, et al. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann Neurol. 1998;44(3):415–422. doi: 10.1002/ana.410440324. [DOI] [PubMed] [Google Scholar]
- 98.Streit WJ. Microglial senescence: does the brain’s immune system have an expiration date? Trends Neurosci. 2006;29(9):506–510. doi: 10.1016/j.tins.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 99.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 100.Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Sci Transl Med. 2011;3(77):77sr1. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ghiso J, et al. Genetic alterations of the BRI2 gene: familial British and Danish dementias. Brain Pathol. 2006;16(1):71–79. doi: 10.1111/j.1750-3639.2006.tb00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dickson DW, et al. Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol. 2009;8(12):1150–1157. doi: 10.1016/S1474-4422(09)70238-8. [DOI] [PubMed] [Google Scholar]
- 103.Cookson MR. alpha-Synuclein and neuronal cell death. Mol Neurodegener. 2009;4:9. doi: 10.1186/1750-1326-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hardy J, Cai H, Cookson MR, Gwinn-Hardy K, Singleton A. Genetics of Parkinson’s disease and parkinsonism. Ann Neurol. 2006;60(4):389–98. doi: 10.1002/ana.21022. [DOI] [PubMed] [Google Scholar]
- 105.Martin I, Dawson VL, Dawson TM. Recent advances in the genetics of Parkinson’s disease. Annu Rev Genomics Hum Genet. 2011;12:301–325. doi: 10.1146/annurev-genom-082410-101440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Giasson BI, Van Deerlin VM. Mutations in LRRK2 as a cause of Parkinson’s disease. Neurosignals. 2008;16(1):99–105. doi: 10.1159/000109764. [DOI] [PubMed] [Google Scholar]
- 107.Samaranch L, et al. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain. 2010;133(pt 4):1128–1142. doi: 10.1093/brain/awq051. [DOI] [PubMed] [Google Scholar]
- 108.Paisan-Ruiz C, et al. Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging. 2012;33(4):814–823. doi: 10.1016/j.neurobiolaging.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012;11(11):986–998. doi: 10.1016/S1474-4422(12)70190-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chartier-Harlin MC, et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet. 2011;89(3):398–406. doi: 10.1016/j.ajhg.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Benvenga S. Conformational mutations in neuroserpin and familial dementias. Lancet. 2002;360(9346):1696. doi: 10.1016/S0140-6736(02)11626-6. [DOI] [PubMed] [Google Scholar]
- 112.Kwiatkowski TJ, Jr, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 113.Vance C, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 115.van Slegtenhorst M, Lewis J, Hutton M. The molecular genetics of the tauopathies. Exp Gerontol. 2000;35(4):461–471. doi: 10.1016/S0531-5565(00)00114-5. [DOI] [PubMed] [Google Scholar]
- 116.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95(13):7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hutton M, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 118.Hoglinger GU, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011;43(7):699–705. doi: 10.1038/ng.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 120.Kabashi E, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40(5):572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 121.Van Deerlin VM, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7(5):409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Deng HX, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477(7363):211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Maruyama H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465(7295):223–226. doi: 10.1038/nature08971. [DOI] [PubMed] [Google Scholar]