

Abstract
Heterozygous loss-of-function SMAD3 (Mothers against decapentaplegic homolog 3) mutations lead to aneurysm-osteoarthritis syndrome (AOS). In the present study, we found that mice lacking Smad3 had a vascular phenotype similar to AOS, marked by the progressive development of aneurysms. These aneurysms were associated with various pathological changes in transmural inflammatory cell infiltration. Bone marrow transplants from Smad3–/– mice induced aortitis and aortic root dilation in irradiated WT recipient mice. Transplantation of CD4+ T cells from Smad3–/– mice also induced aortitis in Smad3+/+ recipient mice, while depletion of CD4+ T cells in Smad3–/– mice reduced the infiltration of inflammatory cells in the aortic root. Furthermore, IFN-γ deficiency increased, while IL-17 deficiency decreased, disease severity in Smad3+/– mice. Cytokine secretion was measured using a cytokine quantibody array, and Smad3–/– CD4+ T cells secreted more GM-CSF than Smad3+/+ CD4+ T cells. GM-CSF induced CD11b+Gr-1+Ly-6Chi inflammatory monocyte accumulation in the aortic root, but administration of anti–GM-CSF mAb to Smad3–/– mice resulted in significantly less inflammation and dilation in the aortic root. We also identified a missense mutation (c.985A>G) in a family of thoracic aortic aneurysms. Intense inflammatory infiltration and GM-CSF expression was observed in aortas specimens of these patients, suggesting that GM-CSF is potentially involved in the development of AOS.
Introduction
Aortic aneurysm is a common cardiovascular illness that has a high mortality rate because of dissections and ruptures. Thoracic aortic aneurysms and dissections (TAAD) can be inherited in an autosomal dominant manner with variable clinical manifestations (1), such as Marfan syndrome (MFS), which is caused by FBN1 mutations (2), and Loeys-Dietz syndrome (LDS), which is caused by TGFBR1 or TGFBR2 mutations (3). TAAD can also be autosomal recessive, as in the case of cutis laxa type I (AR-CL), which is caused by FBLN4 mutations (4).
The TGF-β cytokine pathway is involved in aortic aneurysm formation (5, 6). TGF-β modulates proliferation and differentiation and is widely expressed in various cell types. In canonical signaling, TGF-β binds to the type II receptor, which connects to the type I receptor to form the TβRI/II complex. This complex phosphorylates receptor-activated Smad2 and Smad3, which then form a complex with Smad4, translocate to the nucleus, and regulate target gene transcription (7). In addition, TGF-β induces noncanonical pathways, including RhoA and MAPKs, which include ERK, JNK, and p38 MAPK (8–10).
Vascular tissue obtained from patients with thoracic aortic aneurysms at surgery or autopsy have enhanced TGF-β signaling, as demonstrated by nuclear accumulation of pSMAD2 in VSMCs and increased expression of connective tissue growth factor (CTGF), which is a TGF-β gene product (11). Moreover, in Marfan mice, the disease is attenuated or prevented by administering neutralizing anti–TGF-β antibodies or a noncanonical pathway inhibitor (12, 13). This study tests the hypothesis that different molecular mutations induce unique pathogenetic sequences to enhance TGF-β signaling (mainly by noncanonical pathways) and contribute to aneurysm formation.
However, some issues require further elucidation. First, there is no direct evidence demonstrating that aortic dilation is attenuated by TGF-β antagonism in other aortic aneurysm models. Second, most LDS-related TGF-βRI/II mutations are located in the intracellular receptor kinase domain and thus theoretically reduce TGF-β–mediated signaling. Furthermore, resistance to Ang-II–induced aneurysm formation in normocholesterolemic C57BL/6 mice is disrupted by systemic treatment with neutralizing anti–TGF-β antibodies (14). This is the first evidence, to our knowledge, of a link between the antiinflammatory properties of TGF-β and aneurysm disease progression. Indeed, examination of pathological specimens from patients afflicted with MFS revealed decreased inflammatory cell infiltration in the aortic wall, as manifested by a normal inflammatory cell response to increased TGF-β. These data suggest that TGF-β has biphasic roles and functions in a cell-type–dependent manner in aneurysm pathogenesis.
Recently, heterozygous loss-of-function SMAD3 mutations were shown to induce aneurysm-osteoarthritis syndrome (AOS), which is characterized by arterial aneurysms, arterial tortuosity, and osteoarthritis at a young age as well as by the paradoxical enhancement of aortic wall TGF-β signaling (15–18).
Here, we show that Smad3-deficient mice have progressive aging-induced aortic root and ascending aorta dilation and die from aneurysm rupture and aortic dissection. These aneurysms display various pathological changes in transmural inflammatory cell infiltration.
Results
Murine Smad3 deficiency causes aneurysm rupture or aortic dissection–induced death.
A previous report demonstrated that 50%–70% of Smad3–/– mice die 3 months after birth because of infections adjacent to the mucosal surface (19). The remaining mutant mice overcame infection and died suddenly after appearing healthy. To determine the cause of their unexplained death, we performed a necropsy on a Smad3–/– mouse that died suddenly at 103 days of age and found evidence of vascular compromise, with hemopericardium causing cardiac tamponade (Figure 1A). Dramatic ascending aortic dilation with an aortic diameter increase of at least 2-fold was observed in Smad3–/– mice compared with age- and sex-matched Smad3+/+ mice (Figure 1, A and B). The results from direct examination by necropsy of a group of mice that did not show signs of infection indicated that a large proportion (15/21) of the Smad3–/– mice died from a ruptured aneurysm at up to 8 months of age (Figure 1C). Serial aortic sectioning also revealed the dilation of aortic root and aortic dissection (Figure 1, D and E). Careful examination of the images exhibited inflammatory cell accumulation within the adventitia and medial infiltration (Figure 1, D–F) that was concurrent with medial SMC loss and focal, intense elastin degradation (Figure 1F). Immunohistochemistry demonstrated abundant CD4+ T cells, macrophages, and neutrophils in the vessel wall (Figure 1, G and H). CD19+ B cells, CD8+ T cells, and mast cells were rarely found (data not shown). Foam cells, which are cells that are derived from macrophages and cause atherosclerosis, were not observed. Thus, this Smad3–/– mouse model replicates clinical vascular aspects of AOS.
Figure 1. Smad3 deficiency induced death due to aneurysm rupture and aortic dissection in mice.

(A) Aneurysm rupture in a Smad3–/– mouse and a Smad3+/+ mouse at 103 days of age. Arrow indicates location of laceration. (B) Comparison of thoracic aorta with that of age- and sex-matched Smad3+/+ mice. Arrow indicates location of laceration. (C) Survival curves of WT (n = 22), Smad3+/– (n = 20), and Smad3–/– (n = 19) mice, which were grossly normal. (D) Representative images of H&E staining of transverse sections of the proximal ascending aorta from Smad3–/– mice dead from aneurysm rupture and normal age- and sex-matched Smad3+/+ mice. (E) H&E staining of transverse sections of the ascending aorta near the aortic arch from WT mice showing normal vascular structures and aneurysm dissection in the ascending aorta from Smad3–/– mice. adv, adventitia; m, media. Original magnification, ×40; ×400 (left insets); ×200 (right insets). (F) H&E staining, EVG staining, and IHC staining for α-SMA of proximal ascending aorta from Smad3–/– mice showed focal inflammatory cell infiltration and some regions of aorta tissue were relatively normal. Original magnification, ×40; ×400 (magnified panels). (G) Sections were serially immunostained for CD68, Gr-1, and CD4 to visualize monocytes/macrophages, neutrophils, and CD4+ T cells in the aortic tissues from age- and sex-matched Smad3–/– and Smad3+/+ mice. Original magnification, ×400. (H) Data are presented as mean ± SEM. **P < 0.001 Smad3–/– versus Smad3+/+ (Mann-Whitney test).
Smad3–/– mice undergo progressive aortic root and ascending aortic dilation and aneurysm-associated pathological changes.
We initiated a study to elucidate the mechanisms underlying this previously unrecognized vascular phenotype. At 2 weeks, 2 kinds of Smad3–/– mice were easily identified. Some mice were obviously smaller prior to weaning, and they developed wasting syndrome and had multifocal pyogenic abscesses. The other mice were grossly normal and no infection or inflammation was found in most of their organs (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI67356DS1). To eliminate possible negative influences on the result, we chose the Smad3–/– mice with normal appearance for the ensuing study. We first subjected a group of mice (n = 28) to Doppler ultrasound imaging and another group (n = 16) to direct examination by thoracotomy at 1, 2, 4, and 8 months of age. All of these Smad3–/– mice subjected to Doppler ultrasound imaging (28/28) underwent progressive aortic root dilation. The dilation became pronounced and was evident as early as 2 months of age (Figure 2, A and B). The aortic root diameter at 1 month was 1.481 ± 0.35 mm (n = 7) in the Smad3–/– mice versus 1.506 ± 0.28 mm in the Smad3+/+ mice (n = 9) (P = 0.075, not significant), and at 2 months, these diameters had increased to 1.971 ± 0.15 mm (n = 7) in the Smad3–/– mice versus 1.529 ± 0.229 mm (n = 9) in the Smad3+/+ mice, respectively (P ïιΤ 0.001; Figure 2, A and B). By 4 and 8 months of age, dilation or rupture began to occur in the ascending aorta of Smad3–/– mice, whereas the diameter of the ascending aorta in the Smad3–/– mice increased between 2 and 4 months of age. At 2 months of age, the diameter of the ascending aorta was 1.432 ± 0.146 mm (n = 7) in Smad3–/– mice versus 1.451 ± 0.218 mm in the Smad3+/+ mice (n = 9) (P = 0.092), and at 4 months, the diameter of the ascending aorta was 1.671 ± 0.313 mm (n = 7) in Smad3–/– mice versus 1.469 ± 0.029 mm (n = 9) in the Smad3+/+ mice (P ïιΤ 0.001; Figure 2, A and B). A detailed examination of heterozygous Smad3+/– mice revealed delayed aortic root dilation (Supplemental Figure 2), which indicates a gene dosage effect. We also found that the tendency for aneurysm rupture was not related to the aortic diameter. Observation of serial sections of the whole heart did not reveal defects in the cardiac outflow tract. Histological analysis demonstrated inflammatory cell infiltration with parallel worsening of aortic internal elastic lamina and lumen diameters (Figure 2C and Figure 3A). The dilated aortas were less translucent, which corresponded to an architectural change in the thickening vessel wall (Figure 3B) that generally spread to the right atrial root (Figure 2A). Immunostaining with mouse α-actin and Western blot analysis did not identify a difference at early stages (Figure 3C), but unbalanced SMC hyperplasia and aortic cross-sectional area reduction were observed at later stages (Figure 3D). Neovascularization plays an essential role in AAA formation; we found dense CD31+ cells at the Smad3–/– aortic root (Figure 3E). AOS patient aneurysms were characterized by a paradoxical enhancement of TGF-β signaling in the aortic wall (15). Therefore, we dynamically assessed the timing of TGF-β signaling and vascular distribution of the signals in Smad3+/+ and Smad3–/– mice. First, by immunohistochemical (IHC) staining, we detected the expression of phosphorylated Smad3 (p-Smad3) in the aortic root of Smad3+/+ mice and found that at 2 weeks of vascular growth, p-Smad3 was strongly positive while it was relatively weaker at 4 and 8 weeks (Supplemental Figure 4). These findings indirectly showed that TGF-β signals might play a role at an early stage of vascular growth. In fact, previous reports have demonstrated that absence of TGF-β on vessels might have an impact on the development of vessels (20). Nevertheless, absence of Smad3 molecules on vessels alone is not sufficient to cause obvious structural abnormality at an early stage of development. As demonstrated in our study, no evident dilation was found at the aortic root and the ascending aorta of mice that were 1 month old. Then we detected total LAP–TGF-β (Latency Associated Peptide–), p-Smad2, p-ERK1/2, and p-JNK1 in aortic roots and ascending aortas from mice at 1 month and 4 months old. We found that positive staining for LAP–TGF-β, p-Smad2, p-ERK1/2, and p-JNK1 was predominantly located in the infiltrating inflammatory cells at the aortic root of Smad3–/– mice and that no conspicuous positive staining was observed in relative intact SMCs of 1-month-old Smad3+/+ and Smad3–/– mice (Figure 4A). On the other hand, at the late stage (4 months), when aortas of Smad3–/– mice experienced structural changes, intense LAP–TGF-β, p-Smad2, p-ERK1/2, and p-JNK1 staining was found in the SMCs in dilated areas (the root and ascending section of aorta) (Figure 4A). Western blot analysis also showed upregulation of p-Smad1/5, p-Smad2, p-ERK1/2 and p-JNK1 in the proximal ascending aorta from Smad3–/– mice at 4 months of age (Figure 4B). Smad3 deficiency did not affect the expression of Smad2, ERK, and JNK in SMCs (Figure 4B). So we are inclined to believe that upregulated TGF-β in SMCs at the later stage might be a repairing mechanism of aorta in response to inflammatory damage and that the infiltration of inflammatory cells at the early stage was due to increased invasiveness of inflammatory cells caused by disordered signaling of TGF-β in the cells, while focal loss of SMCs might be caused by degradation as a result of intense local inflammation and breakage of aortic media layer due to the vascular stress.
Figure 2. Smad3–/– mice undergo progressive aortic root and ascending aorta dilation.

(A) Representative photographs and ultrasound imaging of the aortic root and ascending aorta in Smad3–/– mice at different ages. Arrows in the photographs of 2-month-old mice identify areas of neovascularization. (B) Aortic root and ascending aortic diameter, measured by echocardiography, at different ages in Smad3+/+ (n = 9/time points) and Smad3–/– (n = 7/time points) mice. *P < 0.01; **P < 0.001, Smad3–/– versus Smad3+/+ at the same age. (C) H&E staining showed inflammatory cell infiltration in the aortic roots and ascending aortas of Smad3–/– mice (n = 12/time points) at different ages. EVG staining showed medial elastin degradation in the aortic roots and ascending aortas of Smad3–/– mice (n = 11/time points) at different ages. Original magnification, ×200 (C).
Figure 3. Various pathological changes associated with aneurysms in Smad3–/– mice.

(A) Inflammatory cell infiltration and elastin degradation were analyzed according to grade indicated in Supplemental Figure 3. (B) Progression of thickening of the aortic media in Smad3–/– mice. *P < 0.01; **P < 0.001, Smad3–/– versus Smad3+/+ at the same age. (C) Sections from the aortic tissue of Smad3–/–mice (2 months) and Smad3+/+ mice were stained for α-SMA to show SMCs. Western blot analysis showed no difference expression of α-SMA at the early stage (2 months). P = 0.16, Smad3–/– versus Smad3+/+. (D) Unbalanced SMC hyperplasia and aortic cross-sectional area reduction appeared in sections from aortic tissue of 4-month-old Smad3–/– mice. (E) CD31+ microvessel contents in aortas from Smad3–/– and WT mice at 2 months. **P < 0.001, Smad3–/– versus Smad3+/+. Representative CD31 staining of aortas from the 2-month time point are shown in the right panels. Original magnification, ×200 (C, E); ×40 (D, upper left panel); ×400 (D, remaining panels).
Figure 4. Evidence for excessive TGF-β signaling in Smad3–/– mouse aortas.

(A) Immunostaining for LAP–TGF-β, p-Smad2, p-ERK1/2 and p-JNK1 on slides of aortic root and ascending aorta from Smad3+/+ and Smad3–/– mice aged 1 or 4 months. IHC analysis reveals that the positive staining of LAP–TGF-β, p-Smad2, p-Erk1/2, and p-JNK1 are predominantly located in the infiltrating inflammatory cells in the aortic root of 1-month-old Smad3–/– mice, while there are increased expression and nuclear translocation of p-Smad2, p-ERK1/2, and p-JNK1 in the aortic media of 4-month-old Smad3–/– mice. Original magnification, ×400. (B) Representative Western blot showing Smad1/5, Smad2, ERK1/2, JNK1, p-Smad1/5, p-Smad2, p-ERK1/2, and p-JNK1 levels in proximal ascending aortas from Smad3+/+ and Smad3–/– mice (1 and 4 months of age). The ratio between Smad1/5, Smad2, ERK1/2, JNK1, p-Smad1/5, p-Smad2, p-ERK1/2, and p-JNK1 to GAPDH levels is shown. *P < 0.01; **P < 0.001.
Aneurysms occur in the context of chronic inflammation.
We demonstrated that aortic dilation in Smad3–/– mice is initiated in the aortic root. Although fragmentation and degradation of the intact elastic lamina is found at late stages, visualization with the naked eye did not reveal obvious differences in the early stages (1 month); however, dense transmural inflammatory cells infiltrated the aortic root when elastic fiber integrity was changed (Figure 5A). Over a period of several months, this grade of inflammatory cell infiltration was associated with aortic root and ascending aorta dilation. Inflammation also developed in the coronary arteries, as manifested by varying degrees of stenosis/occlusion or dilation. The aortic valve was frequently infiltrated by monocytes in the Smad3–/– mice (Supplemental Figure 5). However, it remains unclear whether inflammation is a cause or a consequence of aortic dilation, and the specific inflammatory mediators that are involved in this process are also unknown.
Figure 5. Aneurysms occur in the context of chronic inflammation.

(A) Representative H&E staining of longitudinal and transverse sections of the ascending aorta from 1-month-old Smad3+/+ and Smad3–/– mouse showing inflammatory cell infiltration in the Smad3–/– aortic root and milder infiltration in that of Smad3+/+ mice. Original magnification, ×100; ×400 (magnified insets). (B) Severity of inflammatory cell infiltration, elastin degradation, and aortic root size in the chimeric mice. The aortic root of Smad3+/+ recipient mouse receiving BM cells from Smad3–/– mouse showed severe inflammatory cell infiltration, elastin degradation, and enlargement of aortic root compared with that of Smad3+/+ recipient mouse receiving BM cells from Smad3+/+ mouse. The aortic root of Smad3–/– mice showed significantly ameliorated pathological changes after receiving Smad3+/+ BM cells. *P < 0.01; **P < 0.001. (C) Representative H&E staining of transverse sections of the aortic root from chimeric mice. Of note, WT recipient mouse receiving BM cells from Smad3–/– mouse revealed a substantially increased accumulation of inflammatory cells within the adventitia and infiltration of inflammatory cells into the media. Original magnification, ×400.
To investigate the role of hematopoietic cells in aneurysm onset in Smad3–/– mice, we generated chimeras in which irradiated 6-week-old Smad3+/+ mice or Smad3–/– mice were reconstituted with Smad3–/– or Smad3+/+ BM after 3 months. The mice were subjected to Doppler ultrasound imaging, and the aortas were removed for histology. Although aortic root dilation and inflammatory and elastic lamina degradation were worse in Smad3–/– animals that had been reconstituted with Smad3–/– marrow, inflammation and elastic lamina degradation were suppressed in Smad3–/– mice that received Smad3+/+ marrow, which is consistent with a role for hematopoietic cells in aneurysm onset (Figure 5B). Furthermore, isolated Smad3–/– hematopoietic cells were sufficient to initiate spontaneous inflammation and aortic root dilation in Smad3+/+ littermates (Figure 5B). The aortic root in Smad3+/+ mice with Smad3–/– mouse marrow had similar transmural inflammatory characteristics and relatively mild dilation when compared with the aortic root in Smad3–/– mice that were reconstituted with Smad3–/– marrow (Figure 5C).
CD4+ T cells are responsible for aneurysm development.
Based on the results from the previous section, we decided to further investigate the relationship between inflammation and aneurysm development in this mouse model. A previous report demonstrated that mice lacking IL-1Ra have a similar spontaneous aortic root inflammation phenotype and that CD4+ T cells are responsible for aortitis development (21, 22). Disturbed TGF-β signaling is associated with autoimmunity (23). The observation of abundant CD4+ cell infiltration at sites of arterial inflammation in Smad3–/– mice also supports this notion. Thus, we examined the role of T cells in aortitis development by transplanting peripheral CD4+ T cells from Smad3–/– mice into Smad3+/+ littermates. T cell transplantation from Smad3–/– mice induced aortitis in Smad3+/+ littermates (Table 1), but not in Smad3+/+ mice that had received Smad3+/+ CD4+ T cells, which indicates that T cells are involved in aortitis development. To assess whether the depletion of CD4+ T cells in Smad3–/– mice would prevent the infiltration of inflammatory cells in the aortic root, we injected CD4-depleting mAb (GK1.5) at a dose of 0.1 mg on 3 days per week into Smad3–/– mice at 2 weeks of age, when the aorta was relatively normal. Six weeks later, the aortas were observed, and it was found that the infiltration of inflammatory cells was significantly reduced as compared with that in mice injected with control IgG (Figure 6, A and B), indicating that lack of CD4+ T cells in Smad3–/– mice might ameliorate the inflammation to a certain extent.
Table 1.
Aortitis in mice following CD4+ T cell transplantation
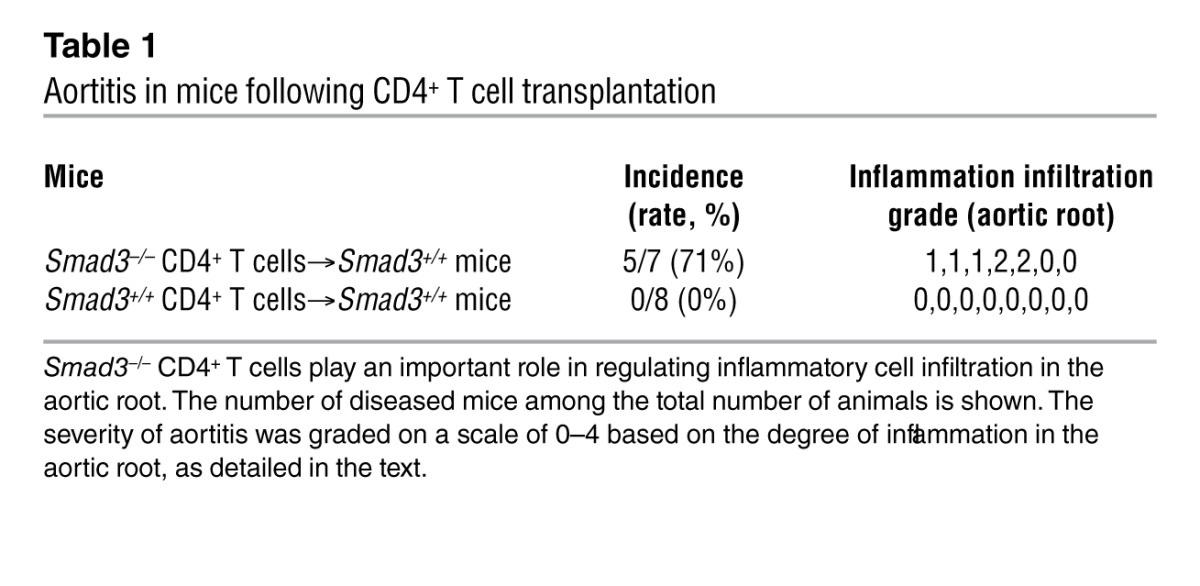
Figure 6. CD4+ T cell depletion inhibits inflammatory aneurysms in Smad3–/– mice.

(A) Two-week-old Smad3–/– mice were given CD4-depleting mAb (GK1.5), control IgG, or nothing. Four weeks later, changes in the aortic root were examined. *P < 0.01 versus control. (B) Representative images of H&E staining of transverse sections of aortic roots from mice from these 3 groups. Original magnification, ×200.
Distinct inflammation patterns may influence the clinical course of arterial disease, such as aneurysm or stenotic atherosclerotic lesions. Therefore, we employed a vascular transplantation model that reflects CD4+ T cell–mediated adaptive immunology. Vascular transplants from Balb/c donors to Smad3+/– hosts (that were crossed into a C57BL/6 background for at least 6 generations) resulted in profound aneurysm formation. The gross appearance (Figure 7, A and B) and histologic appearance (Figure 7, C–F) of transverse aortic allograft sections from Smad3+/+ or Smad3+/– littermate recipients 12 weeks after transplantation are depicted. Allografts in Smad3+/– hosts also demonstrated a markedly fragmented elastic lamina in the aortic media that was not observed in Smad3+/+ littermate recipient allografts (Figure 7, E and F), which means that infiltration of CD4+ T cells lacking Smad3 may induce aortic dilation.
Figure 7. Aortic allografts from Smad3+/– recipients showing aneurysm formation.

(A and B) Gross appearance of aortas from Smad3+/+ recipients and Smad3+/– littermate recipients 12 weeks after transplantation showing aneurysm formation in the latter. (C and D) H&E staining of transverse sections of aortic allograft from Smad3+/+ and Smad3+/– littermate recipients showing greater diameter in the latter than the former. (E and F) EVG staining showing markedly fragmented elastic lamina in the aortic media of transplant from Smad3+/–recipients, which was not observed in the allografts from Smad3+/+ littermate recipient mice. Original magnification, ×40; ×400 (magnified insets).
Smad3–/– CD4+T cells secrete GM-CSF, which is important in AOS.
Given that CD4+ T cells are responsible for aortitis development, we sought to determine which cells or inflammatory mediators are involved in this disease. Since CD4+ T cells are divided into Th1, Th2, Th17, Th22, and Th9 (24), and most studies focused only on Th1 (secreting IFN-γ) and Th17 (secreting IL-17), which are believed to predominantly contribute to the pathological changes in autoimmune and inflammatory aneurysm (25, 26), we selected IL-17 and IFN-γ for use in this study. First, we performed immunohistochemistry on our mouse samples and determined that IFN-γ and IL-17 were expressed in the inflammatory cells (Supplemental Figure 6A).Then, to investigate whether these cytokines play a role in our model, we crossed our mice into a C57BL/6 background for at least 6 generations and generated IFN-γ– and IL-17–deficient Smad3+/– mice. Both the IFN-γ– and the IL-17–deficient littermates grew normally and did not exhibit inflammation. Smad3+/– mice exhibited inflammation and progressive aortic root dilation at 8 months. Smad3+/–Ifng–/– mice exhibited increased inflammation and aortic dilation, whereas Smad3+/–Il17–/– mice were improved when compared with the Smad3+/– mice (Supplemental Figure 6B). CD4+ T cells initiated aortic root inflammation, and the loss of IFN-γ exacerbated this inflammation and aortic dilation; therefore, we examined CD4+ T cell–secreted cytokines. We isolated CD4+ T cells (Supplemental Figure 6C) and detected more activated phenotypes in Smad3–/– CD4+ T cells than in Smad3–/– CD4+ T cells. Then we used a 21-cytokine detection panel to survey cytokine secretion by CD4+ T cells in vitro under neutral priming conditions. We determined that T cells from Smad3–/– and Smad3–/– mice produced similar levels of Th1-related cytokines including IFN-γ and most of the Th-2–related cytokines (IL-4, IL-5, and IL-9) (Figure 8A). A significant increase was detected in IL-2, IL-6, IL-13, and GM-CSF in Smad3–/– CD4+ T cells (Figure 8A). IL-2 and IL-6 regulate T cell proliferation, which has been reported in Smad3–/– mice. IL-13 is an important Th2-related cytokine. We were interested in GM-CSF, which is a growth and differentiation factor for hematopoietic progenitor cells, but can also function as a proinflammatory mediator in a range of pathological conditions. GM-CSF is produced by a wide variety of cell types, including T cells. GM-CSF–deficient T cells have been shown to induce EAE (27), myocarditis (28), and arthritis (29). IL-1β or IFN-γ deficiency has also been shown to significantly induce GM-CSF expression (27, 30, 31), which may explain the disease severity observed in Smad3+/–Ifng–/– mice. Smad3–/– CD4+ T cells produced more GM-CSF than Smad3+/+ CD4+ T cells, which was confirmed by flow cytometry and immunofluorescence (Figure 8B). In splenocytes under neutral priming conditions, IFN-γ deficiency increased GM-CSF production by Smad3+/– CD4+ T cells, whereas IL-17 suppressed it (Supplemental Figure 6, D and E). We did not detect differences in other subsets (Supplemental Figure 7A). We also isolated CD4+ T cells from WT mice and investigated the role of the TGF-β/Smad3 axis in these cells upon activation or transformation under GM-CSF priming conditions, as described previously. Fewer transformed GM-CSF+ CD4+ T cells were obtained from WT mice in the presence of TGF-β and more transformed GM-CSF+ CD4+ T cells in the presence of Smad3 inhibitor (SIS3) (Supplemental Figure 7, B and C), as confirmed by the detection of GM-CSF in the supernatants (Supplemental Figure 7C). Given the evidence linking aortic root inflammation severity to GM-CSF, we administered anti–GM-CSF mAbs or control IgG to 2-week-old Smad3–/– mice and measured aortic root inflammation to determine whether GM-CSF neutralization inhibits inflammatory cell accumulation in the aortic root. We found that neutralizing GM-CSF with anti–GM-CSF mAbs for 6 weeks resulted in significantly less inflammation (Figure 8, C and D).
Figure 8. Smad3–/– CD4+ T cells secrete GM-CSF, which is an important cytokine in AOS.

(A) Analysis of cytokine secretion by CD4+ T cells from Smad3–/– and Smad3+/+ mice (n = 5) using a mouse cytokine antibody array. *P < 0.05; **P < 0.01. (B) Smad3–/– CD4+ T cells produced more GM-CSF than Smad3+/+ CD4+ T cells, which was confirmed by flow cytometry and immunofluorescence staining. This difference was analyzed by comparing the percentage of CD4+GM-CSF+ T cells or counting their number in a high-power field. **P < 0.01. HPF, high-power field. Original magnification, ×400. (C) One-month-old Smad3–/– mice were given anti–GM-CSF mAb, control IgG, or nothing. Four weeks later, changes in the aortic root were examined. **P < 0.01 versus control. (D) Representative images of H&E staining of transverse sections of the aortic roots from mice from these 3 groups. Original magnification, ×200.
Infiltrated cells were predominantly GM-CSF–regulated CD11b+Gr-1+cells.
As previously described, GM-CSF stimulates myeloid cell mobilization from the BM and increases the number of CD11b+Gr-1+ cells at the inflammatory site (32). Many tumor cell types lose TGF-β responsiveness and secrete GM-CSF, which recruits Gr-1+CD11b+ myeloid cells to promote metastasis (33). In Smad3–/– mice, the cells that accumulated in the aortic root were predominantly monocytes. To gain insight into whether these cells are regulated by GM-CSF, we used flow cytometry to analyze their phenotype. We first used a modified collagenase/elastase digestion method to dissociate the cellular aortic components for quantitative, multiparametric flow cytometry analysis (34). To isolate monocytes within the dissociated aortic cell population, cells were subjected to flow cytometry analysis by gating for CD11b expression (Figure 9A). The CD11b+ leukocytes were then gated for Gr-1 and Ly6C. We determined that approximately 60% of CD11b+ cells expressed Gr-1, and these cells could be divided into 2 distinct populations: CD11b+Gr-1+Ly-6Chi inflammatory monocytes (48.1% ± 3.6%) and CD11b+Gr-1+Ly-6Clo inflammatory neutrophils (8.1% ± 2.1%) (Figure 9A). Expression of CCR2 in the CD11b+Gr-1+Ly-6Chi inflammatory monocytes was confirmed by additional immunofluorescence staining (Figure 9B). Isolated aortic mononuclear cells were subjected to Wright-Giemsa staining, which revealed cells with a typical inflammatory monocyte shape (Figure 9B). To evaluate CD11b+Gr-1+ cell proliferation in situ, we employed immunohistochemistry and found that approximately 80% of the inflammatory cells were positive for Ki-67 (Figure 9B). GM-CSF promotes DC proliferation and activation; therefore, we analyzed CD11b+ cells for MHC-II and CD11c expression, and found that CD11b+ cells isolated from aortas of Smad3+/+ mice had lower MHC-II and CD11c expression than those from Smad3–/– mice (Figure 9A). Although CD11b+ cells were present, no Gr-1+ cells were detected in the Smad3+/+ mice aortic root (Figure 9A). These results demonstrate that GM-CSF may be involved in the recruitment of myeloid cells into the aortic root and the regulation of myeloid cell proliferation in situ. Tumor-bearing mice exhibited a high percentage of blood CD11b+Gr-1+ cells. A previous study using the same mouse model demonstrated elevated levels of blood neutrophils and monocytes; thus, we counted CD11b+Ly-6Chi cells in the BM, blood, spleen, and LNs. We found that the percentage and number of CD11b+Ly-6Chi cells in the BM, blood, and spleen were elevated in Smad3–/– mice compared with Smad3+/+ mice (Supplemental Figure 8, A and B). This increased cell number and concentration disrupted the splenic structure and increased the fraction of CD11b+ cells (Supplemental Figure 8C). Infusion of GM-CSF or SIS3 into WT mice significantly increased the number of CD11b+Ly-6Chi cells, and treatment with a GM-CSF antibody reversed this increase (Supplemental Figure 8D). We then isolated the inflammatory monocytes from the blood of Smad3+/+ mice by flow cytometry and cultured them with or without GM-CSF or M-CSF (Supplemental Figure 8E). M-CSF effectively induced the maturation of inflammatory monocytes and their transformation into fusiform wall-adherent cells and they no longer expressed Ly-6C (Supplemental Figure 8E). GM-CSF maintained the most traits of the cells and promoted their proliferation. Without addition of any cytokines, the inflammatory monocytes hardly proliferated (Supplemental Figure 8E). This finding indicates that Smad3-induced GM-CSF inhibition promotes the proliferation of CD11b+Ly-6Chi cells.
Figure 9. The infiltrated cells were predominantly CD11b+Gr-1+ cells, which were regulated by GM-CSF.

(A) Dissociated aortic cells were gated on CD11b; CD11b+ cells were gated on Gr-1 and Ly-6C, which contained CD11b+Gr-1+Ly-6Chi inflammatory monocytes (55.2% ± 4.9%, n = 5) and CD11b+Gr-1+Ly-6Cmid inflammatory granulocytes (7.1% ± 2.3%, n = 5). Histograms showed that the CD11b+Gr-1+Ly-6Chi cells expressed CCR2, which was confirmed by comparison to an isotype control. (B) Wright-Giemsa staining of separated mononuclear cells from aortic tissue revealed the typical inflammatory monocyte shape of these cells. Immunofluorescence staining for CCR2 confirmed the infiltrated cells were inflammatory monocytes. Ki-67 immunostaining of the aortic wall (1-month-old Smad3–/– mice). Approximately 80% of the inflammatory cells stained positive for Ki-67. Original magnification: ×400 (top), ×1000 (top, magnified inset); ×200 (middle), ×400 (middle, magnified inset); ×200 (bottom), ×400 (bottom, magnified inset).
Increased matrix-degrading activity in Smad3–/– mouse aortas and its abrogation by GM-CSF antibodies.
A previous study evaluated TGF-β–mediated suppression of MMPs in monocytes/macrophages and the role of high MMP expression and activity in monocytes/macrophages (14, 35). To evaluate the expression of MMPs in the aorta, we first used the IHC test, which revealed that, in monocytes/macrophages, the expression of MMP9 was stronger than that of MMP2 and MMP12 (Figure 10A). Then we extracted the total protein from the proximal aorta and performed Western blotting for MMP9 using an MMP9-specific mouse monoclonal antibody. We found that the expression of MMP9 was higher in Smad3–/– aorta than that in Smad3+/+ aorta (Figure 10, B and C). Because the IHC test, due to its limitations, didn’t allow the detection of relatively lower MMP9 in SMCs and ECs and its activity, we zymographically analyzed the total protein extracted from the proximal aorta of Smad3–/– mice and showed that the level of activated MMP9 was higher than that of MMP2 and MMP12 (Figure 10, B and D). This finding suggested that MMP9 might be a major member of the 3 MMPs that induced the matrix degradation. We believe that the SMCs/ECs exhibit MMP9 activity in the Smad3–/– aortic root. In fact, in situ zymographical examination showed that MMP activity was much higher in the medial layers of aortas from Smad3–/– mice as compared with Smad3+/+ mice (Figure 10F). Administration of GM-CSF antibody reduced the expression of MMP9 and abrogated MMP9/total MMP activity in aortas of Smad3–/– mice (Figure 10, B–F). These results confirmed that MMP9 expression and gelatinase activity were elevated in a GM-CSF–dependent manner in aortas of Smad3–/– mice.
Figure 10. Smad3–/– mouse aorta showed GM-CSF–dependent increase in MMP9 expression and gelatinase.

(A) MMP2, MMP9, and MMP12 immunostaining of the aortic wall (2-month-old Smad3–/– and Smad3+/+ mice received GM-CSF antibody or Ig-G). Original magnification, ×400. (B) Representative sample of 5 separate experiments showed gelatinolytic MMP2 and MMP9 activities in aortas from Smad3+/+ and Smad3–/– mice that received GM-CSF antibody or Ig-G (top). Representative Western blot showing MMP9 levels in aortas from Smad3+/+ and Smad3–/– mice received GM-CSF antibody or Ig-G (bottom). (C–E) Semiquantitative analysis of gelatinase activity (MMP9 or all) and MMP9 expression. **P < 0.01 versus Smad3+/+; ##P < 0.01 versus Smad3–/– mice that received Ig-G. (F) Representative samples of all gelatinase activity (green) in aortas of Smad3+/+ mice; Smad3–/– mice received GM-CSF antibody or Ig-G. Original magnification, ×400.
GM-CSF expression in surgical aorta specimens of AOS patients.
To detect the inflammatory infiltration and GM-CSF expression in aortas of human AOS patients, we sequenced all SMAD3 exons in 124 individuals with TAAD (35 % of which had a positive family history) and identified a missense mutation (c.985A>G) in a family of TAAD (FTTA) (Figure 11, A and B, and Supplemental Table 1), which resulted in substitution of alanine for threonine (Figure 11C). Then we screened 700 unrelated healthy individuals serving as controls. No mutation was found in the corresponding controls. Besides, further analysis also showed that the mutation p.Thr329Ala was of pathogenic nature: (a) the mutation is highly conserved within different species (Figure 11D); (b) we employed 2 classical computer programs to predict the possible impact of the mutation on the structure and function of SMAD3, and they both showed that the mutation was probably pathogenic (SIFT score = 0.06, PolyPhen score = 0.943); and (c) the mutation was in the MH1 domain of SMAD3 (Figure 11C), a very important functional domain, and could cause some comformational change of the protein. To investigate whether the inflammatory infiltration and GM-CSF expression take place in aortas of human AOS patients, we conducted an IHC test and found that a large number of CD45+ inflammatory cells infiltrated into the media of aorta from case 2 and a number of these inflammatory cells expressed GM-CSF (Figure 11E). In control aorta specimens, no positive staining for CD45 and GM-CSF was detected (Figure 11E). Therefore, GM-CSF might be potentially involved in the development of AOS.
Figure 11. SMAD3 mutations in a family with AOS and the aorta tissue from 1 case showed inflammatory infiltration and increased GM-CSF expression.

(A) Pedigrees with SMAD3 mutations. Squares, males; circles, females. Filled symbols indicate individuals with thoracic aortic aneurysm and/or dissection. Symbols represent individuals with a normal or unknown phenotype. (B) The sequence chromatogram of SMAD3 shows a heterozygous mutation. (C) Secondary structures of SMAD3, which are described by Jpred3 ( http://www.compbio.dundee.ac.uk/www-jpred/). The mutation (p.Thr329Ala) is located in β-folding near H2, which is in the MHI domain of SMAD3. (D) Cross-species protein conservation of SMAD3 around the mutation (p.Thr329Ala) which is described by the IBIVU Server ( http://www.ibi.vu.nl/programs/pralinewww/). (E) H&E staining and immunohistochemistry staining of transverse sections of aorta from a control (donor) and a patient with SMAD3 mutation. It showed inflammatory infiltration and increased GM-CSF expression in the aorta tissues from case 2. Original magnification, ×100 (top panels); ×400 (middle and bottom panels).
Discussion
Since 2009, several reports have examined the phenotypic spectrum and cardiovascular consequences of AOS (15–18), which resembles other disorders such as MFS and LDS. Because AOS was discovered recently, the little existing information regarding disease mechanisms and progression was derived from studies on limited patient specimens. In the present study, we found that mice lacking Smad3 had a vascular phenotype similar to AOS, marked by the progressive development of aneurysms. Similar to MFS, LDS, and AOS, aortic dilation started at the aortic root, predominantly in the coronary sinus, although the ascending aorta subsequently became involved. Defects in elastin fibers and smooth muscle cytoskeletal elements were found in aneurysm tissue. Careful examination of the Smad3–/– mouse vessel wall structure revealed preserved elastic fiber integrity. Elastic fiber fragmentation occurred later in life, when the lamellar structure was already established. This finding suggests that the defects observed in the Smad3 –/– mouse aortas did not arise from a developmental defect in elastic fiber organization, which is also supported by findings obtained from examination of the ascending aorta when the aortic root was enlarged. However, in fibulin-4–deficient mice, elastic lamina disruption was already evident as early as P1, and ascending aortic dilation appeared immediately after birth (4, 36, 37). Although in both mice, the pathological changes were characterized by the proliferation of SMCs, thickening of vascular walls, and infiltration of inflammatory cells, fibulin-4–deficient mice suffered from substantial structural changes in the elastic laminae of the aorta, including a granular look of elastin in the outer layers of the aorta, and the elastic fibers of the inner layer were fragmented and disorganized, which took place before the inflammation infiltration. In contrast, in fibulin-4–deficient mice, the infiltration of inflammatory cells occurred prior to the architectural change of aorta. We believe, in fibulin-4–deficient mice, the imbalance between the assembly/synthesis and degradation of the elastic fibers before birth was responsible for the aforementioned changes. Meanwhile, we hold that upregulation of p-ERK, one of the noncanonical TGF-β pathways, might be the common mechanism shared by 2 types of mice. p-ERK1/2 is upregulated at an early stage in fibulin-4–deficient mice, and p-ERK1/2 is elevated when the ascending aorta of Smad3–/– mice is dilated. It has been shown that NF-κB activation caused by upregulated p-ERK1/2 can trigger the inflammatory response of SMCs. In fibulin-4–deficient mice, MMP9 was substantially upregulated, which was similar to what we found in Smad3–/– mice.
We consistently found a focal loss of medial SMCs in Smad3–/– mice. The total SMC cross-sectional area was increased, which indicated SMC proliferation and may, along with periarteriolar fibrosis thickening in the wall, explain why aneurysm rupture was not related to diameter. Careful laceration examination revealed that focal inflammation induced elastin degradation or medial SMCs at the site of the weak adventitia. All of these findings are similar to those found in inflammatory abdominal aortic aneurysms.
In the aortic root, we determined that the expression of p-Smad1/5, p-Smad2, p-ERK1/2 and p-JNK1 was predominant in inflammatory cells at 1 month and appeared in SMCs at 4 months. At 1 month of age, the aortic root and ascending aorta were relatively normal. We hypothesize that excess TGF-β production by SMCs occurs at a later developmental stage and might be a compensatory mechanism/repairing mechanism of aorta in response to inflammatory damage induced by inflammation-related factors. We believe that the activated JNK1 and ERK1/2 could promote the dilation of aorta, as in Marfan mice. But in our mouse model, since the inflammatory cells infiltrate the aortic root and chronic inflammation could also lead to aberrant proliferation and thickened vascular walls, it was hard for us to distinguish between the effect of disordered TGF-β signals and those of inflammatory infiltration on the functions of SMCs in our model. It has been reported that GM-CSF induced the upregulation of activin A, a member of the TGF-β superfamily, in human proinflammatory macrophages (38). Activin A is involved in the regulation of several biological processes, including cell differentiation and proliferation, and more importantly, it could activate Smad2 (39). Therefore, to determine whether and how the SMC defect contributes to aortic dilation, a conditional knockout of Smad3 in SMCs would be a useful model.
TGF-β plays a pivotal role in the regulation of immune responses. Disruption of the mouse TGF-β gene results in severe multifocal autoimmune disease (40). Deficiency in Smad3, which is an important signaling molecule, causes death in mice after 3 months because of infections adjacent to the mucosal surface (19). Although we examined the inflammatory infiltration principally in the subgroup of mice without infection, we had compared the aortas from the 2 subgroups (mice with and without overt infections) in advance. We found that the aortas from the 2 subgroups had similar inflammatory changes. We could not exclude lower levels of infection in Smad3–/– mice without abscesses, but the inflammation of similar degree in both subgroups indicated that the infection may not be a critical factor that dictates the inflammatory infiltration. Although we could not eliminate the possibility that low-level infection increased the number of inflammatory cells in the vascular system, we are inclined to think that the change of inflammatory cells per se led to their accumulation at the aortic root.
In MFS, there are fewer inflammatory cells, which preserves the integrated signaling pathway response to TGF-β. However, in LDS or AOS, it remains unclear whether impaired immune cell TGF-β signaling induces autoimmune responses, as no related symptoms have been reported in LDS patients. AOS is characterized by early-onset osteoarthritis, in which the inflammatory features are unclear. In our mice, inflammation appeared in the aortic root, coronary arteries, and aortic valves, which is consistent with the cardiovascular phenotype of Kawasaki syndrome, which is an autoimmune disease. Indeed, a recent study demonstrated SMAD3 genetic variants; haplotypes were consistently and reproducibly associated with Kawasaki’s disease (KD) susceptibility, coronary artery aneurysm formation, and aortic root dilation (41). We have demonstrated that some peripheral CD4+ T cells from Smad3–/– mice showed that activated phenotype could cause aortitis in Smad3+/+ mice, suggesting that T cell–intrinsic dysfunction rather than abnormality of positive-negative selection of T cells in the thymus was responsible for the development of aortitis. Based on the finding that the aortic root but not the region of the coronary artery near the ostium was infiltrated by inflammatory cells and that in other parts of heart and other organs such as lungs, liver, and kidney, no obvious inflammation was observed, we thought that autoimmune responses against specific antigens on vessel walls might be induced. Aortic root infiltration can be explained 2 ways. First, the aortic root is susceptible to TGF-β signaling because of the embryonic origin of the vascular cells (42). Second, blood flow can form an eddy within the sinus cavity and generate turbulence that possibly leads to EC damage (43). It is unclear how these immune responses influence each other. In this study, we investigated GM-CSF levels as a potential mediator of the cooperation between the adaptive and innate immune responses. It is widely believed that Th17 cells are responsible for autoimmune inflammation (44). However, a recent report demonstrated that autoreactive helper T cells lacking GM-CSF failed to initiate neuroinflammation despite their IL-17A or IFN-γ expression (27, 30). Although targeted disruption of the mouse Tgf1 or Smad3 gene results in severe multifocal autoimmune disease, the signature cytokines that are responsible remain unknown. Here, we report that GM-CSF may be responsible for this disease phenotype. We found that GM-CSF neutralization with anti–GM-CSF mAbs resulted in marked less inflammation in the aortic roots of Smad3–/– mice. IFN-γ deficiency exacerbated the disease, which is consistent with a report that IFN-γ inhibition induces aneurysms in allograft aortas or angiotensin II–infused ApoE–/– mice (45, 46). IFN-γ deficiency induced GM-CSF secretion by activating CD4+ T cells in vitro. Additionally, inflammasome-derived IL-1β is required for GM-CSF production (31). IL-1R–deficient mice are protected from LCWE-induced (L. casei cell wall extract-) coronary lesions in a mouse model of Kawasaki disease (47). IL-1Ra–/– mice, in which CD4+ T cells are excessively activated by IL-1β, spontaneously develop aortitis, aneurysms, and inflammatory arthropathy and resemble the phenotype of the mice that were used in the present study (21). Thus, we hypothesized that GM-CSF is a common link in the development of inflammatory aneurysms. Although a previous report demonstrated that GM-CSF neutralization ameliorated myocardial infarction, atherosclerosis, and vascular injury, to our knowledge, no report has evaluated GM-CSF neutralization in aneurysm formation, with the exception of a study demonstrating that GM-CSF administration to ApoE–/– mice induced abdominal aortic aneurysms (48). Therefore, GM-CSF has a potential pathogenic effect on inflammatory aneurysms.
Pathological matrix remodeling, elastin degradation, and excessive SMC accumulation have been implicated in vascular injury development. To explain how mutation of TGF-β signal–related molecules, especially in inflammatory cells, leads to damage or remodeling of the vascular system, we can use the mechanism of hereditary hemorrhagic telangiectasia (HHT). HHT is caused by mutations of several TGF-β signal–related molecules, including ALK1, endoglin, and Smad4 (49, 50), and mutation of Smad4, an obligate partner of Smad3, could induce the dilation of aorta (49). At the early stage of HHT development, lymphocytes, including monocytes, surrounded these dilated vessels (51). We believe pathological changes vary with different subsets of monocytes because there are 2 subsets of monocytes in the peripheral blood of mice, i.e., Ly-6C+ and Ly-6C–, with the former being responsible for degradation and the latter for repairing. Previous work showed impaired homing of repairing monocytes from the patients with HHT-1 toward ischemic tissue (51), which was consistent with what we observed in mice. At the aortic root of Smad3–/– mice, we found a large number of degrading Ly-6C+ cells. However, whether disordered TGF-β signaling in inflammatory cells affects the functions of these 2 kinds of cells cannot be known without further study. Further studies that evaluate the immune response in patients with AOS and LDS are necessary to provide more information on this subject.
In conclusion, we have demonstrated what we believe to be a novel pathogenic process for aneurysm development in Smad3-deficient mice. The results of this study emphasize a link between the antiinflammatory properties of TGF-β and aneurysm progression. In LDS (TβRI/II) or AOS (SMAD3), heterozygous loss-of-function mutations result in a failure of cells to transmit signals, which may affect the immune system. Thus, the results of this study may be useful for developing new drugs to inhibit aneurysm progression or rupture in LDS or AOS.
Methods
Animal care and use.
Smad3–/– mice were generated as previously described (19). Heterozygous females (Smad3+/–) were mated with heterozygous males (Smad3+/–) to generate Smad3+/+, Smad3+/–, and Smad3–/– offspring. WT C57BL/6 mice were provided by the Institute of Laboratory Animal Sciences of the Chinese Academy of Medical Sciences (Beijing, China), and C57BL/6 Il17–/– mice were provided by Yoichiro Iwakura (University of Tokyo, Tokyo, Japan). C57BL/6 Ifng–/–mice were obtained from Jackson Laboratories. Smad3+/–Ifng–/– and Smad3+/–Il17–/– mice were backcrossed to C57BL/6 by crossbreeding. Offspring genotypes were determined by PCR analysis of tail DNA. Control mice were age- and sex-matched littermates.
Aortic transplantation.
Eight- to twelve-week-old WT Balb/c mice, Smad3+/– mice backcrossed to C57BL/6 for at least 6 generations, and Smad3+/+ littermates were used as donors or recipients. Aortic transplantation was performed as described (52).
Echocardiography.
All of the echocardiograms were obtained using a high-resolution Vevo 2100 Microimaging System with a 30-MHz transducer (Visualsonic). The aortas were imaged in the parasternal long-axis view, and 3 measurements of the sinuses of Valsalva or ascending aorta were obtained at the indicated time points. Echocardiograph acquisition and analysis were performed by an experienced echocardiographer who was blinded to the genotype and treatment.
Drug administration.
Based on a study by Jinniu M et al., 2.5 mg/kg–1/d–1 SIS3 or an equivalent vehicle volume was delivered by an implanted Alzet osmotic pump (Durect Corp.) for 1 week (53). Recombinant GM-CSF (10 μg/kg–1/d–1), anti-GM-CSF (300 μg), anti-CD4 (1 mg/d), or isotype-matched antibody was injected intraperitoneally every other day.
Microscopic examination.
For morphometric analysis, hearts, aortas, and spleens were harvested from mice at each time point. These samples were fixed overnight in 10% buffered formalin. Tissue was paraffin embedded and sectioned at 4 μm; H&E/elastic Von Gienson (EVG)/Masson staining was performed using routine techniques. The wall thickness at 12 different locations was measured by an observer blinded to the genotype of each mouse and the corresponding treatment. Elastin degradation and inflammatory cell infiltration at 8 sites were assessed by at least 2 blind observers. Elastin degradation and inflammatory cell infiltration were graded according to the grading keys shown in Figure 2B and Figure 4B. For immunohistochemistry, sections were incubated with p-Smad3, p-Smad2, p-JNK1, p-ERK1/2 (Cell Signaling Technology), LAP–TGF-β (R&D ),CD45,CD4, CD68 (Dako), α-SMA, Ki-67, GM-CSF (Abcam), MMP9 (R&D), CTGF, MMP2, or MMP12 (Gene Tex) primary antibodies. Negative controls were included in which the primary antibody was omitted. Then 6-μm–thick frozen sections were applied to poly-l-lysine microscope slides and fixed with cold acetone for 10 minutes for immunofluorescence. The slides were incubated with blocking serum, then with CD4, CD11b, GM-CSF, CCR2 (Abcam), or CD11b (Abcam) primary antibodies and secondary antibodies (Invitrogen), followed by confocal microscopy. The total number of CD4/GM-CSF double-positive cells in each section was counted manually and blindly. MMP activities were determined by gelatin zymography according to the manufacturer’s instructions. Image-Pro Plus 6.0 software was used for image processing.
Cell suspensions.
LNs and spleens were homogenized and passed through a cell strainer to obtain cell suspensions. BM cells were taken from the femurs and tibias of 5- to 6-week-old Smad3+/+ or Smad3–/– mice, and blood was collected in EDTA-coated vials. Erythrocytes were lysed using rbc lysis solution (Invitrogen) for 5 minutes at room temperature. Cells were centrifuged at 600 g for 3 minutes to remove the rbc lysis solution, and the leukocyte pellet was resuspended and washed in cell isolation buffer, followed by centrifugation at 400 g for 10 minutes. CD4+ T cells were isolated by negative selection using a CD4+ T cell isolation kit (Miltenyi Biotec) and an autoMACS machine (Miltenyi Biotech). Aortic infiltrated cells were isolated according to the method used by Tieu et al (34). Briefly, aortic tissue was minced into 3- to 5-mm pieces and digested in digestion solution containing collagenase at room temperature for 1–2 hours with agitation. After digestion, cells were washed in FACS buffer (0.5% BSA, 0.02% NaN3 in PBS) at 300 g for 5 minutes. After washing and counting, cells were submitted to flow cytometry analysis. The collected cells were then centrifuged over Ficoll-Isopaque (GE Health) for Wright-Giemsa staining. After washing and counting, the cells were smeared onto the slide for staining.
CD4+ T cell and BM cell transplantation.
Smad3+/+ or Smad3–/– CD4+ T cells (2 × 107 cells/mouse) isolated from 8-week-old mice and BM cells isolated from 5- to 6-week-old Smad3+/+ or Smad3–/– mice (2 × 107 cells/mouse) were resuspended in 0.2 ml of PBS. CD4+ T cells were transplanted intravenously into 4-week-old WT sex-matched littermates. BM cells were transplanted intravenously into 4-week-old irradiated (7.5 Gy), sex-matched littermates. The recipient mice were histologically examined after 12 weeks.
Flow cytometry.
Murine Fc receptors were blocked using Abs against mouse CD16/32 antigens (eBioscience) for 10 minutes on ice; cells were then washed and resuspended in 100 μl of FACS buffer. Fluorochrome-conjugated Abs (APC-Cy7-CD11b, PE-Cy7-Gr-1, Percp-Cy5.5-Ly-6c, PE-CD11c, FITC-MHC-II, APC-CCR2, or FITC-CD4) were added for 30 to 45 minutes at room temperature. Cytokine intracellular staining was performed on cells that had been stimulated with phorbol myristate acetate (50 ng/ml; Sigma-Aldrich) and ionomycin (1 μM; Sigma-Aldrich) in the presence of GolgiStop (BD Biosciences) for 6 hours, and cells were stained with APC–IL-22, PE-CY7–IL-17, Percp-Cy5.5–IFN-γ, APC–IL-4 (eBioscience), PE–GM-CSF (R&D Systems), or PE–IL-9 (BD Biosciences) mAbs after permeabilization and fixation. Samples were then washed in FACS buffer, centrifuged at 300 g for 5 minutes, fixed in 0.5% PFA, and analyzed with FACS Canto II (BD). The corresponding isotype control Abs were added to “isotype samples” at the same concentrations as the Abs of interest.
Cell culture.
Splenocytes or purified CD4+ T cells (5 × 105) were cultured in 0.3 ml of complete medium in 96-well plates and stimulated with plate-bound anti-CD3 (1 μg/ml) and soluble anti-CD28 mAbs (1 μg/ml) for 48 hours. Anti–IL-4 (10 μg/ml), anti–IFN-γ (10 μg/ml), recombinant mouse IL-1-β (10 ng/ml), IL-23 (10 ng/ml), IL-6 (20 ng/ml) (PeproTech), and TGF-β (10 ng/ml) (R&D Systems). To study monocyte proliferation, WT splenocytes (5 × 106) were cultured in 2 ml of complete medium in 24-well plates. In some experiments, cells were pretreated with 3 μM SIS3 (Sigma-Aldrich) or an equivalent volume of vehicle.
ELISA.
For cytokine production measurements, CD4+ T cell supernatants were assayed for cytokine levels using a mouse cytokine quantibody array (RayBiotech). GM-CSF in the supernatants was measured using a specific ELISA kit from RayBiotech according to the manufacturer’s instructions.
Western blot.
Aortic tissues were lysed with sodium dodecyl sulfate (SDS) sample loading buffer. Lysates were boiled in sample buffer (200 mmol/l Tris [pH 6.8], 20% glycerol, 2% SDS, 0.1% bromophenol blue, and 10% β-ME), and the same amount of total protein from each lysate was loaded onto a 10% SDS-PAGE gel. After electrophoresis, proteins were transferred onto a nylon PVDF membrane and blocked overnight with 4% nonfat dry milk in TBS-T (100 mmol/l Tris, pH 7.5, 0.9% NaCl, 0.1% Tween-20). Thereafter, the membrane was incubated for 1 hour at room temperature with rabbit polyclonal anti-mouse Smad1/5, Smad2, ERK1/2, JNK1, p-Smad1/5, p-Smad2, p-Smad3, p-ERK1/2, p-JNK1 (Cell Signaling Technology), and MMP9 antibodies (R&D). Membranes were washed and incubated for 1 hour with horseradish peroxidase–labeled goat anti-rabbit IgGs (Cedarlane). Blots were developed using ECL (GE Healthcare).
Blood sample collection and screening for mutation of SMAD3.
Genomic DNA was isolated from blood samples of 124 individual with TAAD using standard protocols with the Wizard Genomic DNA Purification Kit (Promega Corporation). We designed primers to amplify all SMAD3 exons and exon-intron boundaries by general PCR. The general PCR was performed in a total of 25 μl volume containing 5 pmol of each primer, 25 ng of genomic DNA, 2.5 μl of 10× PCR buffer with 1.5 mmol/l MgCl2, 5 mmol dNTP, and 1 U of Taq polymerase. DNA sequence analysis was performed with either or both of the forward and reverse primers using the BigDye Terminator v3.1 Cycle Sequencing Kits on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems). The primers used for sequencing the mutation of c.985A>G were as follows: forward primer, CTGCTGTTCTGCCTCCTTTG; reverse primer, CCTGTGCGGCTCGTTTAC. Sequence analyses were performed on Sequence Scanner v1.0.
Statistics.
The values are expressed as the percentage or the mean ± SEM in bar graphs. Statistical tests include Student’s t test (2 tail, assuming unequal variances) and the Mann-Whitney test. Kaplan-Meier survival curves were constructed and analyzed using the log-rank (Mantel-Cox) test. P < 0.05 was considered to be significant.
Study approval.
Animal experiments were approved by the Institutional Animal Care and Use Committee at Huazhong University of Science and Technology. The study protocol for screening the mutation of SMAD3 in patients was approved by the Medical Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology and informed consent was obtained from all patients.
Supplementary Material
Acknowledgments
This work was supported in part by the National Nature Science Foundation of China (81070205, 81130056, 81202335, 81100176, and 81102240).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(5):2317–2331. doi:10.1172/JCI67356.
References
- 1.Lindsay ME, Dietz HC. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature. 2011;473(7347):308–316. doi: 10.1038/nature10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dietz HC, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352(6333):337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 3.Loeys BL, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37(3):275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 4.Dasouki M, et al. Compound heterozygous mutations in fibulin-4 causing neonatal lethal pulmonary artery occlusion, aortic aneurysm, arachnodactyly, and mild cutis laxa. Am J Med Genet A. 2007;143A(22):2635–2641. doi: 10.1002/ajmg.a.31980. [DOI] [PubMed] [Google Scholar]
- 5.Dietz HC. TGF-beta in the pathogenesis and prevention of disease: a matter of aneurysmic proportions. J Clin Invest. 2010;120(2):403–407. doi: 10.1172/JCI42014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin F, Yang X. TGF-beta signaling in aortic aneurysm: another round of controversy. J Genet Genomics. 2010;37(9):583–591. doi: 10.1016/S1673-8527(09)60078-3. [DOI] [PubMed] [Google Scholar]
- 7.Kang JS, Liu C, Derynck R. New regulatory mechanisms of TGF-beta receptor function. Trends Cell Biol. 2009;19(8):385–394. doi: 10.1016/j.tcb.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 8.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 9.Lee MK, et al. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007;26(17):3957–3967. doi: 10.1038/sj.emboj.7601818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell. 2008;31(6):918–924. doi: 10.1016/j.molcel.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gomez D, et al. Syndromic and non-syndromic aneurysms of the human ascending aorta share activation of the Smad2 pathway. J Pathol. 2009;218(1):131–142. doi: 10.1002/path.2516. [DOI] [PubMed] [Google Scholar]
- 12.Neptune ER, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33(3):407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 13.Holm TM, et al. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332(6027):358–361. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, et al. TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J Clin Invest. 2010;120(2):422–432. doi: 10.1172/JCI38136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van de Laar IM, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43(2):121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 16.van de Laar IM, et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J Med Genet. 2012;49(1):47–57. doi: 10.1136/jmedgenet-2011-100382. [DOI] [PubMed] [Google Scholar]
- 17.Regalado ES, et al. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res. 2011;109(6):680–686. doi: 10.1161/CIRCRESAHA.111.248161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Linde D, et al. Aggressive cardiovascular phenotype of aneurysms-osteoarthritis syndrome caused by pathogenic SMAD3 variants. J Am Coll Cardiol. 2012;60(5):397–403. doi: 10.1016/j.jacc.2011.12.052. [DOI] [PubMed] [Google Scholar]
- 19.Yang X, et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 1999;18(5):1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development. 1995;121(6):1845–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- 21.Matsuki T, et al. Involvement of tumor necrosis factor-alpha in the development of T cell-dependent aortitis in interleukin-1 receptor antagonist-deficient mice. Circulation. 2005;112(9):1323–1331. doi: 10.1161/CIRCULATIONAHA.105.564658. [DOI] [PubMed] [Google Scholar]
- 22.Horai R, et al. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. 2000;191(2):313–320. doi: 10.1084/jem.191.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Del GG, Crow MK. Role of transforming growth factor beta (TGF beta) in systemic autoimmunity. Lupus. 1993;2(4):213–220. doi: 10.1177/096120339300200402. [DOI] [PubMed] [Google Scholar]
- 24.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma AK, et al. Experimental abdominal aortic aneurysm formation is mediated by IL-17 and attenuated by mesenchymal stem cell treatment. Circulation. 2012;126(11 suppl 1):S38–S45. doi: 10.1161/CIRCULATIONAHA.111.083451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galle C, et al. Predominance of type 1 CD4+ T cells in human abdominal aortic aneurysm. Clin Exp Immunol. 2005;142(3):519–527. doi: 10.1111/j.1365-2249.2005.02938.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Codarri L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12(6):560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- 28.Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M. GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. J Exp Med. 2008;205(10):2281–2294. doi: 10.1084/jem.20071119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cook AD, Braine EL, Campbell IK, Rich MJ, Hamilton JA. Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (GM-CSF): requirement for GM-CSF in the effector phase of disease. Arthritis Res. 2001;3(5):293–298. doi: 10.1186/ar318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.El-Behi M, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12(6):568–575. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lukens JR, Barr MJ, Chaplin DD, Chi H, Kanneganti TD. Inflammasome-derived IL-1beta regulates the production of GM-CSF by CD4(+) T cells and gammadelta T cells. J Immunol. 2012;188(7):3107–3115. doi: 10.4049/jimmunol.1103308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bayne LJ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822–835. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang L, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13(1):23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tieu BC, et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest. 2009;119(12):3637–3651. doi: 10.1172/JCI38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen J, Knapnougel P, Lesavre P, Bauvois B. Inhibition of matrix metalloproteinase-9 by interferons and TGF-beta1 through distinct signalings accounts for reduced monocyte invasiveness. FEBS Lett. 2005;579(25):5487–5493. doi: 10.1016/j.febslet.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 36.Horiguchi M, et al. Fibulin-4 conducts proper elastogenesis via interaction with cross-linking enzyme lysyl oxidase. Proc Natl Acad Sci U S A. 2009;106(45):19029–19034. doi: 10.1073/pnas.0908268106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang J, et al. Fibulin-4 deficiency results in ascending aortic aneurysms: a potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circ Res. 2010;106(3):583–592. doi: 10.1161/CIRCRESAHA.109.207852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sierra-Filardi E, et al. Activin A skews macrophage polarization by promoting a proinflammatory phenotype and inhibiting the acquisition of anti-inflammatory macrophage markers. Blood. 2011;117(19):5092–5101. doi: 10.1182/blood-2010-09-306993. [DOI] [PubMed] [Google Scholar]
- 39.Chen YG, Wang Q, Lin SL, Chang CD, Chuang J, Ying SY. Activin signaling and its role in regulation of cell proliferation, apoptosis, and carcinogenesis. Exp Biol Med (Maywood). 2006;231(5):534–544. doi: 10.1177/153537020623100507. [DOI] [PubMed] [Google Scholar]
- 40.Gorelik L, Flavell RA. Transforming growth factor-beta in T-cell biology. Nat Rev Immunol. 2002;2(1):46–53. doi: 10.1038/nri704. [DOI] [PubMed] [Google Scholar]
- 41.Shimizu C, et al. Transforming growth factor-beta signaling pathway in patients with Kawasaki disease. Circ Cardiovasc Genet. 2011;4(1):16–25. doi: 10.1161/CIRCGENETICS.110.940858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Topouzis S, Majesky MW. Smooth muscle lineage diversity in the chick embryo. Two types of aortic smooth muscle cell differ in growth and receptor-mediated transcriptional responses to transforming growth factor-beta. Dev Biol. 1996;178:430–445. doi: 10.1006/dbio.1996.0229. [DOI] [PubMed] [Google Scholar]
- 43.Davies PF, Remuzzi A, Gordon EJ, Dewey CJ, Gimbrone MJ. Turbulent fluid shear stress induces vascular endothelial cell turnover in vitro. Proc Natl Acad Sci U S A. 1986;83(7):2114–2117. doi: 10.1073/pnas.83.7.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6(11):1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shimizu K, Shichiri M, Libby P, Lee RT, Mitchell RN. Th2-predominant inflammation and blockade of IFN-gamma signaling induce aneurysms in allografted aortas. J Clin Invest. 2004;114(2):300–308. doi: 10.1172/JCI19855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.King VL, et al. Interferon-gamma and the interferon-inducible chemokine CXCL10 protect against aneurysm formation and rupture. Circulation. 2009;119(3):426–435. doi: 10.1161/CIRCULATIONAHA.108.785949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee Y, et al. Interleukin-1beta is crucial for the induction of coronary artery inflammation in a mouse model of Kawasaki disease. Circulation. 2012;125(12):1542–1550. doi: 10.1161/CIRCULATIONAHA.111.072769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haghighat A, Weiss D, Whalin MK, Cowan DP, Taylor WR. Granulocyte colony-stimulating factor and granulocyte macrophage colony-stimulating factor exacerbate atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2007;115(15):2049–2054. doi: 10.1161/CIRCULATIONAHA.106.665570. [DOI] [PubMed] [Google Scholar]
- 49.Andrabi S, Bekheirnia MR, Robbins-Furman P, Lewis RA, Prior TW, Potocki L. SMAD4 mutation segregating in a family with juvenile polyposis, aortopathy, and mitral valve dysfunction. Am J Med Genet A. 2011;155A(5):1165–1169. doi: 10.1002/ajmg.a.33968. [DOI] [PubMed] [Google Scholar]
- 50.Fernandez-L A, Sanz-Rodriguez F, Blanco FJ, Bernabeu C, Botella LM. Hereditary hemorrhagic telangiectasia, a vascular dysplasia affecting the TGF-beta signaling pathway. Clin Med Res. 2006;4(1):66–78. doi: 10.3121/cmr.4.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marchuk DA, Srinivasan S, Squire TL, Zawistowski JS. Vascular morphogenesis: tales of two syndromes. Hum Mol Genet. 2003;1:R97–R112. doi: 10.1093/hmg/1.2.97. [DOI] [PubMed] [Google Scholar]
- 52.Shimizu K, et al. Host bone-marrow cells are a source of donor intimal smooth- muscle-like cells in murine aortic transplant arteriopathy. Nat Med. 2001;7(6):738–741. doi: 10.1038/89121. [DOI] [PubMed] [Google Scholar]
- 53.Jinnin M, Ihn H, Tamaki K. Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-beta1-induced extracellular matrix expression. Mol Pharmacol. 2006;69(2):597–607. doi: 10.1124/mol.105.017483. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.