

Abstract
For decades, peripartum cardiomyopathy has remained an enigma. Despite extensive research, our understanding of how a previously healthy woman can develop lethal heart failure in the context of pregnancy remains vague. Recent work suggests that inadequacy of the cardiac microvasculature may be the primary abnormality and has implicated an antiangiogenic fragment of the nursing hormone prolactin as playing an important role. In this issue of the JCI, Halkein et al. explore signaling downstream of this prolactin fragment and demonstrate that miR-146a is a critical mediator of the antiangiogenic effects in endothelial cells. In addition, the study uncovers unexpected exosomal transfer of this microRNA to cardiomyocytes that may affect myocardial metabolism.
Peripartum cardiomyopathy: definition of a new syndrome
Reports of heart failure in association with pregnancy date back to 1849, but it was not until 1971 that the more specific syndrome known as peripartum cardiomyopathy (PPCM) was recognized (1). PPCM is defined as the development of heart failure resulting from systolic dysfunction, with onset between one month before and five months after childbirth, that cannot be attributed to another etiology (2). The incidence of this syndrome has been reported as between 1:300 and 1:10,000, the wide variation likely reflecting differing susceptibilities among racial groups (in descending order of susceptibility: African, Asian, mixed European descent, Hispanic). Predisposing factors include multiple pregnancies, carriage of more than one fetus, hypertension, and preeclampsia. While PPCM can be lethal, a striking difference compared with other dilated cardiomyopathies is that a significant percentage of patients recover cardiac function over months to several years (3). Subsequent pregnancies, however, confer increased risk for recurrent disease.
Early studies of pathogenesis
The etiology of PPCM is unknown (4). Over the past several decades, involvement of a variety of pathological processes has been investigated, including myocarditis, abnormalities of innate and adaptive immunity, hormonal imbalances, nutritional deficiencies, and mutations of genes implicated in other cardiomyopathies. While a role for these cannot be excluded, the existing data linking them to PPCM are largely correlative, precluding definitive conclusions. Another consideration in pathogenesis is the hemodynamic changes of pregnancy, which are characterized by increased plasma volume, decreased systemic vascular resistance, and a physiological form of hypertrophy. On the one hand, these changes are considered compensatory, and they substantially precede the typical onset of PPCM, raising questions as to their role in pathogenesis. On the other hand, our knowledge of the factors that maintain physiological versus pathological hypertrophy is incomplete. Moreover, cardiac-specific transgenic expression of Gαq, a protein that transduces multiple stimuli of pathological hypertrophy, elicits a PPCM-like syndrome (5). Accordingly, the role of hemodynamics in the pathogenesis of PPCM remains an open question.
A new paradigm
In the past few years, studies from two independent groups made new headway in the field, suggesting a radically different paradigm for PPCM (6, 7). In this model, the vasculature, rather than the heart muscle, is the primary driver of pathogenesis. The mechanism involves complex interactions among several tissue compartments, including cardiomyocytes, endothelial cells, the interstitial space, the anterior pituitary gland, and the placenta (Figure 1). While important aspects remain to be tested, the general schema appears to explain some key features of PPCM.
Figure 1. Interactions among multiple tissue compartments collude to cause PPCM.
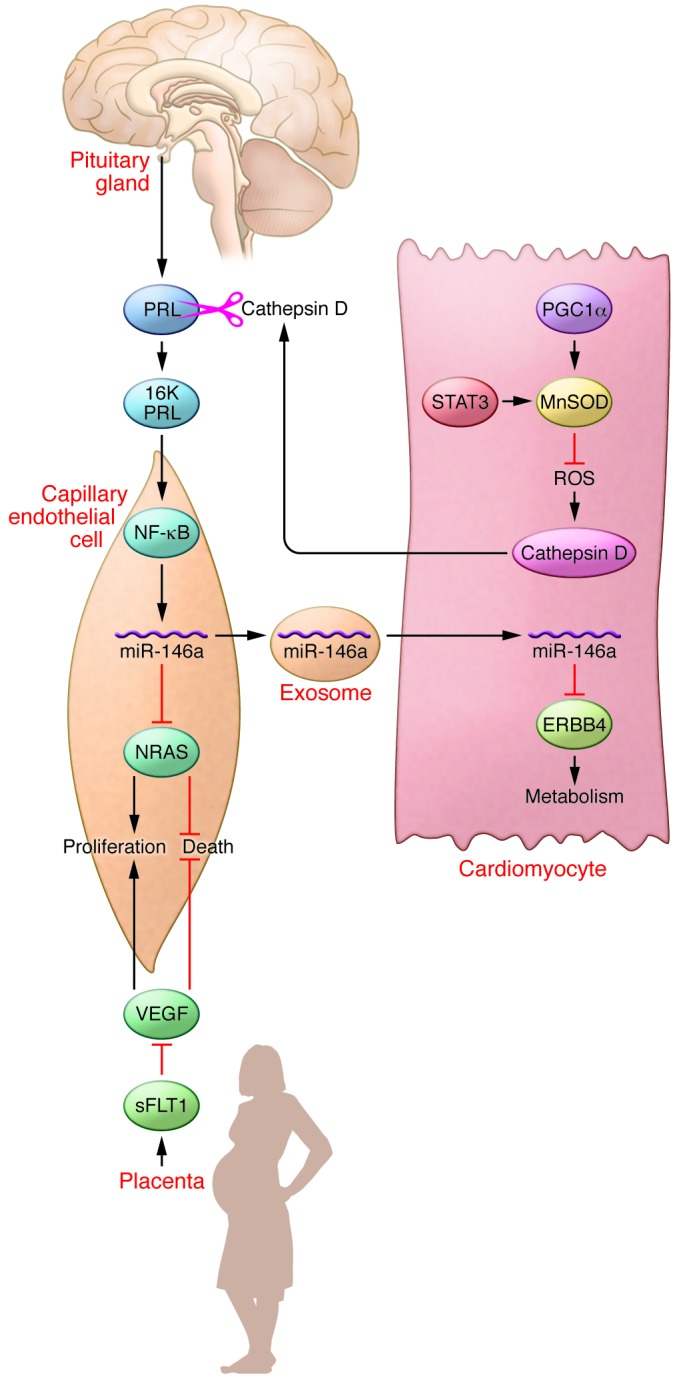
Mice lacking the transcription factor STAT3 or the transcriptional coactivator PGC1α in cardiomyocytes provide models of PPCM. While absence of STAT3 or PGC1α in cardiomyocytes may not be inciting events in human PPCM, these mouse models have helped to reveal the underlying molecular pathogenesis. Absence of these proteins results in decreased expression of the ROS scavenger MnSOD in these cells. Decreased MnSOD levels result in ROS accumulation, increasing and activating Cathepsin D, which is secreted from cardiomyocytes into the interstitial space. Cathepsin D cleaves PRL to generate an antiangiogenic 16-kDa fragment, 16K PRL. 16K PRL stimulates endothelial cells through an unknown mechanism to activate NF-κB, which increases levels of miR-146a. miR-146a decreases levels of NRAS, inhibiting angiogenesis. In addition, miR-146a transits to cardiomyocytes in exosomes to decrease levels of ERBB4, resulting in slowed cardiac metabolism. VEGF levels are decreased by the absence of PGC1α in cardiomyocytes (not shown), and VEGF is antagonized by the secretion of sFLT1 from the placenta, both mechanisms inhibiting angiogenesis. Triggering events in human PPCM include generation of 16K PRL and increases in miR-146a and sFLT1.
The first group, led by Hilfiker-Kleiner (also a senior author of the current study in the JCI; ref. 8), observed that mice with cardiomyocyte-specific knockout of the transcription factor STAT3 (referred to herein as STAT3-CKO mice) manifest a syndrome closely resembling PPCM (6). A proportion of these mice die of heart failure within the first 3 weeks after delivery, and the death toll continues to rise with each subsequent pregnancy. Since STAT3 promotes the transcription of genes encoding antioxidant (e.g., MnSOD) and anti–cell death (e.g., BCL-XL) proteins (9, 10), an obvious conclusion might be that heart failure in postpartum STAT3-CKO mice is related to cell-autonomous effects of STAT3 absence in cardiomyocytes. While this may be part of the story, Hilfiker-Kleiner and colleagues made the critical observation that postpartum STAT3-CKO mice exhibit striking reductions in myocardial capillary density compared with both postpartum wild-type mice and nonpregnant STAT3-CKO mice (6). The investigators linked this vascular phenotype to PPCM through the hormone prolactin (PRL), which is secreted from the anterior pituitary gland. PRL has multiple effects in addition to stimulating milk production, one of which is the promotion of angiogenesis (11). However, the serum of patients with PPCM contains increased levels of a 16-kDa N-terminal PRL cleavage fragment (16K PRL) that is strongly antiangiogenic. The investigators delineated a pathway in which the absence of STAT3 in cardiomyocytes decreases the abundance of MnSOD in these cells, resulting in the accumulation of ROS and increased expression and activity of Cathepsin D, which is secreted from cardiomyocytes into the interstitial space, where it cleaves PRL (Figure 1). Evidence that decreased capillary density is largely responsible for heart failure in PPCM is provided by experiments demonstrating that cardiomyopathy in STAT3-CKO mice is prevented by treatment with bromocriptine, a drug that inhibits PRL secretion from the pituitary. This, in turn, decreases the concentration of 16K PRL, thereby maintaining myocardial capillary density.
Using an independent approach, a second group, led by Arany, arrived at a similar conclusion that insufficient angiogenesis is the primary defect in PPCM (7). These investigators were studying the effects of cardiomyocyte-specific knockout of the transcriptional coactivator PGC1α (PGC1α-CKO mice). At baseline, the absence of PGC1α in cardiomyocytes is well tolerated because of compensation from PGC1β (12). However, similar to the STAT3-CKO mice, PGC1α-CKO mice manifest lethal PPCM accompanied by a paucity of myocardial capillaries. In wild-type mice, PGC1α promotes the expression of proangiogenic factors (e.g., VEGF) in cardiomyocytes that act in a paracrine manner to promote angiogenesis, and PGC1α-CKO hearts contain decreased levels of these proangiogenic factors. Notably, in a sizeable proportion of humans with PPCM, angiogenic signaling downstream of PGC1α is inhibited by soluble FLT1 (sFLT1), a VEGF inhibitor that is secreted from the placenta (Figure 1).
Mechanisms of action for 16K PRL
In the present study, Halkein et al. (in a group led by Struman and Hilfiker-Kleiner) delved deeper into the mechanisms by which 16K PRL promotes PPCM (8). Because the antiangiogenic effects of 16K PRL are known to be dependent on NF-κB (13), the investigators treated endothelial cells with 16K PRL and assayed for changes in the abundance of microRNAs known to be regulated by NF-κB. The expression of miR-146a was found to be induced by 16K PRL in these cells. Using loss- and gain-of-function approaches, miR-146a was shown to inhibit proliferation and promote death of endothelial cells. These antiangiogenic effects were mediated through decreases in levels of neuroblastoma RAS viral oncogene homolog (NRAS), a newly identified target for this microRNA. However, the effects of miR-146a were not limited to endothelial cells. Halkein et al. also found that exosomes loaded with miR-146a transit from endothelial cells into cardiomyocytes, which do not express this microRNA (8). Within cardiomyocytes, miR-146a decreases the abundance of its target, ERBB4, to slow cardiomyocyte metabolism. As predicted, levels of miR-146a were increased, while those of its targets (NRAS, ERBB4, and others) were decreased, in cardiac tissue from the STAT3-CKO mouse model of PPCM. Treatment of STAT3-CKO mice with locked nucleic acid–modified antisense oligonucleotides or cholesterol-modified antago-miRs to miR-146a ameliorated PPCM, but interestingly, these did not interfere with lactation, a function of full-length PRL. Finally, the authors showed that patients with PPCM, but not other forms of dilated cardiomyopathy, had increased levels of miR-146a in serum and heart (8). These data indicate that miR-146a mediates antiangiogenic effects of 16K PRL in endothelial cells and slowing of metabolism in cardiomyocytes. In addition, this microRNA appears to be a specific biomarker of PPCM.
Questions and future directions
As with any good research, this work opens the door to additional questions. Some of these pertain to particulars of the pathway. For example, are the effects of 16K PRL on endothelial cells mediated by a specific receptor? Does the 16K PRL/miR-146a axis mediate effects on other vascular cell types? In addition to influencing quantity of angiogenesis, does this system also regulate the quality of the vessels? What specific aspects of cardiac metabolism are affected by miR-146a? In addition, it is unclear how Cathepsin D, which cleaves PRL to the 16K PRL form, is itself regulated.
More broadly, the overall PPCM paradigm illustrated in Figure 1 raises some larger conceptual questions. The most obvious one is why only a small percentage of pregnant women develop PPCM. The combined work of the two groups of investigators identifies triggers, such as cleavage of PRL and increases in sFLT1. However, what factors at the molecular level predispose some women to these inciting events? As discussed above, it is possible that other poorly understood factors, ranging from inflammation to hemodynamics, may be involved in conferring this susceptibility to disease.
The data discussed herein shine a spotlight on microvascular insufficiency as a cause of cardiomyopathy. While not a new concept (14), this is not a mechanism that has traditionally been considered, except in special cases such as diabetic cardiomyopathy (15). One wonders whether vascular insufficiency is an important factor in the pathogenesis of a broad range of cardiomyopathies and, if so, what functions — in addition to nutrition — the vasculature serves to promote normal cardiac function.
From the perspective of patients with PPCM and the physicians who care for them, the most exciting aspects of this work are its potential implications for therapy. Bromocriptine has been tested in a limited number of patients with promising results, but the randomized trials now in progress will be required to assess this potential therapy. The advantages of a treatment that specifically targets the miR-146a pathway without compromising the functions of full-length PRL (e.g., lactation) are obvious, but multiple issues, such as efficacy, must first be assessed.
Acknowledgments
This work was supported by NIH grants 5R01HL060665-14, 1R03DA031671-02, 5U01HL099776-04, P60DK020541-32, 3P30CA013330-39, 5T32HL007675-23, and 3R01HL060665-14S1 and by the Harrington Discovery Institute. R.N. Kitsis is supported by The Dr. Gerald and Myra Dorros Chair in Cardiovascular Disease of the Albert Einstein College of Medicine. We thank the Wilf family for their ongoing generosity and support.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2013;123(5):1925–1927. doi:10.1172/JCI69286.
See the related article beginning on page 2143.
References
- 1.Demakis JG, et al. Natural course of peripartum cardiomyopathy. Circulation. 1971;44(6):1053–1061. doi: 10.1161/01.CIR.44.6.1053. [DOI] [PubMed] [Google Scholar]
- 2.Elkayam U. Clinical characteristics of peripartum cardiomyopathy in the United States: diagnosis, prognosis, and management. J Am Coll Cardiol. 2011;58(7):659–670. doi: 10.1016/j.jacc.2011.03.047. [DOI] [PubMed] [Google Scholar]
- 3.Felker GM, et al. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med. 2000;342(15):1077–1084. doi: 10.1056/NEJM200004133421502. [DOI] [PubMed] [Google Scholar]
- 4.Sliwa K, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of peripartum cardiomyopathy: a position statement from the Heart Failure Association of the European Society of Cardiology Working Group on peripartum cardiomyopathy. Eur J Heart Fail. 2010;12(8):767–778. doi: 10.1093/eurjhf/hfq120. [DOI] [PubMed] [Google Scholar]
- 5.Hayakawa Y, et al. Inhibition of cardiac myocyte apoptosis improves cardiac function and abolishes mortality in the peripartum cardiomyopathy of Galpha(q) transgenic mice. Circulation. 2003;108(24):3036–3041. doi: 10.1161/01.CIR.0000101920.72665.58. [DOI] [PubMed] [Google Scholar]
- 6.Hilfiker-Kleiner D, et al. A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell. 2007;128(3):589–600. doi: 10.1016/j.cell.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 7.Patten IS, et al. Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature. 2012;485(7398):333–338. doi: 10.1038/nature11040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halkein J, et al. MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. J Clin Invest. 2013;123(5):2143–2154. doi: 10.1172/JCI64365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Negoro S, et al. Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation. 2001;104(9):979–981. doi: 10.1161/hc3401.095947. [DOI] [PubMed] [Google Scholar]
- 10.Bolli R, et al. A murine model of inducible, cardiac-specific deletion of STAT3: its use to determine the role of STAT3 in the upregulation of cardioprotective proteins by ischemic preconditioning. J Mol Cell Cardiol. 2011;50(4):589–597. doi: 10.1016/j.yjmcc.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 11.Reuwer AQ, et al. Functional consequences of prolactin signalling in endothelial cells: a potential link with angiogenesis in pathophysiology? J Cell Mol Med. 2012;16(9):2035–2048. doi: 10.1111/j.1582-4934.2011.01499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai L, et al. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008;22(14):1948–1961. doi: 10.1101/gad.1661708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tabruyn SP, Nguyen NQ, Cornet AM, Martial JA, Struman I. The antiangiogenic factor, 16-kDa human prolactin, induces endothelial cell cycle arrest by acting at both the G0-G1 and the G2-M phases. Mol Endocrinol. 2005;19(7):1932–1942. doi: 10.1210/me.2004-0515. [DOI] [PubMed] [Google Scholar]
- 14.Sano M, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446(7134):444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- 15.Yoon YS, et al. Progressive attenuation of myocardial vascular endothelial growth factor expression is a seminal event in diabetic cardiomyopathy: restoration of microvascular homeostasis and recovery of cardiac function in diabetic cardiomyopathy after replenishment of local vascular endothelial growth factor. Circulation. 2005;111(16):2073–2085. doi: 10.1161/01.CIR.0000162472.52990.36. [DOI] [PubMed] [Google Scholar]