

Abstract
Background
Life-threatening cardiac arrhythmia is a major source of mortality worldwide. Besides rare inherited monogenic diseases such as long-QT or Brugada syndromes, which reflect abnormalities in ion fluxes across cardiac ion channels as a final common pathway, arrhythmias are most frequently acquired and associated with heart disease. The mineralocorticoid hormone aldosterone is an important contributor to morbidity and mortality in heart failure, but its mechanisms of action are incompletely understood.
Methods and Results
To specifically assess the role of the mineralocorticoid receptor (MR) in the heart, in the absence of changes in aldosteronemia, we generated a transgenic mouse model with conditional cardiac-specific overexpression of the human MR. Mice exhibit a high rate of death prevented by spironolactone, an MR antagonist used in human therapy. Cardiac MR overexpression led to ion channel remodeling, resulting in prolonged ventricular repolarization at both the cellular and integrated levels and in severe ventricular arrhythmias.
Conclusions
Our results indicate that cardiac MR triggers cardiac arrhythmias, suggesting novel opportunities for prevention of arrhythmia-related sudden death.
Keywords: aldosterone, arrhythmia, hormones, ion channels, mineralocorticoid receptors
Sudden death related to cardiac arrhythmia is a major health problem for which conventional therapies have yielded disappointing results. Myocardial infarction, hypertrophic cardiomyopathy, heart failure, and medications are common risk factors for life-threatening arrhythmia, but the mechanisms of acquired arrhythmias are not fully understood.1 Several voltage-dependent ion channels act in concert to generate action potentials (APs) that propagate through the myocardium and couple excitation to muscle contraction. Most inherited arrhythmias associated with monogenic diseases involve defects in cardiac ion channel proteins. New pathways affecting the properties of partners of channel proteins involved in the generation of cardiac excitability have been recently identified.2 Human genetics and studies on mouse models led to the discovery of multiple molecular defects responsible for arrhythmogenesis, including connexins (Cx) (gap junction protein subunits Cx40 and Cx43),3 accessory proteins required for channel trafficking to the cell membrane (KChIP2),4 or adaptor proteins that anchor ion transporters to a specialized membrane domain (ankyrin B).5 Transcription factors (Nkx2.5, HF-1b, Tbx5) are thought to control the expression of genes involved in cardiac rhythm (eg, channels, metabolic pathways), supporting the idea of arrhythmia as a polygenic disease.2 Hence, reversing the genetic reprogramming induced by transcription factors may be relevant for arrhythmia generation and/or treatment in diseases associated with a risk of sudden death.
Increasing attention has been devoted to the analysis of the pathophysiological role of the mineralocorticoid hormone aldosterone in the cardiovascular field. Aldosterone binds to the mineralocorticoid receptor (MR), which belongs to the nuclear receptor superfamily and acts as a ligand-dependent transcription factor.6 Aldosterone participates in the control of sodium reabsorption in the kidney, thus playing a key role in the regulation of blood volume and blood pressure.7 MR is also expressed in nonepithelial cells such as cardiomyocytes of both animals and humans.8,9 Two clinical trials recently indicated that MR blockade, in addition to standard therapy, reduces the rate of sudden death among patients with severe heart failure or acute myocardial infarction complicated by left ventricular dysfunction.10,11 Because aldosterone displays pleiotropic effects, MR inhibition can directly antagonize cardiac aldosterone effects but also affects other pathways such as Na/K homeostasis that could secondarily reduce the occurrence of cardiac arrhythmia. Therefore, it has been difficult to define the origins of the beneficial effects of MR inhibition on cardiac function.
To address the pathophysiological role of MR in the heart independently of other aldosterone-sensitive organs, we designed a mouse model to control MR expression exclusively in cardiomyocytes. This conditional model allows control of MR expression specifically in the selected cells (cardiomyocytes) without changes in other aldosterone-target cells, particularly in the kidney. Moreover, because transgene expression is inducible, it allows precise tuning of MR expression over time. We show here that targeted MR activation in cardiomyocytes induced defects in heart function, leading to early sudden death. Surviving animals exhibited major ECG abnormalities with prolonged ventricular repolarization and spontaneous and triggered ventricular arrhythmias. This was associated with ion channel remodeling, leading to an increase in AP duration (APD) and in Ca transient amplitude.
Methods
Generation of the Human MR Conditional Model
The human MR (hMR) coding sequence (sharing 97.5% similarity at the protein level with the mouse MR) was used to generate the hMR conditional model. The AflII-XmaIII fragment of hMR3750 plasmid12 containing 2995 bp of hMR cDNA (accession number NM_000901) from position 203 to 3198, including the full-length coding sequence, was blunt ended and inserted into the EcoRV site located between the tetO promoter and the rabbit β-globin polyadenylation sequence of the bidirectional tet-inducible pBI4 vector,13 resulting in the tetO-hMR construct. This construct was functionally tested ex vivo in a previously characterized tet-ON renal epithelial cell model14; its transactivation activity is induced by aldosterone (10 nmol/L), an effect abolished in the presence of the MR antagonist RU26752 (Figure S1 in the online-only Data Supplement). Seven founder transgenic mice were obtained after pronuclear injections of a 4230-bp AseI fragment of the tetO-hMR construct that includes the bidirectional tetO promoter, the hMR cDNA, and 1180 bp of the rabbit β-globin polyadenylation sequence into (B6D2)F2 fertilized eggs at the Service d’Expérimentation Animale et de Transgénèse laboratory (Villejuif, France). Two independent tetO-hMR mouse strains, called L1 and L2, were used for phenotypic analyses. Double-transgenic (DT) mice were obtained by crossing the tetO-hMR mice with the α-MHCtTA transactivator mouse strain.15 The MHCtTA transactivator mouse strain allows conditional, cardiac-specific expression of the transgene as early as E9.5.16 Phenotypic analysis indicated that the tetO-hMR, αMHC-tTA, and wild-type (WT) littermates were not different: they had the same survival rate, ECG parameters, and APD (Figure S2 in the Data Supplement). Thus, they were used interchangeably as controls. The data were pooled when male and female animals exhibited the same phenotype. The use of animals was in compliance with the guidelines of the European Community and approved by our Institutional Animal Care and Use Committee.
Pharmacological Treatments
All mice were fed standard chow (A03, Scientific Animal Food Engineering), and untreated animals drank tap water ad libitum. When required, drugs were given as solution in the drinking water to mothers from breeding until weaning and/or to the offspring. Treatments were as follow: (1) doxycycline (Dox) 2 mg/mL (D-9891, Sigma-Aldrich) in 2% sucrose to mask the bitter taste of Dox; (2) spironolactone 20 mg · kg−1 · d−1 (aldactone, Pharmacia SAS & Pfizer). Propranolol 10 mg · kg−1 · d−1 (Astrazeneca) was administered to the pregnant mothers until their death (E14.5). These doses have been previously demonstrated to exhibit significant blockage of mineralocorticoid and β-adrenergic receptors in mice.17–19
MR mRNA Detection
Total RNA was extracted from cardiac ventricle cells according to the TRIzol reagent protocol (Invitrogen). For RNase protection assay, an hMR 274 nucleotide riboprobe, generating a 218-bp protected fragment (kindly provided by M. Lombes),20 was used to detect hMR expression on 20 μg total RNA with the RNase protection assay III kit (Ambion) according to the manufacturer’s instructions. GAPDH was used as an internal standard (protected fragment, 164 bp) and cohybridized during the assay. For convenience and high sensitivity, hMR expression was also analyzed by reverse-transcriptase polymerase chain reaction (RT-PCR) with hMR sense (5′-AAC TAA GTG TCC CAA CAA TT-3′) and β-globin rabbit antisense (5′-CCA CAC CAG CCA CCA CCT TC-3′) specific primers. The 18S internal control was used to test RT-PCR efficiency with the 18S sense (5′ CCCTGCCCTTTGTAC-ACACC-3′) and 18S antisense (5′-CGATCCGAGGGCCTCATCA-3′) specific primers. Thirty-five cycles RT-PCR were performed as according to the manufacturer’s specifications (Invitrogen). Real-time PCR was carried out on the iCycler iQ apparatus (Biorad Laboratories, LifeSciences, France) with SYBR Green I detection. PCR was performed in triplicate for each sample using a qPCR Core Kit for SYBR Green I (Eurogentec, Belgique) containing 300 nmol/L primers, 200 μmol/L dNTPs, 3.5 mmol/L MgCl2, 1× buffer, SYBR Green I diluted (1/66 000), fluorescein 10 nmol/L, 0.025 U/μL Hot Gold Star Taq polymerase, and 3 μL template cDNA in 25 μL total volume. The following primers were used: mouse MR (mMR): forward, 5′-TCA CAT TTT TAA CAT GTG ACG GC-3′, and reverse, 5′-CTT AGT CAG CTC AGG CTT GCC-3′; hMR: same as above. HPRT was used as reference gene: forward, 5′-CTC AAC TTT AAC TGG AAA GAA TGT C-3′, and reverse, 5′-TCC TTT TCA CCA GCA AGC T-3′. PCR conditions consisted of an activation step of the Hot Gold Start Taq DNA polymerase (95°C) for 10 minutes, followed by 40 cycles of 10 seconds at 95°C (denaturation step) and 1 minute at 60°C (primer annealing, extension, and fluorescence acquisition). Serial dilution of pooled cDNA was used in each experiment to assess PCR efficiency. Expression of hMR relative to mMR was estimated by the ratio of the quantitative relative value for each gene. Quantitative relative values were calculated by DeltaDeltaCt method (Analysis of relative gene expression data using real-time quantitative PCR and the 2[−delta delta C(T)] method,21 after assessment that PCR efficiency was 100%.
Echocardiographic Analysis
Echocardiography was performed on lightly anesthetized adult mice (isoflurane, Abbot, in oxygen) (age, 1 to 3 months; body weight, 15 to 30 g) as previously described.22
ECG Recording
Surface ECG recordings were obtained in anesthetized 2- to 3-month-old adult male mice as previously described.23 The QTr and QT intervals were corrected for heart rate by this formula: QTc=QT/(RR/100),1/2 established for mice with QT and RR expressed in milliseconds. ECG recordings were obtained under baseline conditions and 10 minutes after injection of atropine sulfate (0.5 mg/kg IP) and propranolol (1 mg/kg IP) to block the autonomic nervous system. ECG readings over 24 hours were obtained after abdominal implantation of the telemetry transmeter (TA10EA-F20, Data Science International). ECG recordings were stored as digital data via the IOX 1.585 program (EMKA Technologies) and analyzed with the ECG-Auto program (EMKA Technologies). Arrhythmias were semiautomatically detected and manually validated as premature ventricular beat (PVB), couplets, and runs of ventricular tachycardia (VT). Risk of arrhythmia was assessed by the measurement of heart rate variability (HRV) as the SD of RR intervals over the recorded period (SDNN). Heart rate turbulence was computed when PVBs were recorded as the turbulence onset (To HRT) as (RR+1+RR+2)−(RR−1+RR−2)/(RR−1+RR−2)×100, where RR−1 and RR−2 are RR intervals of the 2 complexes preceding the PVB and RR+1 and RR+2 are RR intervals of sinusal complexes following the PVB.24
For intracardiac recording and pacing, mice were anesthetized with etomidate (8 mg/kg IP) and pentobarbital (30 mg/kg IP). An octopolar 2F electrode catheter with an electrode spacing of 0.5 mm (Cordis Webster) was introduced into the right atrium and ventricle through the right internal jugular vein. Using this catheter, we performed simultaneous atrial and ventricular pacing and recording. Surface ECG (lead I) and intracardiac electrograms were recorded and analyzed as described above. Intracardiac pacing was performed with a Biotronik UHS20 stimulator, modified by the manufacturer to pace at short coupling intervals. Standard pacing protocols with 1 to 3 extrastimuli were used to induce ventricular arrhythmias. Extra-stimuli were imposed under sinus rate and after trains of 6 paced beats at a cycle length of 100 ms.
Cellular Electrophysiology and Ca2+ Imaging
Experiments were conducted on age- and sex-matched littermates. Cardiac ventricular myocytes were isolated as previously described.25 APs and ICa were recorded using solutions and protocols previously described.25 Outward K+ currents were evoked during 300-ms depolarizing voltage steps to potentials between −70 and 60 mV (10-mV increments) from a holding potential of −80 mV. Internal solution was the same as for AP recordings; external solution was modified by equimolar replacement of NaCl with 138-mmol/L choline Cl, 1-mmol/L CoCl2, and 1 mmol/L BaCl2. Ito values were obtained by subtracting corresponding current records with and without a 100-ms inactivating prepulse to 30 mV. For Ca2+ imaging, field-stimulated and fluo-3AM–loaded cells were viewed with a confocal microscope (LSM 510 Zeiss) as described elsewhere.26 [Ca2+]i was derived from the fluorescent signal.27
Statistical Analysis
Data are expressed as mean±SEM. Groups were compared by use of an unpaired Student t test or χ2 test with the StatView software (Abacus Concepts Inc).
Results
Conditional hMR Overexpression in Cardiomyocytes Leads to Sudden Death Without Cardiac Structural Alteration
Conditional expression of hMR was achieved in transgenic mice with the tet-Off inducible system28 (Figure 1A). DT mice—DT1 and DT2, respectively—were obtained by crossing L1 or L2 tetO-hMR mice with the previously described αMHC-tTA transactivator mouse strain.15 Cardiac hMR mRNA as measured by RNase protection assay and RT-PCR was expressed in DT only (Figure 1B and 1C), showing that the system is not leaky and that hMR is not expressed in monotransgenic tetO-hMR mice. hMR expression was prevented when Dox was administered to DT (Figure 1C) and restricted to the heart, not to kidney, colon, or other tissues (Figure 1D). The apparent ratio of exogenous to endogenous MR mRNA expression, as estimated by qualitative PCR, was ≈4/1 (ratio of hMR to mMR, 4±0.9; n=5). Taken together, these findings indicate that hMR expression is tightly controlled in vivo. Substantial efforts to measure MR expression at the protein level were made using currently available anti-MR antibodies; unfortunately, no specific signal could be obtained in mouse tissues (including kidney).
Figure 1.

In vivo cardiac expression of hMR. A, hMR conditional system. Tetracycline-inducible system (tet system) is a bigenic system allowing inducible expression of the gene of interest. To obtain cardiac-specific, Dox-controlled transgene expression, α MHC-tTA transgenic mouse line (transactivator strain, α MHC-tTA), expressing tTA transactivator under control of cardiac-specific α MHC promoter, is mated with hMR transgenic mice (acceptor strain, tetO-hMR), leading to DT conditional and cardiac-specific model. B, hMR mRNA, as determined by RNAse protection assay, is expressed in heart of 1-month-old DT mice; no expression was detectable in monotransgenic littermates used as controls. C, hMR mRNA, as determined by RT-PCR, is expressed in heart of 10-week-old DT animals; no expression was detectable in monotransgenic tetO-hMR littermates used as controls (1 mouse per lane). Expression in DT is prevented when animals received Dox (+Dox). D, hMR expression, as determined by RT-PCR, is restricted to heart of DT. Expression of the transgene is absent in brain, colon, kidney, liver, lung, muscle, and surrenal gland.
DT offspring were not present at weaning with the expected mendelian ratio (Figure 2A), pointing to embryonic and/or early postnatal lethality. Fifty percent of the DT2 and female DT1 mice died before weaning; male DT1 mice exhibited mortality later. Embryonic lethality occurred between E12.5 and E14.5; the ratios of DT2 embryos collected at E10.5 were 32% and 25% at E12.5 (ie, close to the expected mendelian ratio), whereas it was only 11% at E14.5 (P<0.001, χ2 test). Lethality of DT could be prevented when pregnant mothers received either Dox to suppress hMR expression or the MR antagonist spironolactone (Figure 2B). These results demonstrate that death was specifically related to hMR overexpression and activation. Histological analysis showed that hearts of the E13.5 DT embryos were normal (data not shown), indicating that embryonic death occurred in absence of major structural defects. After birth, 31% of the surviving DT1 females and 50% of the surviving DT1 males died between week 3 and 6. Plasma aldosterone concentrations were normal in 2-month-old DT (control: 324±95 pg/mL, n=5; DT: 269±64 pg/mL, n=6; P=NS) and corticosterone (control: 266±45 ng/mL, n=4; DT: 322±53 ng/mL, n=4; NS). As expected from the mouse model (hMR expression solely in the heart, not in the kidney), plasma electrolytes such as Na, K, Ca, and Mg were not altered in DT versus controls (Table S1 in the Data Supplement). Mild cardiomyopathy was occasionally observed with an alteration in the left ventricle contractility in about one fourth of the surviving DT, whereas in other DTs, cardiac function was normal (see individual values in Figure 3A). The variable penetrance of the DT phenotype in the mixed genetic background studied here may reflect the effect of modifier genes. Because aldosterone has been previously proposed to induce cardiac fibrosis and inflammation, specific attention was focused on this point. Fibrosis, inflammatory cell infiltrate, and apoptosis were not observed in hearts of 2-month-old and 1-year-old DTs (data not shown).
Figure 2.

Cardiac and Dox-controlled expression of hMR-induced early lethality. A, Cardiac hMR overexpression induced lethality of DT mice vs control littermates. Only 12 of 125 female DT1, 12 of 110 male DT2, and 12 of 94 female DT2 animals were alive at time of genotyping (3 weeks). Both strains were affected, but gender effect was observed in DT1; ratio of DT1 males (29 DT1/143) is not significantly different from the 25% expected mendelian ratio. B, Lethality did not occur when hMR expression was prevented by administration of Dox (49DT2/201) vs nontreated animals (24DT2/204) or when hMR activity was antagonized in vivo by spironolactone (spiro) (26DT2/102). β-Adrenergic receptor antagonist propranolol (propra) (26DT2/84) also prevented embryonic lethality when given to pregnant mother. *P<0.001, **P<0.0005, χ2 test; numbers in bars are numbers of surviving DT at 3 weeks of age.
Figure 3.

In vivo cardiac phenotypic characterization of hMR-overexpressing mice. A, Most DT mice had normal cardiac function and no cavity dilation as assessed with ejection fraction (EF; top) and left ventricular end-diastolic diameter/body weight (LVEDD/BW; bottom) measured by Doppler echocardiography. Cardiac hMR overexpression was occasionally associated with signs of heart failure with decrease in ejection fraction. Note in individual data that only 3 DT animals were affected among 12 at 5 weeks, reflecting variability of this phenotype. n=25 control mice; n=12 DT. B, Surface ECG in 2-month-old DT animals. Spontaneous ventricular arrhythmias such as PVB (top) or VT (bottom) were observed in DT mice only. C, Pacing-induced arrhythmias. Typical recordings obtained in WT and DT mice. Top trace depicts lead I of surface ECG; bottom trace, intracardiac electrogram. In DT, VT was induced by train of 9 stimuli at cycle length of 100 ms, followed by extrastimulus (S2) with coupling interval of 40 ms. In control mouse, pacing had no effect. D, Recordings obtained in same DT. Pacing protocol consisted of trains of 9 stimuli (cycle length, 100 ms), followed by 2 extrastimuli: first with a coupling interval of 40 ms and second with coupling decreasing from 52 ms (first salvo) to 44 ms (last salvo). *P<0.01, Student t test.
Conditional Overexpression of hMR in Cardiomyocytes Is Associated With Ventricular Arrhythmia
The lethal phenotype observed in DT embryos and young animals in absence of structural cardiac alterations was suggestive of a functional heart defect such as fatal arrhythmia. In keeping with this hypothesis, administration of the β-adrenergic receptor blocker propranolol (a class II antiarrhythmic drug) to the pregnant mother prevented embryonic lethality (Figure 2B). Surface ECGs recorded under anesthesia revealed that surviving adult DTs had prolonged AV (PQ interval) and ventricular (QRS interval) conduction time (Figure S3 in the Data Supplement). The early and late phases of ventricular repolarization (QTr and QT intervals) were increased, although the prolongation of total repolarization was significant only after blockade of the autonomic nervous system (the Table). Seventy percent of DT mice had spontaneous PVBs on short term (15 to 30 minutes) ECG recording, whereas PVBs were not observed in control littermates. Couplets and short VT runs occurred spontaneously in DTs (Figure 3B).
Table.
ECG Characteristics of WT and DT1 Mice Under Baseline Conditions and After Blockade of the Autonomic Nervous System
| RR, ms | P, ms | PQ, ms | QRS, ms | QTr, ms | QT, ms | QTrc, ms | QTc, ms | |
|---|---|---|---|---|---|---|---|---|
| WT | ||||||||
| Baseline | 136±17 | 19±1 | 34±1 | 15±1 | 26±1 | 62±3 | 23±1 | 55±3 |
| ANS– | 200±12 | 21±1 | 43±1 | 16±1 | 30±1 | 75±2 | 22±1 | 54±3 |
| DT | ||||||||
| Baseline | 139±11 | 19±1 | 46±2‡ | 17±1† | 33±2† | 68±5 | 29±2* | 57±3 |
| ANS– | 185±16 | 21±1 | 51±2† | 20±1‡ | 40±2‡ | 93±4* | 29±1‡ | 68±1† |
RR indicates RR interval duration; P, P wave duration; PQ, PQ interval duration; QRS, QRS complex duration; QTr, QTr interval duration; QT, QT interval duration; QTrc and QTc, corrected QTr and QT interval duration; and ANS–, after blockade of the autonomic nervous system.
Data are expressed as mean±SEM of 8 WT and 7 DT mice, except for the QT and QTc intervals in DT mice (n=6 mice under baseline and 5 under ANS–).
P<0.05,
P<0.01,
P<0.001, DT vs WT.
Implantable telemetry was used to record long-term ECG in conscious surviving adults. To assess arrhythmia susceptibility, we measured heart rate turbulence onset (To HRT) and HRV, which are both recognized as risk markers of arrhythmia and sudden death in humans. DTs had a smaller HRV over the recorded period as assessed with SDNN (control: 36.8±4.25 bpm; DT: 28.07±1.9 bpm; n=8 in each group; P<0.05) and exhibited a positive heart rate turbulence onset, at variance with WT, in which it was negative (control: −6.45±3.15%; DT: 14.3±4.7%; n=8 in each group; P<0.01). These features are in agreement with an increased risk of arrhythmia in DTs. The susceptibility to the induction of arrhythmia was gauged by programmed intracardiac stimulation. Ventricular pacing-induced non-sustained VT in 6 of 7 DTs but in only 1 of 7 controls (P<0.05, DT versus control) (Figure 3C). All mice with inducible VT could be reinduced at least once (Figure 3D). Taken together, although cardiac structure and function were normal, DT mice disclosed several features suggestive of arrhythmogenic disease, namely QT prolongation and spontaneous and triggered ventricular arrhythmias.
Cardiac hMR Overexpression Leads to the Prolongation of AP
Changes in the QRS and QT ECG intervals and the occurrence of spontaneous and triggered arrhythmias suggest alterations in myocyte APs. APs were recorded in isolated ventricular cardiomyocytes using the conventional whole-cell current clamp technique (Figure 3A). The APD was significantly prolonged at 20%, 50%, and 90% of repolarization in DT compared with WT (4.1±0.6 versus 1.2±0.1 ms at 20%, P<0.005; 14.1±1.8 versus 3.9±0.6 ms at 50%, P<0.005; and 80.9±4.5 versus 39.8±6.3 ms at 90%, P<0.0005, DT versus WT cells, respectively), whereas neither the resting membrane potential nor the AP amplitude was different. AP lengthening was specific for hMR overexpression because electrophysiological characteristics of monotransgenic littermates (ie, not expressing hMR) were similar to those of WT (Figure S2 in the Data Supplement). Moreover, Dox administration during pregnancy and to offspring prevented APD increase in DTs (Figure 4B, +Dox). To assess whether changes in APD also occurred if hMR expression is started solely in adult mice but not during embryonic development, we analyzed 2-month-old mice that had received Dox until 1 month of age and expressed hMR from that time point. APD prolongation was obvious compared with control littermates submitted to the same protocol (Figure 4B, Dox retrieval). In vivo hMR antagonism with spironolactone prevented APD lengthening in DT mice (Figure 4B, +spiro). APD was normalized in DT animals and was not different between controls and DT mice treated with spironolactone.
Figure 4.
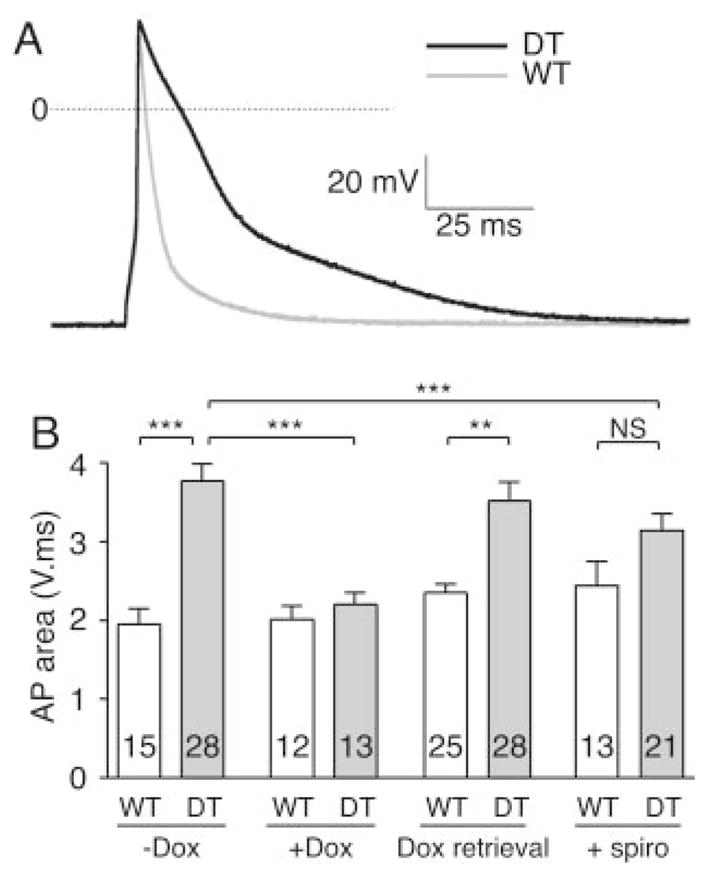
Overexpression of hMR leads to prolongation of AP repolarization in isolated cardiomyocytes. A, Superimposed representative examples of APs recorded in ventricular myocytes isolated from littermate WT (gray line) and DT (black line) mice. APD was significantly lengthened from 20% of repolarization. B, AP prolongation is specific for hMR overexpression. Mean AP area was evaluated by integration of voltage variation over time. Because amplitudes or zero current voltages are not modified, increase in AP area represents lengthening of APD. AP area of isolated myocytes from DT was increased vs WT (−Dox). APD prolongation was prevented in Dox-treated DT mice (+Dox). Dox treatment had no effect in WT vs nontreated WT mice. APD lengthening was also present when hMR overexpression was started in adult mice only (Dox retrieval). In vivo hMR antagonism with spironolactone (+spiro) blunted APD lengthening in DT. Numbers in bars represent numbers of patches from 2 to 4 mice in each group. **P<0.005, ***P<0.0005, Student t test.
Thus, cardiac hMR overexpression induced a specific APD prolongation, which most probably underlies the QT interval prolongation and the link to arrhythmogenesis in DTs. Indeed, early afterdepolarization occurred in 18% of DT myocytes, whereas it was never seen in controls (Figure S4 in the Data Supplement). This alteration may be 1 of the causal mechanisms for induction of arrhythmia. Importantly, the increase in APD also occurred when hMR was overexpressed in adulthood only, suggesting a potential role in the pathogenesis of acquired cardiac arrhythmias.
Electrical Remodeling Underlying APD Lengthening
To identify the mechanisms that result in increased APD, we examined the outward and inward currents that are primarily responsible for the APD. K+ and Ca2+ currents were recorded in isolated ventricular cardiomyocytes, normalized to cell capacitance (Cm), and expressed as densities (pA/pF) (Figure 5A). No differences in Cm of DT (173±11 pF, n=51 cells) and WT (162±8, n=34 cells) myocytes were observed, indicating a lack of cellular hypertrophy. In adult mouse ventricle, outward K+ currents involved in cardiac repolarization consist of 3 components distinguishable by their specific time and voltage dependency and sensitivity to pharmacological agents: (1) a transient current (Ito) that comprises a fast (Ito,f) and a slow (Ito,s) component, (2) a low-concentration 4-aminopyridine–sensitive current (IKur), and (3) a noninactivating steady-state component (ISS). Currents recorded after Ito inactivation were not significantly different in DT and WT ventricular myocytes; at 40 mV in pA/pF, the 250-μmol/L 4-aminopyridine–sensitive IKur density was 5.3±0.5 (n=31) versus 5.4±0.4 (n=15) and the ISS density was 7.1±0.3 (n=34) versus 6.8±0.3 (n=16) for DT versus WT, respectively. In contrast, DT myocytes had a smaller Ito (Figure 5A, left), without change in Ito kinetics (data not shown).
Figure 5.

Electrical remodeling in DT mice. A, K+ transient out-ward current (Ito) is downregulated, whereas L-type Ca2+ current (ICa) is increased in DT myocytes. Left, After Ito amplitude normalization to membrane capacitance (to account for cell size variation), current densities are plotted as function of voltage. There is significant decrease in Ito densities in DT myocytes (●, n=29) vs WT (○, n=18). Right, Current density-voltage relationship for ICa measured in myocytes from 1-month-old WT (○, n=15) and DT (●, n=16) mice, showing significant increase in ICa density in DT myocytes in −20- to 40-mV voltage range. B, hMR overexpression increases [Ca2+]i transients in DT myocytes. Representative line scan images of fluo-3AM fluorescence recorded in WT and DT myocytes. Below images are corresponding [Ca2+]i transients. Changes in limits of cell, at top and bottom edges of image, allowed measurement of contraction. C, Increased pooled peak amplitude of [Ca2+]i transients in DT vs WT littermates. D, Cell shortening analyzed in WT and DT myocytes showing significant stimulation of excitation-contraction coupling. n indicates number of patches from 2 to 4 mice in each group. *P<0.01, **P<0.005, ***P<0.0005, Student t test.
An increase in Ca2+ current in aldosterone-treated adult cardiomyocytes has previously been reported.29 We therefore investigated whether in vivo hMR overexpression modulates Ca2+ signaling. We observed a 1.5-fold increase in whole-cell Ca2+ current (ICa) in DT mice compared with controls (Figure 5A, right), whereas neither time- nor voltage-dependent properties of the current varied (data not shown). In vivo hMR antagonism with spironolactone prevented the increase in whole-cell ICa in DT (Gmax, pA/pF, controls: 186±16 [n=14]; DT: 252±10 [n=14], P<0.005; controls+spironolactone: 199±11 [n=11]; DT+spironolactone: 218±8 [n=10]; P=NS).
Besides its role in AP repolarization, Ca2+ influx into the cardiomyocyte (through ICa) opens the ryanodine receptor responsible for Ca2+ release from the sarcoplasmic reticulum, leading to a transient elevation of cytoplasmic Ca2+ concentration that triggers contraction. Single myocytes isolated from DT mice displayed larger [Ca2+]i transients (Figure 5B and 5C) and cell contraction (percent shortening) (Figure 5B and 5D) than myocytes from littermate controls. The increase in the [Ca2+]i transient in DT mice was not explained by a change in the sarcoplasmic reticulum Ca2+ storage, because the amount of Ca2+ released (estimated after caffeine application) was not altered (DT: 1130±218 nmol/L [n=43]; controls: 1203±224 nmol/L [n=29]; P=NS). Moreover, [Ca2+]i transient decay time in DT mice (115±4 ms, n=72) was similar to that in WT mice (118±4 ms, n=32).
Taken together, these data show that the activity of the Ca2+ recycling process is not altered in DT mice. Thus, the prolonged AP and the corresponding risk of arrhythmia are due to specific ion channel remodeling, namely downregulation of Ito and upregulation of ICa.
Discussion
To gain new insights into the role of the MR in cardiac pathophysiology, we designed a conditional mouse model aimed at specifically controlling MR expression exclusively in cardiomyocytes. This model allows precise analysis of MR function in heart without affecting the concentration of circulating aldosterone or plasma electrolyte composition. We show here that restricted human MR overexpression in cardiomyocytes leads to cardiac arrhythmias. Of note, no fibrosis was observed in the hMR overexpressing model presented here.
Other experimental models have addressed the pathophysiological role of the MR. Two mouse models with constitutive alteration of MR expression have been generated. MR knockout mice die during the first week of life from renal salt wasting, precluding the evaluation of the effects of myocardial MR suppression in adult animals.30 Constitutive hMR overexpression in all MR-expressing tissues, notably the kidney and heart, leads to both renal and mild cardiac alterations without cardiac fibrosis.31 Of interest, during blood pressure measurements, it was noticed that these mice had increased heart rate and dysrhythmia. Such a model is of interest to explore widespread functions of MR in vivo, but it does not allow one to discriminate between direct and secondary effects of hMR overexpression. The targeted approach presented here ruled out the possibility of secondary effects resulting from changes in renal ion homeostasis or general aldosterone-induced disturbances and focused on the specific role of the MR in the cardiomyocytes. We previously documented the consequences of conditional downexpression of the endogenous murine MR in the heart; this model unexpectedly led to profound structural cardiac remodeling associated with dilated cardiomyopathy and fibrosis in the absence of changes in circulating aldosterone.22 On the whole, it is interesting to note that increasing cardiac MR in mice leads to arrhythmia without fibrosis, whereas its underexpression is associated with cardiac remodeling. A phenotype resembling the MR antisense mice has been reported in a transgenic model with cardiac-specific overexpression of the 11 β-hydroxysteroid dehydrogenase type 2.32 Both models resulted in local MR/GR or aldosterone/glucocorticoid imbalance, pointing to a crucial role of MR or GR signaling in cardiac physiology.
Another experimental approach consists of modifying the ligand for the MR rather than the MR itself. A complex rat model including uninephrectomy, high-salt diet, and 1-to 2-month administration of aldosterone indicated that aldosterone excess leads to detrimental effects on cardiovascular functions.33–35 The molecular mechanisms of such effects remain unclear, and it is not known whether the alterations are related to direct cardiac effects of aldosterone, to sodium load, or to consequences of the renal actions of aldosterone on Na+ and K+ homeostasis. In contrast, chronic hyperaldosteronism in a rescued mouse model of ENac gene inactivation failed to induce cardiac remodeling and fibrosis under a normal-salt diet.36 Taken together, these observations indicate that manipulating the ligand concentration (aldosterone), combined with other pathological stresses (high-sodium diet, uninephrectomy, etc), is not equivalent to changes in MR expression or activation.
Our data are of particular interest in view of clinical trials exploring the electrophysiological consequences of inappropriate MR activation in human heart. The synthetic mineralocorticoid fludrocortisone exacerbates37 while spironolactone improves38,39 electrophysiological parameters such as QT interval dispersion. Spironolactone increases heart rate variability38,40 and decreases PVBs and nonsustained VT episode occurrence41,42 in patients with heart failure at rest and during exercise42; consistent with these observations, cardiac hMR overexpression, as reported here, reduced HRV and increased PVB and nonsustained VT episodes.
Increased plasma aldosterone concentration can be associated with prolonged inhibition of ACE, leading to inappropriate MR activation despite renin-angiotensin system blockade in heart failure.43 These clinical data suggested that MR antagonism, on top of standard therapy, might be beneficial. Two large-scale randomized trials confirmed this hypothesis. The RALES study demonstrated a decrease in mortality with spironolactone treatment in patients with severe heart failure,10 and EPHESUS recently showed an impressive benefit of eplerenone, a second-generation MR antagonist, in patients with acute myocardial infarction with left ventricular systolic dysfunction.11 Half of the benefit in both RALES and EPHESUS was attributed to a decrease in sudden cardiac death. The mechanisms responsible for these favorable effects probably rely on both renal changes in electrolyte excretion and myocardial fibrosis inhibition, thereby reducing arrhythmogenic substrates. From the effects of spironolactone observed in our model, we propose that part of the benefit also relies on inhibition of changes in cardiomyocyte electrophysiological properties related to cardiac MR activation by corticosteroid hormones. Indeed, aldosterone per se has previously been linked to ionic channel remodeling in isolated rat adult cardiomyocytes with upregulation of Ca2+ current, a decrease in the Ito K+ current, and lengthening of the late phase of AP repolarization.25,29 In the present study, we demonstrate that cardiac-specific hMR overexpression in vivo leads to similar effects at the cellular level and is associated with severe ventricular arrhythmias, even when hMR expression occurred in adulthood, pointing to MR involvement in acquired disease.
Genetically modified animals may be useful to identify altered gene networks as candidate contributors in polygenic disease, particularly for arrhythmia in humans. Our data strongly support the hypothesis that the MR could be considered a candidate gene for human arrhythmia, acting as an integrator in the control of ion channel expression and/or function and possibly of other factors important in rhythm disturbance. As a ligand-dependent transcription factor, MR may act as a genetic modifier that confers susceptibility to life-threatening cardiac arrhythmias. Therefore, aldosterone/MR targets may represent novel therapeutic targets in disease-associated electrical remodeling. To date, aldosterone-induced proteins have typically been identified through cell culture systems or supraphysiological concentrations of aldosterone. Mouse conditional models may be a key step in identifying relevant targets. Indeed, differential gene expression analysis at various time points before and during onset or remission of the pathology (through controlled upregulation or downregulation of MR expression) should allow identification of altered signaling cascades, avoiding interference with long-term compensatory mechanisms that can preclude identification of relevant targets. Microarray analyses using hearts from transgenic mice with hMR overexpression and normal circulating aldosterone are in progress to define genes specifically regulated by MR in the heart. This could allow the discovery and validation of drugs that affect function of multiple targets (as opposed to channel-specific drugs) and have a broader spectrum of action, adding novel possibilities for arrhythmia prevention or therapy.
Supplementary Material
Acknowledgments
A.O.-P. was supported by the Ministère de la Recherche and the Fondation pour la Recherche Médicale, Y.S.-M. was supported by a CIFRE program of ANVAR and the Nucleis SA company (Lyon, France). G.I.F. is the recipient of a Burroughs-Wellcome Fund Clinical Scientist Award in Translational Research. This work was supported by grants from INSERM, the Ministère de la Recherche et de la Technologie (ACI grant), and the Fondation de France. We thank C. Heymes, A. Meulemans, V. Delage, M. Peuchmaur, and the Centre d’explorations fonctionnelles intégrées of the Bichat Federative Research Institute 02 for help in the phenotypic characterization of the model. We also thank P. Corvol and X. Jeunemaître for critical reading of the manuscript and C. Ferreira for his help in animal care.
Footnotes
The online-only Data Supplement can be found with this article at http://circ.ahajournals.org/cgi/content/full/CIRCULATIONAHA.104.503706/DC1.
References
- 1.Huikuri HV, Castellanos A, Myerburg RJ. Sudden death due to cardiac arrhythmias. N Engl J Med. 2001;345:1473–1482. doi: 10.1056/NEJMra000650. [DOI] [PubMed] [Google Scholar]
- 2.Cheng CF, Kuo HC, Chien KR. Genetic modifiers of cardiac arrhythmias. Trends Mol Med. 2003;9:59– 66. doi: 10.1016/s1471-4914(03)00004-2. [DOI] [PubMed] [Google Scholar]
- 3.Gutstein DE, Morley GE, Tamaddon H, et al. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88:333–339. doi: 10.1161/01.res.88.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuo HC, Cheng CF, Clark RB, et al. A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell. 2001;107:801– 813. doi: 10.1016/s0092-8674(01)00588-8. [DOI] [PubMed] [Google Scholar]
- 5.Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634– 639. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 6.Farman N. Molecular and cellular determinants of mineralocorticoid selectivity. Curr Opin Nephrol Hypertens. 1999;8:45–51. doi: 10.1097/00041552-199901000-00008. [DOI] [PubMed] [Google Scholar]
- 7.Bonvalet JP. Regulation of sodium transport by steroid hormones. Kidney Int. 1998;65:S49–S56. [PubMed] [Google Scholar]
- 8.Lombes M, Oblin ME, Gasc JM, et al. Immunohistochemical and biochemical evidence for a cardiovascular mineralocorticoid receptor. Circ Res. 1992;71:503–510. doi: 10.1161/01.res.71.3.503. [DOI] [PubMed] [Google Scholar]
- 9.Lombes M, Alfaidy N, Eugene E, et al. Prerequisite for cardiac aldosterone action: mineralocorticoid receptor and 11 β-hydroxysteroid dehydrogenase in the human heart. Circulation. 1995;92:175–182. doi: 10.1161/01.cir.92.2.175. [DOI] [PubMed] [Google Scholar]
- 10.Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure: Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709–717. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 11.Pitt B, Remme W, Zannad F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–1321. doi: 10.1056/NEJMoa030207. [DOI] [PubMed] [Google Scholar]
- 12.Arriza JL, Weinberger C, Cerelli G, et al. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science. 1987;237:268–275. doi: 10.1126/science.3037703. [DOI] [PubMed] [Google Scholar]
- 13.Baron U, Freundlieb S, Gossen M, et al. Co-regulation of two gene activities by tetracycline via a bidirectional promoter. Nucleic Acids Res. 1995;23:3605–3606. doi: 10.1093/nar/23.17.3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puttini S, Beggah AT, Ouvrard-Pascaud A, et al. Tetracycline-inducible gene expression in cultured rat renal CD cells and in intact CD from transgenic mice. Am J Physiol Renal Physiol. 2001;281:F1164–F1172. doi: 10.1152/ajprenal.0360.2000. [DOI] [PubMed] [Google Scholar]
- 15.Yu Z, Redfern CS, Fishman GI. Conditional transgene expression in the heart. Circ Res. 1996;79:691– 697. doi: 10.1161/01.res.79.4.691. [DOI] [PubMed] [Google Scholar]
- 16.Dor Y, Camenisch TD, Itin A, et al. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development. 2001;128:1531–1538. doi: 10.1242/dev.128.9.1531. [DOI] [PubMed] [Google Scholar]
- 17.Knowles JW, Esposito G, Mao L, et al. Pressure-independent enhancement of cardiac hypertrophy in natriuretic peptide receptor A-deficient mice. J Clin Invest. 2001;107:975–984. doi: 10.1172/JCI11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MacDonald P, MacKenzie S, Ramage LE, et al. Corticosteroid regulation of amiloride-sensitive sodium-channel subunit mRNA expression in mouse kidney. J Endocrinol. 2000;165:25–37. doi: 10.1677/joe.0.1650025. [DOI] [PubMed] [Google Scholar]
- 19.Michel F, Ambroisine ML, Duriez M, et al. Aldosterone enhances ischemia-induced neovascularization through angiotensin II– dependent pathway. Circulation. 2004;109:1933–1937. doi: 10.1161/01.CIR.0000127112.36796.9B. [DOI] [PubMed] [Google Scholar]
- 20.Deppe CE, Heering PJ, Viengchareun S, et al. Cyclosporine a and FK506 inhibit transcriptional activity of the human mineralocorticoid receptor: a cell-based model to investigate partial aldosterone resistance in kidney transplantation. Endocrinology. 2002;143:1932–1941. doi: 10.1210/endo.143.5.8821. [DOI] [PubMed] [Google Scholar]
- 21.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402– 408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 22.Beggah AT, Escoubet B, Puttini S, et al. Reversible cardiac fibrosis and heart failure induced by conditional expression of an antisense mRNA of the mineralocorticoid receptor in cardiomyocytes. Proc Natl Acad Sci U S A. 2002;99:7160–7165. doi: 10.1073/pnas.102673599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lande G, Demolombe S, Bammert A, et al. Transgenic mice overexpressing human KvLQT1 dominant-negative isoform, part II: pharmacological profile. Cardiovasc Res. 2001;50:328–334. doi: 10.1016/s0008-6363(01)00232-2. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt G, Malik M, Barthel P, et al. Heart-rate turbulence after ventricular premature beats as a predictor of mortality after acute myocardial infarction. Lancet. 1999;353:1390–1396. doi: 10.1016/S0140-6736(98)08428-1. [DOI] [PubMed] [Google Scholar]
- 25.Benitah JP, Perrier E, Gomez AM, et al. Effects of aldosterone on transient outward K+ current density in rat ventricular myocytes. J Physiol. 2001;537(pt 1):151–160. doi: 10.1111/j.1469-7793.2001.0151k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gomez AM, Kerfant BG, Vassort G, et al. Autonomic regulation of calcium and potassium channels is oppositely modulated by microtubules in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2004;286:H2065–2071. doi: 10.1152/ajpheart.00933.2003. [DOI] [PubMed] [Google Scholar]
- 27.Gomez AM, Schwaller B, Porzig H, et al. Increased exchange current but normal Ca2+ transport via Na+-Ca2+ exchange during cardiac hypertrophy after myocardial infarction. Circ Res. 2002;91:323–330. doi: 10.1161/01.res.0000031384.55006.db. [DOI] [PubMed] [Google Scholar]
- 28.Bujard H. Controlling genes with tetracyclines. J Gene Med. 1999;1:372–374. doi: 10.1002/(SICI)1521-2254(199909/10)1:5<372::AID-JGM61>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 29.Benitah JP, Vassort G. Aldosterone upregulates Ca(2+) current in adult rat cardiomyocytes. Circ Res. 1999;85:1139–1145. doi: 10.1161/01.res.85.12.1139. [DOI] [PubMed] [Google Scholar]
- 30.Berger S, Bleich M, Schmid W, et al. Mineralocorticoid receptor knockout: pathophysiology of Na+ metabolism. Proc Natl Acad Sci U S A. 1998;95:9424–9429. doi: 10.1073/pnas.95.16.9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le Menuet D, Isnard R, Bichara M, et al. Alteration of cardiac and renal functions in transgenic mice overexpressing human mineralocorticoid receptor. J Biol Chem. 2001;276:38911–38920. doi: 10.1074/jbc.M103984200. [DOI] [PubMed] [Google Scholar]
- 32.Qin W, Rudolph AE, Bond BR, et al. Transgenic model of aldosterone-driven cardiac hypertrophy and heart failure. Circ Res. 2003;93:69–76. doi: 10.1161/01.RES.0000080521.15238.E5. [DOI] [PubMed] [Google Scholar]
- 33.Brilla CG, Weber KT. Mineralocorticoid excess, dietary sodium, and myocardial fibrosis. J Lab Clin Med. 1992;120:893–901. [PubMed] [Google Scholar]
- 34.Robert V, Silvestre JS, Charlemagne D, et al. Biological determinants of aldosterone-induced cardiac fibrosis in rats. Hypertension. 1996;26(pt 1):971–978. doi: 10.1161/01.hyp.26.6.971. [DOI] [PubMed] [Google Scholar]
- 35.Young M, Head G, Funder J. Determinants of cardiac fibrosis in experimental hypermineralocorticoid states. Am J Physiol. 1995;269(pt 1):E657–E662. doi: 10.1152/ajpendo.1995.269.4.E657. [DOI] [PubMed] [Google Scholar]
- 36.Wang Q, Clement S, Gabbiani G, et al. Chronic hyperaldosteronism in a transgenic mouse model fails to induce cardiac remodeling and fibrosis under a normal-salt diet. Am J Physiol Renal Physiol. 2004;286:F1178–F1184. doi: 10.1152/ajprenal.00386.2003. [DOI] [PubMed] [Google Scholar]
- 37.Lim PO, Farquharson CA, Shiels P, et al. Adverse cardiac effects of salt with fludrocortisone in hypertension. Hypertension. 2001;37:856– 861. doi: 10.1161/01.hyp.37.3.856. [DOI] [PubMed] [Google Scholar]
- 38.Yee KM, Pringle SD, Struthers AD. Circadian variation in the effects of aldosterone blockade on heart rate variability and QT dispersion in congestive heart failure. J Am Coll Cardiol. 2001;37:1800–1807. doi: 10.1016/s0735-1097(01)01243-8. [DOI] [PubMed] [Google Scholar]
- 39.Akbulut M, Ozbay Y, Ilkay E, et al. Effects of spironolactone and metoprolol on QT dispersion in heart failure. Jpn Heart J. 2003;44:681– 692. doi: 10.1536/jhj.44.681. [DOI] [PubMed] [Google Scholar]
- 40.MacFadyen RJ, Barr CS, Struthers AD. Aldosterone blockade reduces vascular collagen turnover, improves heart rate variability and reduces early morning rise in heart rate in heart failure patients. Cardiovasc Res. 1997;35:30–34. doi: 10.1016/s0008-6363(97)00091-6. [DOI] [PubMed] [Google Scholar]
- 41.Barr CS, Lang CC, Hanson J, et al. Effects of adding spironolactone to an angiotensin-converting enzyme inhibitor in chronic congestive heart failure secondary to coronary artery disease. Am J Cardiol. 1995;76:1259–1265. doi: 10.1016/s0002-9149(99)80353-1. [DOI] [PubMed] [Google Scholar]
- 42.Ramires FJ, Mansur A, Coelho O, et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idiopathic dilated or to ischemic cardiomyopathy. Am J Cardiol. 2000;85:1207–1211. doi: 10.1016/s0002-9149(00)00729-3. [DOI] [PubMed] [Google Scholar]
- 43.Lee AF, MacFadyen RJ, Struthers AD. Neurohormonal reactivation in heart failure patients on chronic ACE inhibitor therapy: a longitudinal study. Eur J Heart Fail. 1999;1:401– 406. doi: 10.1016/s1388-9842(99)00046-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.