

Abstract
Background
Heterotrimeric guanine nucleotide binding proteins of the G12/13 subfamily, which includes the α-subunits Gα12 and Gα13, stimulate the monomeric G protein RhoA through interaction with a distinct subset of Rho-specific guanine nucleotide exchange factors (RhoGEFs). The structural features that mediate interaction between Gα13 and RhoGEFs have been examined in crystallographic studies of the purified complex, whereas a Gα12:RhoGEF complex has not been reported. Several signaling responses and effector interactions appear unique to Gα12 or Gα13, despite their similarity in amino acid sequence.
Methods
To comprehensively examine Gα12 for regions involved in RhoGEF interaction, we screened a panel of Gα12 cassette substitution mutants for binding to leukemia-associated RhoGEF (LARG) and for activation of serum response element mediated transcription.
Results
We identified several cassette substitutions that disrupt Gα12 binding to LARG and the related p115RhoGEF. These Gα12 mutants also were impaired in activating serum response element mediated signaling, a Rho-dependent response. Most of these mutants matched corresponding regions of Gα13 reported to contact p115RhoGEF, but unexpectedly, several RhoGEF-uncoupling mutations were found within the N- and C-terminal regions of Gα12. Trypsin protection assays revealed several mutants in these regions as retaining conformational activation. In addition, charge substitutions near the Gα12 N-terminus selectively disrupted binding to LARG but not p115RhoGEF.
Conclusions
Several structural aspects of the Gα12:RhoGEF interface differ from the reported Gα13:RhoGEF complex, particularly determinants within the C-terminal α5 helix and structurally uncharacterized N-terminus of Gα12. Furthermore, key residues at the Gα12 N-terminus may confer selectivity for LARG as a downstream effector.
Keywords: Gα12, Gα13, Heterotrimeric G protein, RhoGEF, Rho, LARG, Serum response element
Background
The G12/13 subfamily of heterotrimeric guanine nucleotide binding proteins (G proteins) is comprised of two α-subunits in mammals, Gα12 and Gα13, that have been implicated in a variety of physiological and pathological cellular responses that include proliferation, cytoskeletal rearrangements, migration, and metastatic invasion [1,2]. A diverse set of putative effector proteins have been identified as direct interactors with one or both G12/13 subfamily members; however, the roles of individual Gα-effector interactions in specific cellular responses remain largely undefined [3]. The most extensively characterized G12/13 target proteins are a subset of Rho-specific guanine nucleotide exchange factors (RhoGEFs) that activate the monomeric G protein Rho via tandem Dbl-homology/pleckstrin-homology domains [4]. The Rho monomeric GTPases are known primarily for their role in regulating actin cytoskeletal dynamics, but these proteins also mediate cell polarity, microtubule dynamics, membrane transport pathways, transcription factor activity, cell growth, and tumorigenesis [5]. The G12/13-RhoGEF-Rho axis mediates critical signaling and developmental pathways in model organisms that include Drosophila melanogaster[6], Caenorhabditis elegans[7], and zebrafish [8]. In addition, direct interaction with RhoGEFs is required for mutationally activated Gα12 to trigger increased invasiveness of breast cancer cells [9].
Activated G12/13 α-subunits trigger Rho activation via binding and stimulation of three distinct RhoGEFs: p115RhoGEF, LARG and PDZ-RhoGEF [10-13]. This interaction is mediated primarily by a domain, located near the N-terminus of each RhoGEF, that is closely related to the regulator of G protein signaling (RGS) domain that defines the growing family of RGS proteins [14,15]. Although p115RhoGEF, LARG and PDZ-RhoGEF are highly similar in this “RGS homology“ (RH) domain [16], these proteins appear to be activated by different mechanisms and play non-redundant roles in G12/13 subfamily-mediated signaling. Purified p115RhoGEF binds Gα12 and Gα13 and accelerates GTPase activity for both proteins, but only Gα13 can stimulate p115RhoGEF to activate RhoA in vitro[10,17]. Interaction of Gα12 or Gα13 with purified LARG can trigger its activation of RhoA; however, stimulation by Gα12 requires prior phosphorylation of LARG by the nonreceptor tyrosine kinase Tec [13]. Furthermore, studies utilizing small interfering RNA to hinder expression of specific RhoGEFs show that LARG is a specific downstream effector of thrombin receptor-mediated signaling, whereas signaling through the lysophosphatidic acid (LPA) receptor is attenuated by blocking PDZ-RhoGEF expression [18]. These results are compelling in light of a separate report that the thrombin receptor shows preferential coupling to Gα12, whereas the LPA receptor preferentially utilizes Gα13 as a conduit to downstream signaling [19]. Although it is possible that Gα12 stimulates a post-translationally modified form of p115RhoGEF or PDZ-RhoGEF in cells, the evidence to date suggests LARG as the most likely RhoGEF serving as a physiological effector for Gα12. Gains in our understanding of the specificity of RhoGEF engagement within the G12/13 subfamily should provide insights into the non-overlapping functions of Gα12 and Gα13 in signal transduction.
Crystallographic studies have revealed important structural aspects of the interaction between Gα13 and the RH domain of p115RhoGEF, including numerous residues in both proteins that provide contact points [20,21]. Initially, purification of Gα13 for crystallography required that it be engineered as a chimera in which amino acid sequence within several regions, including the N- and C-termini, was replaced by corresponding sequence from the Gi subfamily protein Gαi1[20]. The structure of the Gα13:p115RhoGEF-RH complex was later refined in crystallographic studies that utilized a Gα13 chimera harboring Gαi1 sequence only at the N-terminus. Because the Gα N-terminus was unstructured in this crystallized complex, any role of this region in RhoGEF interaction remains to be determined. Although the region of Gα13 downstream of the Switch III region harbors several residues critical for RhoGEF engagement, notably Glu273, Thr274, Asn278, and Arg279 within the α3 helix and α3-β5 loop, other regions closer to the Gα13 C-terminus do not emerge in the crystal structure as providing key RhoGEF contact points [21].
In contrast to Gα13, a structure of Gα12 in complex with a RhoGEF target has not been reported, although a chimeric Gα12 harboring the N-terminus of Gαi1 has been crystallized [22]. To examine the full sequence of Gα12 for structural features mediating its interaction with RhoGEFs, we engineered a series of cassette substitutions within constitutively activated Gα12 and examined these variants for in vitro binding to the RH domains of LARG and p115RhoGEF, as well as ability to drive the Rho-dependent process of serum response element (SRE) mediated transcription in cells [23]. Our results reveal unexpected regions of Gα12 as harboring determinants of its functional interaction with RhoGEFs, and also identify key charged amino acids near the Gα12 N-terminus that may confer selective binding to LARG.
Results
Myc-tagged Gα12 retains RhoGEF binding, Rho-mediated signaling, and conformational activation
To identify mutants of Gα12 impaired in RhoGEF binding, we first sought to establish an in vitro system in which Gα12 mutants could be expressed ectopically in cultured cells, rendered soluble in a detergent extract, and detected without interference from endogenous Gα12. We engineered the constitutively active Gln229Leu variant of Gα12 (Gα12QL) to harbor a myc epitope tag, flanked by linkers of the sequence SGGGGS and positioned between residues Pro139 and Val140. This insertion site was chosen due to its approximate alignment with the position of green fluorescent protein in Gαq in a prior study [24]. We expressed myc-tagged and untagged Gα12QL in HEK293 cells, prepared detergent-soluble extracts, and analyzed these by immunoblotting. As shown in Figure 1A, myc-tagged Gα12QL was detected by both anti-myc and anti-Gα12 antibodies, with the latter generating a much stronger signal while avoiding an off-target 37 kDa band detected in all samples by the anti-myc antibody. Also, the myc-tagged protein (~45 kDa) was readily discernible from endogenous Gα12 and untagged Gα12QL (~43 kDa). Next, we subjected myc-Gα12QL to pulldown experiments using an immobilized GST fusion of the p115RhoGEF RH domain, as described in Methods. Myc-tagged and untagged Gα12QL bound to p115-RH with similar affinity (Figure 1B), and comparison with mock-transfected cells indicated the ~45 kDa band detected by anti-Gα12 was dependent on transfection with the myc-Gα12QL plasmid. Furthermore, LARG-RH and p115-RH showed similar ability to co-precipitate myc-tagged Gα12QL (Figure 1C). To ascertain that myc-Gα12 is functional as a mediator of cellular signal transduction through Rho, we measured transcriptional activation of a luciferase reporter gene positioned downstream of the serum response element (SRE), a component of the c-fos promoter that provides a readout of Gα12-mediated Rho activation [23]. Myc-tagged and untagged Gα12QL exhibited similar ability to stimulate this response in HEK293 cells co-transfected with SRE-luciferase (Figure 1D). Furthermore, trypsin digestion of HEK293 cell lysates harboring myc-Gα12QL yielded a protected fragment of ~40 kDa, comparable to results observed previously with GTPγS-loaded, purified Gα12[25]. An inactive, constitutively GDP-bound (Gly228Ala) variant of myc-tagged Gα12 did not yield this ~40 kDa fragment when digested with trypsin (Figure 1E). Taken together, these results suggest myc-Gα12QL undergoes conformational activation and retains normal signaling through the RhoGEF:Rho pathway. Because of the superior sensitivity of anti-Gα12 antibody in detecting myc-Gα12QL, and the easily discernible gel shift of Gα12 caused by the myc tag and linkers (see Figures 1A and B), we chose to utilize anti-Gα12 to detect myc-Gα12QL in subsequent protein binding experiments.
Figure 1.

Effector binding and conformational activation of myc-tagged, constitutively activated Gα12. Molecular weight markers (in kDa) are indicated at right of panels where applicable. All results shown are representative of two or more independent experiments. (A) Expression and solubilization of Gα12QL (12QL) and myc-tagged Gα12QL (myc-12QL) transiently expressed in HEK293 cells. Cells transfected with the vector pcDNA3.1 are included as a negative control (vector). Detergent-soluble extracts were prepared by high-speed centrifugation and subjected to SDS-PAGE and immunoblotting, using either anti-myc (Zymed) or anti-Gα12 (Santa Cruz Biotechnology) antibodies as described in Methods. (B) In vitro binding of myc-tagged and untagged Gα12QL by p115RhoGEF. HEK293 cells extracts containing myc-Gα12QL were subjected to protein interaction assays (see Methods) using an immobilized GST fusion of the RH domain of p115RhoGEF (RGS) or GST alone (GST). Samples were washed, separated by SDS-PAGE, and analyzed by immunoblotting using antibodies described above. (C) Specificity of myc-Gα12QL detection in interaction assays. HEK293 cells transfected with either myc-Gα12QL (myc-12QL) or the empty pcDNA3.1 plasmid (vect) were lysed and assayed for binding to GST fusions of the RH domain of p115RhoGEF (p115) or LARG, or GST alone (GST). Immunoblot analysis was performed using anti-Gα12 antibody as described above. (D) Serum response element (SRE) luciferase activation by myc-Gα12QL. HEK293 cells grown in 12-well plates were co-transfected with the plasmids SRE-L (0.2 μg) and pRL-TK (0.02 μg), plus 0.1 μg of the plasmid indicated on the X-axis. Y-axis values show firefly luciferase signal normalized for Renilla luciferase signal within each sample. (E) Trypsin protection assays of myc-tagged Gα12. Lysates from HEK293 cells transfected with myc-Gα12QL (myc-12QL) or the constitutively GDP-bound Gly228Ala mutant of wildtype Gα12 (myc-12G228A) were subjected to trypsin digests as described in Methods. Immunoblot analysis was performed using J169 antibody [25] at 1:700 dilution.
Mutations that uncouple Gα12 from RhoGEF binding and Rho-mediated signaling
To scan Gα12 for regions participating in its interaction with RhoGEFs, we utilized a comprehensive panel of mutants in which sextets of consecutive amino acids in myc-Gα12QL are replaced by the sextet Asn-Ala-Ala-Ile-Arg-Ser (Figure 2 shows the native amino acid sextet and alphabetical designation for each mutant). This strategy of “NAAIRS” cassette substitutions was chosen due to prediction of this motif being tolerated in the three-dimensional structure of proteins [26], prior use of this approach in mapping functional regions of both retinoblastoma and the telomerase catalytic subunit [27,28], and our previous success employing this strategy to identify Gα12 determinants of binding to the scaffolding subunit of protein phosphatase-2A and the cytoplasmic tail of polycystin-1 [29,30]. Variants of Gα12 were expressed in HEK293 cells and tested for interaction with immobilized LARG-RH, as described in Methods. As shown in Figure 3, myc-Gα12QL was co-precipitated by a GST fusion of LARG-RH but not by GST alone. Many of these cassette mutants yielded a moderate-to-robust signal in the LARG-precipitated fraction; however, a subset displayed a weak or absent signal (Figure 3). To assess impairment of LARG binding for each myc-Gα12QL variant, we quantified the band intensity for each precipitated sample (pulldown), and divided this by the band intensity in the starting cellular extract (load). These calculations generated a “pulldown:load ratio” for each mutant, and also for the positive control myc-Gα12QL that was tested in each experiment. Nearly all cassette mutants were solubilized by our detergent conditions and detected by immunoblotting; exceptions were mutant W, which we did not engineer due to overlap with the insertion site of the myc tag (see Figure 2), and mutant CC due to low expression levels that produced inconclusive results (data not shown). As shown in Table 1, the majority of cassette mutants exhibited pulldown:load ratios greater than 40% of the ratio determined for myc-Gα12QL. However, a number of mutants exhibited lower pulldown:load ratios (<20% of positive control) with a subset generating a ratio less than 10% of the positive control. For all samples, precipitation by immobilized GST yielded no Gα12 signal (Figure 3), indicating these mutants were not merely binding the GST-glutathione-sepharose complex nor forming insoluble aggregates under these in vitro conditions. Also, we examined the full panel of Gα12 cassette mutants for interaction with a GST fusion of the N-terminal 252 amino acids of p115RhoGEF (p115-RH). None of the LARG binding-impaired mutants (those with pulldown:load ratio <20% of positive control; see Table 1) yielded a signal intensity in the p115-RH precipitate that exceeded 50% of intensity for the positive control myc-Gα12QL (data not shown).
Figure 2.

Residues replaced in Gα12 cassette mutants. For each mutant, designated in italics (A-Z, AA-ZZ, AAA-KKK), the native amino acid sextet replaced by the sequence Asn-Ala-Ala-Ile-Arg-Ser is shown. An arrow between Pro139 and Val140 indicates the site of myc tag insertion. Mutant W was not produced. The dashed box indicates the native Gln229 mutated to Leu to render Gα12 constitutively active. The native residues replaced in mutant KKK are Lys-Asp-Ile-Met-Leu-Gln and thus partially overlap with mutant JJJ. All cassette mutants contain the activating Q229L mutation, except mutant LL due to its cassette substitution.
Figure 3.

In vitro interaction of Gα12 mutants with LARG. Immunoblot results for all LARG binding-impaired Gα12 cassette mutants and selected other mutants are shown. HEK293 cells were transfected with the indicated plasmids (7.0 μg per 10-cm plate) and lysates were prepared for co-precipitation assays as described in Methods. Prior to this step, 5% of each lysate was set aside as starting material (load). Pulldown experiments were performed on 7–9 mutants per experiment, plus myc-Gα12QL as a positive control, using equal amounts of GST-LARG-RH (LARG) immobilized on glutathione-sepharose. Immobilized GST was utilized in parallel as a negative control. For all experimental samples, 20% of the volume was analyzed by SDS-PAGE and Coomassie blue staining to verify equal amounts of GST-LARG-RH and GST proteins in the precipitates (data not shown). Immunoblots displayed in this figure are representative of at least three trials per cassette mutant, except for mutants A-D, F-H, V, and KKK that showed minimal impairment in LARG binding after two trials. (Inset) Coomassie blue analysis of GST-fusion constructs expressed in bacteria and immobilized on glutathione-sepharose: GST-LARG-RH (LARG), GST-p115-RH (p115), and GST alone. Molecular weight standards (in kDa) are indicated at right.
Table 1.
Gα12 cassette mutants impaired in binding LARG-RH
| 70-100% |
A-D F-H L N O T-V X BB DD-FF II KK XX YY BBB DDD III-KKK |
| 40-70% |
H P GG JJ WW ZZ CCC |
| 20-40% |
Y AA PP |
| 10-20% |
I OO QQ-SS UU VV FFF |
| 0-10% |
E J K M Q R S Z HH LL-NN TT AAA EEE GGG HHH |
| N/D | W CC |
Cassette substitution mutants of myc-Gα12QL (see Figure 2 for alphabetical designations) were expressed in HEK293 cells and subjected to protein interaction assays using a GST-fusion of the RH domain of LARG as described in Methods, and for each mutant a pulldown:load ratio was determined and calculated as a percent (left column) of the same ratio for unmodified myc-Gα12QL assayed in parallel. Each Gα12 mutant was analyzed in three independent experiments, except for mutants that appeared in the 70-100% category in two independent experiments.
The Gα12 cassette mutant designated OO was among those impaired in LARG binding, consistent with our previous work demonstrating its uncoupling from Rho-mediated signaling [31], and several other cassette substitutions within the Switch regions disrupted binding to LARG (mutants HH, LL, MM, NN, QQ, and RR; see Figure 2). However, impaired LARG binding also was caused by substitutions in other regions of Gα12 (Table 1). Prior crystallographic studies identified several residues in Gα13 that serve as contact points with p115-RH [20,21]. Table 2 lists Gα13 residues identified as contact points with p115-RH in these earlier studies, and indicates the corresponding Gα12 cassette mutant for each Gα13 residue. From our in vitro binding results (Table 1), it is apparent that most Gα12 mutants corresponding to RhoGEF-contacting Gα13 residues displayed partial or severe impairment of LARG binding, mutants V, BB and DDD being exceptions. However, several RhoGEF-uncoupling substitutions in Gα12 (cassette mutants E, I, J, K, M, Z, NN, OO, VV, AAA, EEE, FFF, GGG and HHH) replaced amino acids that do not correspond to Gα13 contacts with p115-RH. Gα12 mutants J and K replaced sections of the P-loop, a motif critical in guanine nucleotide binding, and thus would be predicted as impaired in signaling. However, our finding of RhoGEF-uncoupling mutations at the N- and C-termini of Gα12 was unexpected, because these regions either lacked corresponding contact points in the Gα13:p115-RH complex or were disordered in the G12/13 crystal structures (i.e. the N-terminus). To determine whether these N- and C-terminal mutations in Gα12 are impaired in Rho-mediated signaling, we expressed these variants in HEK293 cells and measured stimulation of SRE-luciferase transcription. All N- and C-terminal mutants impaired in RhoGEF binding were poor activators of this reporter gene (Figure 4A). Several cassette mutants in the N- and C-terminal regions of Gα12 that displayed normal binding to LARG (mutants F, V, and KKK) stimulated SRE-luciferase in a manner comparable to the myc-Gα12QL positive control (Figure 4A). With the exception of mutant VV, immunoblot analysis of HEK293 cell lysates revealed expression levels of these mutants similar to myc-Gα12QL (Figure 4B).
Table 2.
Gα12 cassette mutants corresponding to rgRGS contact points within Gα13
| Gα13 residues in contact with p115-RH | myc-Gα12QL NAAIRS mutant |
|---|---|
| Val98 |
Q |
| Asp101, Ala102 |
R |
| Lys105, Leu106 |
S |
| Thr127, Arg128 |
V |
| Phe168 |
BB |
| Arg200, Pro202, Lys204 |
HH |
| Gln226 |
LL |
| Arg230, Lys231, Phe234 |
MM |
| Met257 |
QQ |
| Arg260 |
RR |
| Asn270 |
SS |
| Ile271, Glu273, Thr274, Ile275 |
TT |
| Asn278, Arg279, Val280 |
UU |
| Arg335 | DDD |
Gα13-native residues previously identified as providing contact points with the RH domain of p115RhoGEF [20,21] are indicated in the left column. Cassette mutants (“NAAIRS” substitution) in which the homologous residue(s) within Gα12 have been altered are indicated in the right column. See Figure 2 for Gα12 mutant designations.
Figure 4.
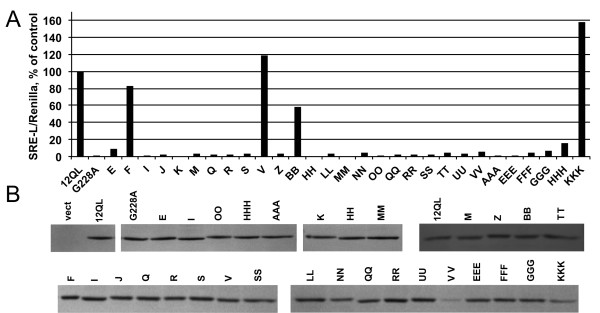
Activation of serum response element mediated transcription by Gα12 mutants. (A) Luciferase reporter assay results of selected cassette mutants. HEK293 cells grown in 12-well plates were co-transfected with the plasmids SRE-L (0.2 μg) and pRL-TK (0.02 μg), plus 1.0 μg of the plasmid encoding each cassette mutant indicated on the X-axis. Firefly luciferase values were normalized for Renilla luciferase values within each sample, and values are presented as a percent of the value calculated for myc-Gα12QL (Y-axis) within the same experiment. Mutationally active (12QL) and inactive (G228A) samples were analyzed in parallel. Results shown are a representative of two experiments performed per Gα12 variant. (B) Expression level of Gα12 mutants. A sample of each lysate was set aside prior to luminometry and analyzed by SDS-PAGE and immunoblotting using anti-Gα12 antibody (Santa Cruz Biotechnology). For all samples, densitometric intensity was determined as described in Methods, then divided by positive control myc-Gα12QL levels within the same experiment, and SRE-L/Renilla values were adjusted to reflect this normalization for protein levels.
Conformational activation of RhoGEF-uncoupled Gα12 mutants
A concern in our experimental approach was that specific “NAAIRS” cassette substitutions could cause global disruption of Gα12 shape, so that a mutant might fail to assume an activated conformation. For RhoGEF-uncoupled Gα12 mutants at the N-terminus (i.e. upstream of the P-loop) and C-terminus, we measured protection against trypsin proteolysis. Exchange of GDP for the activating GTP on Gα proteins triggers a conformational change that conceals a trypsin cleavage site within the Switch II region; this property allows the activated state of the Gα protein to be revealed by resistance to trypsin [25,32]. As shown in Figure 5A, mutants E, I, and HHH yielded a protected fragment of approximately 40 kDa that matched the fragment observed following tryptic digestion of myc-Gα12QL. Results for mutant AAA were difficult to interpret; a band of slightly smaller size than undigested AAA was generated by tryptic digestion, but it was unclear whether this matched the ~40 kDa trypsin-protected fragment in myc-Gα12QL. Other C-terminal mutants we tested− VV, EEE, FFF, and GGG− appeared to match the constitutively inactive myc-Gα12G228A which lacked this ~40 kDa fragment (Figure 5A). These results suggest several C-terminal mutants of Gα12 were sufficiently distorted in shape by the “NAAIRS” substitution to allow trypsin access to proteolytic sites normally not exposed in the GTP-bound state. However, cassette mutants E and I at the N-terminus and HHH at the C-terminus appeared to maintain an activated conformation despite their impairment in RhoGEF binding and SRE stimulation.
Figure 5.

Conformational efficacy of N-terminal and C-terminal Gα12 mutants uncoupled from RhoGEFs. (A) Trypsin protection of selected Gα12 mutants. HEK293 cell lysates expressing the indicated variants of myc-Gα12QL, or unmodified myc-Gα12QL (12QL), or the G228A variant of myc-Gα12 (12G228A) were subjected to trypsin protection assays as described in Methods. Samples were incubated 20 min at 30°C in the presence (+) or absence (−) of TPCK-treated trypsin, and were analyzed by SDS-PAGE and immunoblotting using J169 antibody (1:700 dilution). Small horizontal arrows indicate position of the trypsin-protected fragment in selected lanes. Data presented are representative of two or more independent experiments per sample. (B) Specificity of uncoupling in selected Gα12 variants. For each cassette mutant of myc-Gα12QL (indicated at top), interaction with each Gα12 target (indicated at left) was quantified as a pulldown:load ratio as described in Methods, and was calculated as a percent of the identical ratio determined for myc-Gα12QL within the same experiment. Values are indicated as follows: (++) = >60%, (+) = 20 to 60%, (−) = 0 to 20%. Interacting proteins are GST fusions of the following: RH domain of LARG (LARG), C-terminal 107 amino acids of heat shock protein-90 alpha (Hsp90), protein phosphatase-5 (PP5), scaffolding Aα subunit of protein phosphatase-2A (PP2A), C-terminal 98 amino acids of E-cadherin (E-cad). Values presented indicate the mean of two or more trials per interaction sample.
We also tested whether RhoGEF-uncoupled cassette mutants at the N- and C-termini of Gα12 could interact in vitro with other reported binding partners: heat shock protein-90, protein phosphatase-5, the scaffolding Aα subunit of protein phosphatase-2A, and the cytoplasmic tail of E-cadherin [33-36]. As shown in Figure 5B, each mutant displayed pulldown:load ratios >60% of the positive control, myc-Gα12QL, for at least two of these non-RhoGEF targets. Taken as a whole, these findings reveal a subset of mutations at the N- and C-terminus that selectively uncouple Gα12 from RhoGEFs while preserving conformational activation and ability to bind other downstream proteins.
We next visualized the position of these RhoGEF-interacting regions in the crystal structure of a Gα12 chimera in which the N-terminal 48 residues were replaced by the N-terminus of Gαi1[22]. The native region of Gα12 replaced in cassette mutant E is not ordered in this structure; however, the regions replaced in the C-terminal mutants EEE-HHH are highlighted (Figure 6). The sextet replaced in mutant HHH (highlighted in black) resides in the α5 helix that extends along the Gα12 surface and approaches the C-terminus at the top of the diagram.
Figure 6.

Structural position of Gα12 C-terminal determinants of RhoGEF binding. The structure of N-terminally Gαi1-substituted Gα12 (PDB accession code 1ZCA, [22]) as a GDP•AlF4¯ activated complex was analyzed using PyMOL software. The native Gα12 region substituted for the sequence “NAAIRS” in the C-terminal mutants EEE, FFF, and GGG is highlighted in orange, and the sextet substituted in mutant HHH is highlighted in black. The bound GDP molecule is highlighted in blue. Figure was rendered in The PyMOL Molecular Graphics System, Version 1.5.0.1 Schrödinger, LLC.
Differential uncoupling of Gα12 from LARG and p115RhoGEF
We next sought to identify specific residues within these N- and C-terminal sextets of Gα12 that mediate RhoGEF interaction. To examine putative surface residues, we performed charge substitutions in the native regions corresponding to cassette mutants E, I, and HHH, and examined these variants for SRE-luciferase activation. None of the single-residue charge-reversals in the regions encompassed in mutants I or HHH caused significant decrease in SRE signaling (data not shown). However, a double charge-reversal in the mutant E region, converting Glu31 and Glu33 to Arg residues, caused a near-complete loss of SRE activation in HEK293 cells despite normal levels of protein expression (Figure 7A). We next examined this Gα12 mutant, designated Glu31/33Arg, for binding to the RH domains of LARG and p115RhoGEF. As shown in Figure 7B, a selective loss of RhoGEF binding was observed: the Glu31/33Arg charge-reversals severely disrupted LARG-RH binding relative to non-mutated myc-Gα12QL (pulldown:load ratio ~18% of control) but had minimal effect on p115-RH binding (ratio ~86% of control). In trypsin protection assays, the Glu31/33Arg mutant yielded a protected fragment at the same molecular weight (~40 kDa) as observed for the myc-Gα12QL positive control, suggesting its ability to attain an activated conformation (Figure 7C). The intermediate intensity of this band (approximately a midpoint between activated Gα12 and the constitutively inactive Gly228Ala variant) may be due in part to the mutational introduction of Arg residues providing additional sites for trypsin proteolysis. Taken as a whole, these findings not only provide evidence that the structurally uncharacterized N-terminus of Gα12 plays a role in its functional interaction with RhoGEFs, but also reveal individual charged residues in this region as candidates for conferring specificity of Gα12 for LARG among the RH-containing RhoGEFs.
Figure 7.

Selective RhoGEF uncoupling by N-terminal charge substitutions in Gα12. (A) Luciferase reporter gene assays. Cassette mutant E (see Figure 2) and the double charge substitution mutant Glu31/33Arg were compared to myc-Gα12QL (12QL) in SRE-luciferase assays under the cell transfection conditions described in Figure 4. A constitutively GDP-bound variant of wildtype myc-Gα12 (12G228A) was assayed in parallel as a negative control. Results shown are the mean of three independent experiments, and error bars indicate range. (B) Protein-protein interaction assays. Detergent-soluble extracts from transfected HEK293 cells transfected with myc-Gα12QL, the Glu31/33Arg mutant, or empty pcDNA3.1 plasmid (vector) were subjected to co-precipitation assays as described in Methods, using GST-fusions of either LARG-RH (LARG), p115RhoGEF-RH (p115), the N-terminal domain of the Gα12 target radixin [46], or no adduct (GST). Prior to the precipitation step, 5% of each lysate was set aside as starting material (load). Table values show the pulldown:load ratio for Glu31/33Arg as a percent of the positive control value (12QL), with mean +/- range presented for three independent experiments. (C) Trypsin protection of the Glu31/33Arg mutant, in comparison to constitutively GTP- and GDP-bound Gα12. Assays were performed as described in Methods. Results shown are representative of two independent experiments.
Discussion
The G12 subfamily members Gα12 and Gα13 are well-documented as utilizing RhoGEFs as downstream signaling effectors. Crystallographic studies by Chen et al. [20] and Hajicek et al. [21] have provided intricate structural details of the interaction between Gα13 and the RH domain of p115RhoGEF, identifying a set of Gα13 residues that directly contact this target protein. The structure of Gα12 also has been elucidated, using a chimera comprised of amino acids 49–379 of Gα12 preceded by amino acids 1–28 of Gαi1[22]. However, a Gα12:RhoGEF complex has not been reported. In the current study, we utilized in vitro and cell-based approaches to examine the interaction between Gα12 and two putative target RhoGEFs, LARG and p115RhoGEF. Using immobilized RGS-homology (RH) domains of these RhoGEFs, we identified several substitutions of native amino acids in Gα12 that disrupted its binding to these proteins and blocked its ability to stimulate the Rho-dependent process of SRE-mediated transcription. Although our results indicated that a number of common determinants in Gα12 and Gα13 mediate RhoGEF binding, several RhoGEF-uncoupling mutations in Gα12 did not correspond to regions of RhoGEF contact within Gα13; these include amino acid sextet substitutions in the C-terminal α5 helix as well as the structurally uncharacterized N-terminus. Several of these Gα12 mutants exhibited protection from tryptic digestion as well as unimpeded binding to other, non-RhoGEF targets, indicating their impaired interaction with RhoGEFs is not caused by failure to attain an activated conformation and suggesting the shapes of other effector-binding surfaces in these Gα12 mutants remain intact as RhoGEF interaction is disrupted.
Although Gα12 and Gα13 share 67% amino acid identity and bind several common downstream targets, several functional differences between these Gα proteins suggest their signaling mechanisms are not redundant [1,3]. Both Gα12 and Gα13 bind LARG and p115RhoGEF [10,12], and both of these RhoGEFs accelerate GTPase activity of purified Gα12 and Gα13 in single-turnover assays [13,17]. Whereas Gα13 stimulates both p115RhoGEF and LARG to trigger guanine nucleotide exchange on RhoA in vitro, Gα12 can only stimulate LARG under these experimental conditions, and in a manner dependent on prior phosphorylation of LARG by the tyrosine kinase Tec [10,13]. Also, activated Gα12 is more potent than Gα13 in recruiting the RH domain of p115RhoGEF to the plasma membrane, and specific mutations in p115RhoGEF disrupt Gα12 but not Gα13 in triggering this localization [37]. At the cellular and organismal levels, it is increasingly clear that Gα12 and Gα13 utilize non-overlapping signaling pathways. Mice lacking Gα13 die early in embryogenesis due to defects in vascular development and thrombin-induced cell migration, but mice lacking Gα12 do not display these developmental defects. However, knockout of Gα12 combined with absence of Gα13 causes earlier lethality than Gα13 knockout alone, and in mice lacking one Gα13 allele, at least one Gα12 allele must be present for normal embryonic development [38,39]. Furthermore, LPA-induced activation of mTOR complex 2 leading to activation of PKC-δ requires Gα12 but not Gα13[40]. Because of these differences, plus the increasing list of Gα12-specific effector proteins (including another RhoGEF, AKAP-Lbc, that is activated exclusively by Gα12 within the G12/13 subfamily), we believe the Gα12:RhoGEF interface cannot be defined summarily by structural features of the Gα13:RhoGEF complex.
Among the Gα13 residues that provide contact points with p115RhoGEF in crystallographic studies [20,21], many have corresponding residues within Gα12, and therefore we paid particular attention to Gα12 cassette mutants corresponding to these key Gα13 residues (see Table 2). For example, the Gα12 mutant HH replaced residues corresponding to Gα13 residues Arg200, and Lys204, both of which provide contact points with p115-RH. In another Gα12 cassette mutant, termed RR, a substituted residue corresponds to Arg260 within Gα13; this residue provides a key contact with amino acids within the βN-αN region of p115RhoGEF. Also, Gα12 cassette mutants Q, R, and S contain altered residues in the Gα12 helical domain that correspond to p115-RH interacting residues in Gα13. Among the Gα12 mutants corresponding to p115-RH contact points in Gα13, most showed impaired RhoGEF interaction and poor stimulation of SRE-mediated signaling. However, several differences between Gα12 and Gα13 were noted, particularly in the helical domain. Gα12 cassette mutant V alters residues that correspond to two contact points within the Gα13:p115-RH complex; however, this mutant showed minimal impairment in RhoGEF binding in vitro and stimulated SRE-mediated transcription robustly in cells. Gα12 mutant BB, which removes a Phe corresponding to a Gα13 contact point with p115-RH, displayed a slight impairment in SRE-mediated transcriptional activation and no impairment of RhoGEF binding. In addition, Gα13 utilizes a C-terminal residue (Arg335) as a contact point with p115-RH, but the corresponding Gα12 cassette mutant (DDD) exhibited normal binding to RhoGEFs and only modest impairment in SRE signaling. However, because this cassette mutant preserves the corresponding Arg residue in Gα12 (DRKRRN substituted for NAAIRS), it is possible this Arg in Gα12 participates in RhoGEF binding despite the alteration in adjacent amino acids.
Aside from the N- and C-terminal mutants of Gα12 that show impaired RhoGEF binding, we have identified other RhoGEF-uncoupling mutations in Gα12 that lack corresponding Gα13 contact points for p115-RH (see Tables 1 and 2). None of the native Gα12 residues replaced in cassette mutants M and Z match p115-RH contact points in Gα13, and thus may indicate Gα12-specific determinants of RhoGEF interaction. Impaired RhoGEF binding also was observed in Gα12 mutants J and K; however, this most likely was due to these substitutions disrupting the canonical GXGXXGKS guanine nucleotide binding motif [41]. Although our results suggest a core similarity in the mechanisms utilized by Gα12 and Gα13 to engage RhoGEF targets, it is apparent that several determinants of RhoGEF binding are unique to Gα13. We have identified determinants that may be unique to Gα12 or potentially important for both G12/13 subfamily members in RhoGEF engagement. Studies of Gα13 variants harboring corresponding mutations will be important in distinguishing these possibilities.
A role for the C-terminus of G12/13 subfamily proteins in RhoGEF engagement has been suggested by prior studies. Kreutz et al. [42] engineered chimeras of Gα12 and Gα13 that were interchanged downstream of the Switch III region, and demonstrated the C-terminal 114 amino acids of Gα13 as sufficient for its unique ability to stimulate purified p115RhoGEF to activate RhoA. Also, a chimeric Gα13 in which the region downstream of Switch III was replaced by the corresponding region of Gαi2 displayed loss of ability to stimulate SRE-mediated transcriptional activation [43]. Initial crystallographic studies of Gα13:RhoGEF interaction utilized a chimeric Gα13 harboring Gαi1 sequence at the C-terminus, and determinants of RhoGEF binding were not found downstream of the Switch regions in this protein [20]. Subsequent crystallographic work utilizing Gα13 with native C-terminal sequence did identify residues slightly downstream of the Switch III region as critical for RhoGEF engagement [21], and also revealed a more distal residue in the C-terminal region (Arg335) as providing a contact point with the RH domain of p115RhoGEF. However, no residues at the extreme C-terminus of Gα13, including the α5 helix, were found to mediate RhoGEF binding. Our results suggest differences between Gα12 and Gα13 in the role of the C-terminus, as several substitutions near the extreme C-terminus of Gα12 disrupted RhoGEF interaction, most notably the cassette mutant HHH within the α5 helix.
The N-terminus provides the greatest amino acid sequence divergence between Gα12 and Gα13. Gα subunits utilize this region for interaction with Gβγ [44], and in Gα12 and Gα13 this region confers specificity of coupling to thrombin and LPA receptors, respectively [19]. Importantly, Gα13 is a more potent stimulator of RhoGEF activation in vitro than a chimeric Gα13 harboring the N-terminus of Gαi1, indicating a possible role of the Gα13 N-terminus in RhoGEF activation [21]. However, specific determinants within the N-terminus of G12/13 subfamily proteins that mediate binding to effectors, including RhoGEFs, have not been reported. The 48-residue region at the N-terminus of Gα12 has not been characterized in crystallographic studies, because its replacement by the Gαi1 N-terminus was necessary for obtaining sufficient quantities of purified protein [16,22]. Furthermore, the N-terminus was disordered in crystallographic analysis of both the aforementioned Gαi1/Gα13 hybrid and a more recent structure of full-length Gα13[21], suggesting the Gα12 N-terminus may be refractory to crystallographic analysis even if native sequence is utilized. Our approach of employing cassette substitution mutants throughout the length of Gα12 has provided an indirect means of circumventing this obstacle, and has revealed specific N-terminal regions as possible determinants of RhoGEF interaction. Importantly, our discovery that mutations in this N-terminal region (cassette mutants E and I) cause loss of RhoGEF binding allowed us to focus on putative surface residues in these substituted regions, ultimately revealing Glu31 and Glu33 as critical for Gα12 interaction with LARG and stimulation of SRE-mediated transcription. Our finding that charge substitutions of these N-terminal Gα12 residues disrupted binding to the LARG-RH domain but had minimal effect on interaction with the corresponding domain of p115RhoGEF was intriguing, and suggested these residues play a role in targeting Gα12 preferentially to LARG. It is possible that Gα12 harbors sufficient RhoGEF-interacting surfaces for in vitro binding to p115RhoGEF, but that a functional, physiological interaction (i.e. with LARG) requires this N-terminal region. Our RhoGEF binding results for Gα12 cassette mutant E, as well as the more specific Glu31/33Arg mutant, were surprising in light of earlier findings that RhoGEF binding was preserved in a Gα12 chimera containing the Gαi1 N-terminus [22]. It is possible that “NAAIRS” substitution and particularly the Glu31/33Arg charge-reversals cause a more dramatic change to this RhoGEF binding surface than occurs when Gαi1 sequence is introduced. Cassette mutant E and the Glu31/33Arg mutant are impaired in activating the Rho-dependent readout of SRE-mediated transcriptional activation in cells, and it remains to be determined whether the Gαi1/Gα12 chimera is similarly impaired in stimulating this pathway.
Because previous phosphorylation of LARG by Tec is a requirement for Gα12, but not Gα13, for in vitro activation of Rho, it will be important to determine whether this phosphorylation event regulates interaction of LARG with Gα12, particularly its N-terminus and C-terminal α5 helix. Furthermore, as suggested by Hajicek et al. [21], it is conceivable that post-translational modification of p115RhoGEF in cells modulates its responsiveness to Gα13 or could potentially render it a target of Gα12. A challenge for future studies of Gα12- and Gα13-mediated signaling will be to determine the combinations of G12/13 subfamily α-subunits and RhoGEFs that activate Rho in response to different signaling inputs, and in different cell and tissue types.
Conclusions
Gα12 and Gα13 define the G12/13 class of heterotrimeric G protein α-subunits, which participate in numerous signaling pathways through stimulation of RhoGEFs that subsequently activate Rho. Although these proteins are non-redundant in their stimulation of effectors and their cellular and organismal roles, only Gα13 has been characterized in the structural basis of its interaction with RhoGEF targets. However, the involvement of Gα12 in stimulating SRE-mediated transcription, cell rounding, c-Jun N-terminal kinase activation, cell growth, and metastatic invasion supports a physiological role for a Gα12-RhoGEF-Rho axis in developmental pathways and disease progression [45]. Therefore, an improved understanding of the structural aspects of Gα12:RhoGEF interaction likely will be of broad importance. Our results provide several key additions to this structural model: 1) characterization of the Gα12:RhoGEF interacting surface by identifying regions in Gα12 that mediate binding; 2) unexpected roles of the Gα12 N-terminal region and C-terminal α5 helix in engagement of RhoGEFs; 3) identification of specific residues near the Gα12 N-terminus that may mediate its selectivity for LARG as an effector protein. To date, no structural studies have examined the interaction of Gα12 with RhoGEFs. Our hope is that mutant-based strategies will augment such crystallographic approaches and provide key details toward understanding the structural aspects and biological role of this Gα:effector interaction.
Methods
DNA constructs
Plasmids encoding 1) a fusion of glutathione-S-transferase (GST) to amino acids 320–606 of LARG (GST-LARG-RH), and 2) amino acids 1–252 of p115RhoGEF with an N-terminal myc epitope tag were kindly provided by Tohru Kozasa (Univ. of Ill., Chicago). We used PCR to subclone the p115RhoGEF sequence into pGEX-2T (GE Healthcare) to produce GST-p115-RH. All “NAAIRS” amino acid substitution mutants within myc-tagged Gα12 Gln229Leu (myc-Gα12QL) were engineered as described previously [29]. Single amino acid substitutions were engineered in myc-Gα12QL using the QuikChange II® site-directed mutagenesis system (Agilent Technologies), and this system was used to engineer a constitutively inactive Gly228Ala variant (myc-Gα12G228A) within a plasmid encoding myc-tagged, wildtype Gα12 (provided by Pat Casey, Duke University). The luciferase reporter plasmid SRE-L was a gift from Channing Der (University of North Carolina Chapel Hill).
Expression and immobilization of GST fusion proteins
GST fusion constructs were transformed into BL21(Gold)-DE3 cells (Stratagene). Cells were grown under 75 μg/ml ampicillin selection to OD600 of 0.5−0.7, and recombinant protein expression was induced using 0.5 mM isopropyl-β-D-thiogalactopyranoside (Fisher Scientific). After 3 h, cells were lysed on ice using 0.32 mg/ml lysozyme (MP Biomedicals), and GST fusion proteins were bound to glutathione-sepharose 4B (GE Healthcare) as described previously [31,34]. Following three washes in 50 mM Tris pH 7.7 supplemented with 1 mM EDTA, 1 mM dithiothreitol, and 150 mM NaCl, samples were snap-frozen in aliquots and stored at −80°C.
Preparation of detergent-soluble extracts harboring Gα12 mutants
Human embryonic kidney cells (HEK293) were grown in Dulbecco’s modified Eagle medium (Mediatech, Manassas, VA) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT), penicillin and streptomycin. For myc-Gα12QL and each of its 62 NAAIRS substitution mutants (see Figure 2), 7.0 μg of plasmid DNA was transfected into a 10-cm dish of HEK293 cells grown to approximate 90% confluence, using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. After 36–42 hours, cells were scraped from dishes, washed twice with phosphate-buffered saline, and solubilized in NAAIRS Lysis buffer (50 mM HEPES pH 7.5, 1 mM EDTA, 3 mM dithiothreitol, 10 mM MgSO4, 1% (w/v) polyoxyethylene-10-lauryl ether) containing the protease inhibitors 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (1.67 mM), leupeptin (2.1 μM), pepstatin (1.45 μM), TLCK (58 μM), TPCK (61 μM), and phenylmethylsulfonyl fluoride (267 μM). Samples were centrifuged at 80,000 g for 1 h, and supernatants were snap-frozen in 60-μl aliquots and stored at −80°C.
Protein interaction assays
HEK293 cell extracts were diluted in NAAIRS Lysis buffer lacking polyoxyethylene-10-lauryl ether, using sufficient volume to dilute this detergent in the samples to 0.05% (w/v). Next, sepharose-bound GST fusion proteins were added and allowed to incubate for approximately 2 h at 4°C with continuous inversion. A percentage of the diluted extract was set aside as starting material prior to sepharose addition. Next, samples were centrifuged at 1,300 g, and pellets were washed three times and then subjected to SDS-PAGE and immunoblot analysis using an antibody specific to the Gα12 N-terminus (Santa Cruz Biotechnology) or the myc 9E10 epitope tag (Zymed), followed by alkaline phosphatase conjugated secondary antibodies (Promega). For each variant of myc-Gα12QL, the Gaussian intensity of the ~45 kDa band from the precipitated material and the corresponding band from the starting material were quantified using a Kodak Gel Logic 100 system equipped with Molecular Imaging 5.X software (Carestream Health, New Haven CT).
Reporter gene assays
HEK293 cells grown in 12-well plates were transfected with 0.2 μg SRE-luciferase plasmid (encoding firefly luciferase) and 0.02 μg pRL-TK plasmid encoding Renilla luciferase, plus plasmids encoding variants of myc-Gα12QL. Reporter assays for SRE-mediated transcriptional activation were performed as described previously [31]. Briefly, cells were washed with phosphate-buffered saline and lysed in 1X passive lysis buffer (Promega), and lysates were analyzed using a Dual-luciferase assay system and GloMax 20/20 luminometer (Promega). Light output due to firefly luciferase activity was divided by output from Renilla luciferase activity to normalize samples for transfection efficiency.
Trypsin protection experiments
HEK293 cells grown in 10-cm dishes were transfected with various Gα12 constructs using Lipofectamine 2000 (Invitrogen), and tryptic digestions were performed as a modification of the procedure of Kozasa and Gilman [25]. Briefly, cells were lysed in 50 mM Hepes pH 8.0, 1 mM EDTA, 3 mM dithiothreitol, 1% polyoxyethylene-10-lauryl ether containing the same protease inhibitors as NAAIRS Lysis buffer (see above) but at two-fold lower concentration. Samples were cleared by centrifugation at 70,000 g for 1 h, and supernatants were diluted 20-fold in volume using 50 mM Hepes pH 8.0, 1 mM EDTA, 3 mM dithiothreitol, 10 mM MgSO4. Samples were digested with 10 μg/ml TPCK-treated trypsin (New England Biolabs) for 20 min at 30°C, and proteolysis was terminated by addition of 100 μg/ml lima bean trypsin inhibitor (Worthington, Lakewood NJ). Samples were analyzed by SDS-PAGE and immunoblotting using J169 antisera specific to the Gα12 C-terminus, provided by Tohru Kozasa (Univ. of Ill., Chicago).
Abbreviations
Gα12 and Gα13: Heterotrimeric guanine nucleotide binding protein α-subunits of the G12/13 subfamily; GST: Glutathione-S-transferase; HEK: Human embryonic kidney; LARG: Leukemia-associated RhoGEF; LPA: Lysophosphatidic acid; NAAIRS mutant: Variant of Gα12 in which a consecutive sextet of native residues has been replaced by Asn-Ala-Ala-Ile-Arg-Ser; RGS: Regulator of G protein signaling; RH: RGS homology; RhoGEF: Rho-specific guanine nucleotide exchange factor; SRE: Serum response element.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BJR and WCS participated in design of the study, carried out PCR-based mutagenesis, designed and executed protein interaction screens, and participated in drafting the manuscript. In addition, BJR performed reporter assays and WCS performed 3-D protein imaging and analysis. ERM, ESF, TYC, and CMO engineered various GST-fusion constructs and carried out protein interaction screens. LAF participated in initial design of the study and carried out pilot experiments. TEM conceived of the study, participated in its design, coordination, engineering of constructs and data collection, and drafted the manuscript. All authors have read and approved the final manuscript.
Authors’ information
TEM is an affiliate member of the UNC Lineberger Comprehensive Cancer Center (Chapel Hill, NC).
Contributor Information
Benjamin J Ritchie, Email: britchie@email.unc.edu.
William C Smolski, Email: wsmolsk@ncsu.edu.
Ellyn R Montgomery, Email: epmonty@gmail.com.
Elizabeth S Fisher, Email: fishere@livemail.uthscsa.edu.
Tina Y Choi, Email: tiychoi@gmail.com.
Calla M Olson, Email: cmolson11@gmail.com.
Lori A Foster, Email: nosering@bellsouth.net.
Thomas E Meigs, Email: tmeigs@unca.edu.
Acknowledgements
We are grateful to Tohru Kozasa, Channing Der, and Pat Casey for reagents, and Todd DeMarco, Benjamin Smith, and Joseph Martin for technical assistance. We thank Pat Casey for critical reading of this manuscript, and Dan Kaplan, Juhi Juneja, Pat Kelly, Tim Fields, and Nicole Hajicek for helpful discussions. TEM, WCS, and ERM received support from the Lineberger Comprehensive Cancer Center (Chapel Hill, NC) through a University Cancer Research Fund collaborative award. BJR, ERM, ESF, CMO, and TEM were supported by a grant from the North Carolina Biotechnology Center (BRG-1229). LAF received support from the UNC-Asheville Undergraduate Research Program. TEM received additional support as co-investigator on National Institutes of Health grant CA100869.
References
- Worzfeld T, Wettschureck N, Offermanns S. G12/G13-mediated signalling in mammalian physiology and disease. Trends Pharmacol Sci. 2008;29:582–589. doi: 10.1016/j.tips.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Hajicek N, Kozasa T. Regulation and physiological functions of G12/13-mediated signaling pathways. Neurosignals. 2009;17:55–70. doi: 10.1159/000186690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly P, Casey PJ, Meigs TE. Biologic functions of the G12 subfamily of heterotrimeric G proteins: growth, migration, and metastasis. Biochemistry. 2007;46:6677–6687. doi: 10.1021/bi700235f. [DOI] [PubMed] [Google Scholar]
- Sternweis PC, Carter AM, Chen Z, Danesh SM, Hsiung YF, Singer WD. Regulation of Rho guanine nucleotide exchange factors by G proteins. Adv Protein Chem. 2007;74:189–228. doi: 10.1016/S0065-3233(07)74006-8. [DOI] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- Barrett K, Leptin M, Settleman J. The Rho GTPase and a putative RhoGEF mediate a signaling pathway for the cell shape changes in Drosophila gastrulation. Cell. 1997;91:905–915. doi: 10.1016/S0092-8674(00)80482-1. [DOI] [PubMed] [Google Scholar]
- Yau DM, Yokoyama N, Goshima Y, Siddiqui ZK, Siddiqui SS, Kozasa T. Identification and molecular characterization of the Gα12-Rho guanine nucleotide exchange factor pathway in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2003;100:14748–14753. doi: 10.1073/pnas.2533143100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F, Sepich DS, Chen S, Topczewski J, Yin C, Solnica-Krezel L, Hamm H. Essential roles of Gα12/13 signaling in distinct cell behaviors driving zebrafish convergence and extension gastrulation movements. J Cell Biol. 2005;169:777–787. doi: 10.1083/jcb.200501104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly P, Moeller BJ, Juneja J, Booden MA, Der CJ, Daaka Y, Dewhirst MW, Fields TA, Casey PJ. The G12 family of heterotrimeric G proteins promotes breast cancer invasion and metastasis. Proc Natl Acad Sci USA. 2006;103:8173–8178. doi: 10.1073/pnas.0510254103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, Sternweis PC, Bollag G. Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Gα13. Science. 1998;280:2112–2114. doi: 10.1126/science.280.5372.2112. [DOI] [PubMed] [Google Scholar]
- Fukuhara S, Murga C, Zohar M, Igishi T, Gutkind JS. A novel PDZ domain containing guanine nucleotide exchange factor links heterotrimeric G proteins to Rho. J Biol Chem. 1999;274:5868–5879. doi: 10.1074/jbc.274.9.5868. [DOI] [PubMed] [Google Scholar]
- Fukuhara S, Chikumi H, Gutkind JS. Leukemia-associated Rho guanine nucleotide exchange factor (LARG) links heterotrimeric G proteins of the G(12) family to Rho. FEBS Lett. 2000;485:183–188. doi: 10.1016/S0014-5793(00)02224-9. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Nakamura S, Mano H, Kozasa T. Gα12 activates Rho GTPase through tyrosine-phosphorylated leukemia-associated RhoGEF. Proc Natl Acad Sci USA. 2003;100:733–738. doi: 10.1073/pnas.0234057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollinger S, Hepler JR. Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol Rev. 2002;54:527–559. doi: 10.1124/pr.54.3.527. [DOI] [PubMed] [Google Scholar]
- Siderovski DP, Willard FS. The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. Int J Biol Sci. 2005;1:51–66. doi: 10.7150/ijbs.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Singer WD, Danesh SM, Sternweis PC, Sprang SR. Recognition of the activated states of Gα13 by the rgRGS domain of PDZRhoGEF. Structure. 2008;16:1532–1543. doi: 10.1016/j.str.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC. p115 RhoGEF, a GTPase activating protein for Gα12 and Gα13. Science. 1998;280:2109–2111. doi: 10.1126/science.280.5372.2109. [DOI] [PubMed] [Google Scholar]
- Wang Q, Liu M, Kozasa T, Rothstein JD, Sternweis PC, Neubig RR. Thrombin and lysophosphatidic acid receptors utilize distinct rhoGEFs in prostate cancer cells. J Biol Chem. 2004;279:28831–28834. doi: 10.1074/jbc.C400105200. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Katoh H, Negishi M. N-terminal short sequences of α subunits of the G12 family determine selective coupling to receptors. J Biol Chem. 2003;278:14936–14939. doi: 10.1074/jbc.M301409200. [DOI] [PubMed] [Google Scholar]
- Chen Z, Singer WD, Sternweis PC, Sprang SR. Structure of the p115RhoGEF rgRGS domain-Gα13/i1 chimera complex suggests convergent evolution of a GTPase activator. Nat Struct Mol Biol. 2005;12:191–197. doi: 10.1038/nsmb888. [DOI] [PubMed] [Google Scholar]
- Hajicek N, Kukimoto-Niino M, Mishima-Tsumagari C, Chow CR, Shirouzu M, Terada T, Patel M, Yokoyama S, Kozasa T. Identification of critical residues in Gα13 for stimulation of p115RhoGEF activity and the structure of the Gα13-p115RhoGEF regulator of G protein signaling homology (RH) domain complex. J Biol Chem. 2011;286:20625–20636. doi: 10.1074/jbc.M110.201392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutz B, Yau DM, Nance MR, Tanabe S, Tesmer JJG, Kozasa T. A new approach to producing functional Gα subunits yields the activated and deactivated structures of Gα12/13 proteins. Biochemistry. 2006;45:167–174. doi: 10.1021/bi051729t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromm C, Coso OA, Montaner S, Xu N, Gutkind JS. The small GTP-binding protein Rho links G protein-coupled receptors and Gα12 to the serum response element and to cellular transformation. Proc Natl Acad Sci USA. 1997;94:10098–10103. doi: 10.1073/pnas.94.19.10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TE, Zhang H, Logothetis DE, Berlot CH. Visualization of a functional Gαq-green fluorescent protein fusion in living cells. Association with the plasma membrane is disrupted by mutational activation and by elimination of palmitoylation sites, but not by activation mediated by receptors or AlF4–. J Biol Chem. 2001;276:4227–4235. doi: 10.1074/jbc.M007608200. [DOI] [PubMed] [Google Scholar]
- Kozasa T, Gilman AG. Purification of recombinant G proteins from Sf9 cells by hexahistidine tagging of associated subunits. Characterization of α12 and inhibition of adenylyl cyclase by αz. J Biol Chem. 1995;270:1734–1741. doi: 10.1074/jbc.270.4.1734. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Haft DH, Getzoff ED, Tainer JA, Lerner RA, Brenner S. Identical short peptide sequences in unrelated proteins can have different conformations: a testing ground for theories of immune recognition. Proc Natl Acad Sci USA. 1985;82:5255–5259. doi: 10.1073/pnas.82.16.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers WR, Novitch BG, Miyake S, Heith A, Otterson GA, Kaye FJ, Lassar AB, Kaelin WG. Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor cell growth. Genes Dev. 1998;12:95–106. doi: 10.1101/gad.12.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster BN, Banik SS, Guo C, Smith AC, Counter CM. N-terminal domains of the human telomerase catalytic subunit required for enzyme activity in vivo. Mol Cell Biol. 2001;21:7775–7786. doi: 10.1128/MCB.21.22.7775-7786.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Tate RI, Ruediger R, Meigs TE, Denker BM. Domains necessary for Gα12 binding and stimulation of protein phosphatase-2A (PP2A): Is Gα12 a novel regulatory subunit of PP2A? Mol Pharmacol. 2007;71:1268–1276. doi: 10.1124/mol.106.033555. [DOI] [PubMed] [Google Scholar]
- Yu W, Ritchie BJ, Su X, Zhou J, Meigs TE, Denker BM. Identification of polycystin-1 and Gα12 binding regions necessary for regulation of apoptosis. Cell Signal. 2011;23:213–221. doi: 10.1016/j.cellsig.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meigs TE, Juneja J, DeMarco CT, Stemmle LN, Kaplan DD, Casey PJ. Selective uncoupling of Gα12 from Rho-mediated signaling. J Biol Chem. 2005;280:18049–18055. doi: 10.1074/jbc.M500445200. [DOI] [PubMed] [Google Scholar]
- Miller RT, Masters SB, Sullivan KA, Beiderman B, Bourne HR. A mutation that prevents GTP-dependent activation of the α chain of Gs. Nature. 1988;334:712–715. doi: 10.1038/334712a0. [DOI] [PubMed] [Google Scholar]
- Vaiskunaite R, Kozasa T, Voyno-Yasenetskaya TA. Interaction between the Gα subunit of heterotrimeric G12 protein and Hsp90 is required for Gα12 signaling. J Biol Chem. 2001;276:46088–46093. doi: 10.1074/jbc.M108711200. [DOI] [PubMed] [Google Scholar]
- Meigs TE, Fields TA, McKee DD, Casey PJ. Interaction of Gα12 and Gα13 with the cytoplasmic domain of cadherin provides a mechanism for β-catenin release. Proc Natl Acad Sci USA. 2001;98:519–524. doi: 10.1073/pnas.021350998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Katoh H, Mori K, Negishi M. Gα12 and Gα13 interact with Ser/Thr protein phosphatase type 5 and stimulate its phosphatase activity. Curr Biol. 2002;12:1353–1358. doi: 10.1016/S0960-9822(02)01034-5. [DOI] [PubMed] [Google Scholar]
- Zhu D, Kosik KS, Meigs TE, Yanamadala V, Denker BM. Gα12 directly interacts with PP2A: evidence for Gα12-stimulated PP2A phosphatase activity and dephosphorylation of microtubule-associated protein, Tau. J Biol Chem. 2004;279:54983–54986. doi: 10.1074/jbc.C400508200. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya R, Banerjee J, Khalili K, Wedegaertner PB. Differences in Gα12- and Gα13-mediated plasma membrane recruitment of p115-RhoGEF. Cell Signal. 2009;21:996–1006. doi: 10.1016/j.cellsig.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offermanns S, Mancino V, Revel JP, Simon MI. Vascular system defects and impaired cell chemokinesis as a result of Gα13 deficiency. Science. 1997;275:533–536. doi: 10.1126/science.275.5299.533. [DOI] [PubMed] [Google Scholar]
- Gu JL, Muller S, Mancino V, Offermanns S, Simon MI. Interaction of Gα12 with Gα13 and Gαq signaling pathways. Proc Natl Acad Sci USA. 2002;99:9352–9357. doi: 10.1073/pnas.102291599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan X, Wang J, Wang C, Sommer E, Kozasa T, Srinivasula S, Alessi D, Offermanns S, Simon MI, Wu D. PRR5L degradation promotes mTORC2-mediated PKC-δ phosphorylation and cell migration downstream of Gα12. Nat Cell Biol. 2012;14:686–696. doi: 10.1038/ncb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm HE, Gilchrist A. Heterotrimeric G proteins. Curr Opin Cell Biol. 1996;8:189–196. doi: 10.1016/S0955-0674(96)80065-2. [DOI] [PubMed] [Google Scholar]
- Kreutz B, Hajicek N, Yau DM, Nakamura S, Kozasa T. Distinct regions of Gα13 participate in its regulatory interactions with RGS homology domain-containing RhoGEFs. Cell Signal. 2007;19:1681–1689. doi: 10.1016/j.cellsig.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Vazquez-Prado J, Miyazaki H, Castellone MD, Teramoto H, Gutkind JS. Chimeric Gαi2/Gα13 proteins reveal the structural requirements for the binding and activation of the RGS-like (RGL)-containing Rho guanine nucleotide exchange factors (GEFs) by Gα13. J Biol Chem. 2004;279:54283–54290. doi: 10.1074/jbc.M410594200. [DOI] [PubMed] [Google Scholar]
- Denker BM, Neer EJ, Schmidt CJ. Mutagenesis of the amino terminus of the alpha subunit of the G protein Go. In vitro characterization of αoβγ interactions. J Biol Chem. 1992;267:6272–6277. [PubMed] [Google Scholar]
- Juneja J, Casey PJ. Role of G12 proteins in oncogenesis and metastasis. Br J Pharmacol. 2009;158:32–40. doi: 10.1111/j.1476-5381.2009.00180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaiskunaite R, Adarichev V, Furthmayr H, Kozasa T, Gudkov A, Voyno-Yasenetskaya TA. Conformational activation of radixin by G13 protein α subunit. J Biol Chem. 2000;275:26206–26212. doi: 10.1074/jbc.M001863200. [DOI] [PubMed] [Google Scholar]