

Abstract
Background
Chloroplast is an essential organelle in plants which contains independent genome. Chloroplast genomes have been widely used for plant phylogenetic inference recently. The number of complete chloroplast genomes increases rapidly with the development of various genome sequencing projects. However, no comprehensive platform or tool has been developed for the comparative and phylogenetic analysis of chloroplast genomes. Thus, we constructed a comprehensive platform for the comparative and phylogenetic analysis of complete chloroplast genomes which was named as chloroplast genome analysis platform (CGAP).
Results
CGAP is an interactive web-based platform which was designed for the comparative analysis of complete chloroplast genomes. CGAP integrated genome collection, visualization, content comparison, phylogeny analysis and annotation functions together. CGAP implemented four web servers including creating complete and regional genome maps of high quality, comparing genome features, constructing phylogenetic trees using complete genome sequences, and annotating draft chloroplast genomes submitted by users.
Conclusions
Both CGAP and source code are available at http://www.herbbol.org:8000/chloroplast. CGAP will facilitate the collection, visualization, comparison and annotation of complete chloroplast genomes. Users can customize the comparative and phylogenetic analysis using their own unpublished chloroplast genomes.
Keywords: Chloroplast genomes, Comparative and phylogenetic analysis, Web-based platform
Background
The chloroplast is an essential organelle in plants which performs photosynthesis. Chloroplast contains independent genome derived from a cyanobacterial ancestor [1]. Chloroplast genome typically consists of circular double-stranded DNA molecules of 110–200 kb size, including 100–200 unique genes. Most chloroplast genomes contain two large inverted repeats (IRs) of 6–76 kb which are highly conserved and divide the genomes into one large and one small single-copy region (called LSC and SSC, respectively) [2]. The chloroplast genomes contain important genes involved in photosystems and biosynthetic pathways. Many coding and non-coding sequences of chloroplast genomes have been used for the phylogeny analysis of plants, including: rbcL, matK and psbA-trnH[3,4]. Because of the conserved nature, appropriate size, persistent gene organization and potential ability for plant phylogenetic inference and transgenic expression, chloroplast genomes have been widely sequenced and used for the comparison and phylogeny analysis [5-7].
As the number of sequenced chloroplast genomes increases rapidly, bioinformatics tools become more critical for the analysis of complete chloroplast genomes. GenBank, EMBL and DDBJ are the primary nucleotide sequences databases. The chloroplast genome database (CGDB: http://nar.oxfordjournals.org/content/34/suppl_1/D692.full) and GOBASE (http://gobase.bcm.umontreal.ca/) are specialized chloroplast repositories [8,9]. Dual organelle genome annotator (DOGMA: http://dogma.ccbb.utexas.edu/) is a web-based annotation tool for chloroplast and mitochondrial genomes [10]. GeneOrder (http://binf.gmu.edu:8080/GeneOrder3.0/) and BADGER (http://badger.duq.edu/manual2/models.html) can be used for comparative analysis of gene arrangements in small genomes [11]. GRAPPA (http://www.cs.unm.edu/~moret/GRAPPA/) and MGR (http://grimm.ucsd.edu/MGR/) perform phylogenetic analysis based on gene order changes [12-14]. Several tools offer the option to create chloroplast genome maps (e.g. PlasMapper, CGView and OGDRAW) [15-17]. However, there is no comprehensive platform or tool which can be used for the comparative and phylogenetic analysis of chloroplast genomes. We aim to construct a platform which integrates genome collection, visualization, comparison, phylogenetic analysis and annotation functions together. It will facilitate the comparative and phylogenetic analysis of complete chloroplast genomes.
Implementation
CGAP contains a built-in database and four web servers including visualization of genomes, comparison of genome features, phylogeny analysis and genome annotation. The architecture of the platform was showed in Figure 1. CGAP was implemented using Python programming language and Web2py web framework (http://www.web2py.com). Entire platform was constructed on a machine with 16 GB RAM. The performances of the database and web servers were tested via a variety of web browsers (e.g. IE, Firefox, Chrome and Safari). As of writing this article, CGAP has been running for half a year.
Figure 1.

Architecture of CGAP.
Results and discussion
CGAP collected 284 complete chloroplast genomes from NCBI Organelle Genome Resources (http://www.ncbi.nlm.nih.gov/genomes). According to the annotation information stored in the GenBank format file, CGAP extracted all types of genome features including Gene, CDS, tRNA, rRNA, Exon, Intron, Promoter, RepeatRegion, StemLoop, -10 Signal and -35 Signal. Complete chloroplast genomes and all genome features were stored in CGAP chloroplast database. You can view and download all genomes and features in Fasta format online.
Visualization of genomes
In order to better illustrate chloroplast genomes, CGAP implemented three functions for the visualization of genomes, including the visualization of circular complete genomes and linear regional genomes, the visualization of modified published genomes, and the visualization of user unpublished genomes. Complete and regional genome maps of Populus trichocarpa [GenBank: NC_009143.1] were showed in Figure 2; All functions used Perl modules (including BioPerl, PerlMagick, PostScriptSimple, TestSimple and PerlXML) and OGDRAW to create high quality genome maps [17]. In the genome maps, different features were indicated by different colors, and every feature was annotated using its name. For each genome map CGAP provided five types of figures for viewing and downloading, including TIFF, PNG, JPG, GIF and PS. In order to create maps of the modified published genome, user needs to indicate the genome using its organism name or accession number, and submit a file which contains the modified items of the published genome. Every line contains one modified item which has three fields separated by comma, including FeatureName, the Start and End position. For maps of unpublished genomes, user needs to submit the annotation file of the genome. The first part of the annotation file contains the annotation items, one annotation item per line. Every annotation item has four fields separated by comma, including FeatureType, FeatureName, the Start and End position. The second part of the annotation file contains the complete genome sequence in Fasta format. Model files for test can be found from the website where it is used.
Figure 2.

Complete and regional genome map of Populus trichocarpa.
Comparison of genome features
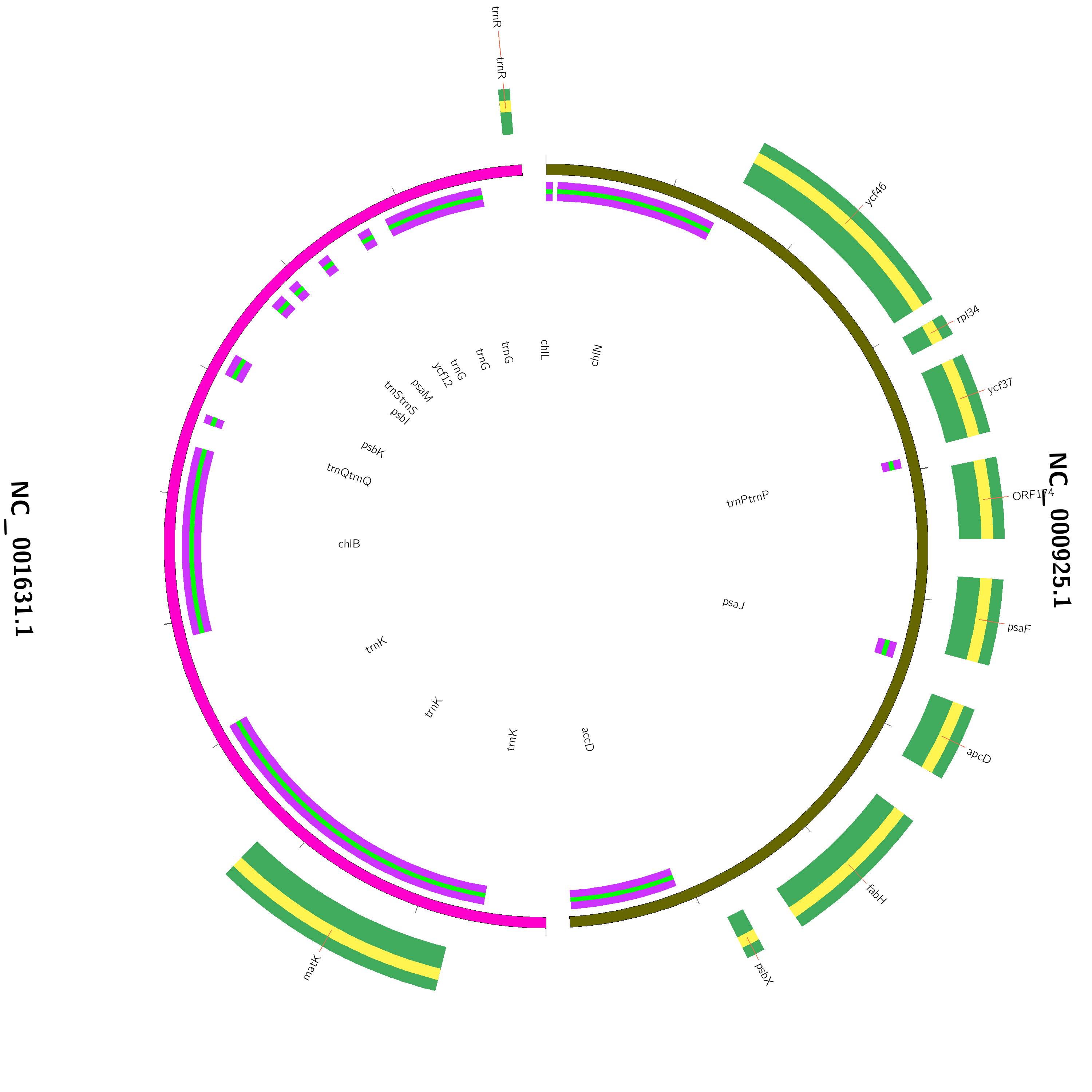
The feature content of chloroplast genome gives detailed information about the composition of the genome. In general, chloroplast genomes differ from each other in feature content. CGAP compared the similarities and differences of the feature content between different genomes, which was implemented based on text mining method and the annotated feature information of the genomes. CGAP also visualized the comparison results in high quality, detailed circular layout using Circos [18]. CGAP implemented two functions for the comparison of feature content, including one by one and one by more comparison. Figure 3 showed the comparison results between Pinus thunbergii [GenBank: NC_001631.1] and Porphyra purpurea [GenBank: NC_000925.1]. Figure 4 showed the comparison results of Medicago truncatula [GenBank: NC_003119.6] versus Gossypium barbadense [GenBank: NC_008641.1], Nuphar advena [GenBank: NC_008788.1], Cuscuta reflexa [GenBank: NC_009766.1], Cuscuta gronovii [GenBank: NC_009765.1], Ephedra equisetina [GenBank: NC_011954.1] and Syntrichia ruralis [GenBank: NC_012052.1]. In both Figures the internal annotations and lines between genome features indicated the same features between the genomes compared, and the external annotations indicated the different features. For both types of comparisons, you can submit your own draft genome and customize the chloroplast genomes used in your comparative analysis. For one by more comparison using only the published genomes, user needs to submit a file which contains the names of organisms or accession numbers of the genomes compared. All names are placed in the first line of the file and separated by comma. For one by more comparison using the user unpublished genome, all names are also placed in the first line and separated by comma, and the first name indicates the user unpublished genome. The second line describes the length of user genome. From the third line to the end of the file, each line contains one focused feature of the user genome. Optionally, you can supply a range of the genome, and then CGAP will compare the genomes and visualize the comparison results only on the focused range of the genome. Results of the regional comparison of genomes can be seen in Additional files 1 and 2.
Figure 3.

Feature content comparison results between Pinus thunbergii and Porphyra purpurea.
Figure 4.
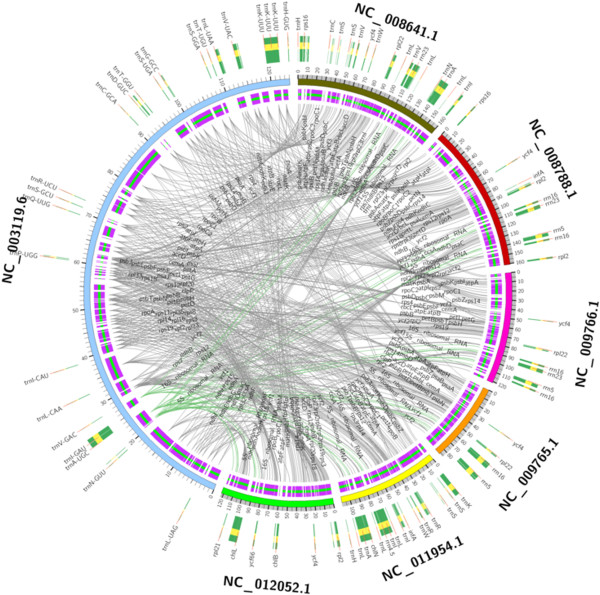
One by more comparison results of feature content.
Phylogeny analysis
Traditional phylogeny analysis is based on multiple sequence alignment. Sequence alignment methods meet huge challenge when dealing with large-scale complete genomes. Thus, various alignment-free methods have been proposed [19,20]. CGAP used a novel sequence feature called base-base correlation (BBC) to characterize the complete chloroplast genome. BBC was first proposed by Liu et al. [21,22]. For each chloroplast genome CGAP extracted one BBC feature vector, and then calculated the distance matrix of the feature vectors using one of the ten distance methods implemented in CGAP. Finally, CGAP constructed the phylogenetic tree based on the distance matrix and neighbor-joining (NJ) method [23]. In order to compare the results of alignment-free method with traditional alignment-based method, CGAP also implemented phylogenetic analysis based on whole genome sequence alignment. The alignment of whole genome sequences was performed using MUMmer, and the distance of genomes was calculated using following formula [24].
Where, Nmat denotes the number of nucleotides matched between genomes A and B, Lmax is the max length of all genomes analyzed.
CGAP saved the distance matrix of the genomes as three kinds of formats, including the standard Nexus format and distance formats used in MEGA and PHYLIP [25,26]. CGAP also drew a tree map for the overview of the phylogenetic relationship (see Additional file 3), and saved the phylogeny tree as standard Newick and Nexus formats. Optionally, you can supply your unpublished genomes and customize the chloroplast genomes used in your phylogeny analysis. In this situation, users need to submit a txt file, the first part of the file contains all names of organisms or accession numbers of the published genomes used in the analysis process, and the second part of the file contains the unpublished complete genomes in Fasta format.
Genome annotation
CGAP annotated new chloroplast genomes based on feature sequences of the chloroplast genomes collected in CGAP database and basic local alignment method (BLAST 2.2.25+: http://blast.ncbi.nlm.nih.gov/) [27]. CGAP identified the potential elements of your genome according to the sequence similarities between the elements and the features in the database. Then, CGAP attached biological information to the elements identified based on the information of the most similar feature [28-32]. Finally, CGAP returned you a list of non-redundant annotated entries which described the potential features on your genome. Every annotated entry for a segment sequence of your genome has 8 fields, including NormalizedFeatureName, Start, End, FeatureName or Location, LengthRatio, Identity, Score and Expectation. The meaning of each field was described in Table 1. CGAP also visualized the genome in high-quality circular map based on the annotations.
Table 1.
Meanings of the fields in annotation entry
| Fields of annotation entry | Meanings of the fields |
|---|---|
| Normalized FeatureName |
Normalized feature name of a segment sequence of the genome |
| Start |
Starting position of the segment sequence on the chloroplast genome |
| End |
Ending position of the segment sequence on the chloroplast genome |
| FeatureName/Location |
Potential feature name or location identifier of the segment sequence of the genome |
| LengthRatio |
|End-Start|/L, L indicated the length of the feature sequence compared to the segment sequence of the genome |
| Identity |
Nmatch/Nmissmatch, Nmatch and Nmissmatch indicated the number of match and mismatch bases of the segment sequence respectively in sequence alignment |
| Score |
Score obtained in sequence alignment |
| Expectation | Expectation value computed in sequence alignment |
Conclusions
CGAP was developed for the comparative analysis of complete chloroplast genomes. It integrated genome collection, visualization, content comparison, phylogeny analysis and annotation functions together. CGAP implemented feature content comparison of chloroplast genomes and a novel alignment-free method for the phylogenetic analysis. Users can customize the comparative and phylogenetic analysis using their own unpublished genomes. To our knowledge, CGAP represents the first comprehensive platform for the comparative analysis of chloroplast genomes. It would facilitate the researches and applications of complete chloroplast genomes.
Availability and requirements
Project name: CGAP
Project home page: http://www.herbbol.org:8000/chloroplast
Operating system(s): Linux for the distributed source code and operating system independent for the web servers
Programming language: Python 2.6
License: Free for academic use
Abbreviations
CGAP: Chloroplast genome analysis platform; IRs: Inverted repeats; LSC: Large single-copy; SSC: Small single-copy; BBC: Base-base correlation; NJ: Neighbor-joining
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JC implemented the programs, and wrote the initial manuscript draft. XZ supplied constructive suggestions for the functions of the platform. GR set up the frame work for the web server. ZL designed the whole study, proposed the alignment-free method and revised the manuscript extensively. All authors read and approved the final manuscript.
Supplementary Material
One by one regional comparison results of genomes.
{kind=link}
One by more regional comparison results of genomes.
{kind=link}
Overview of the phylogenetic tree constructed in phylogeny analysis.
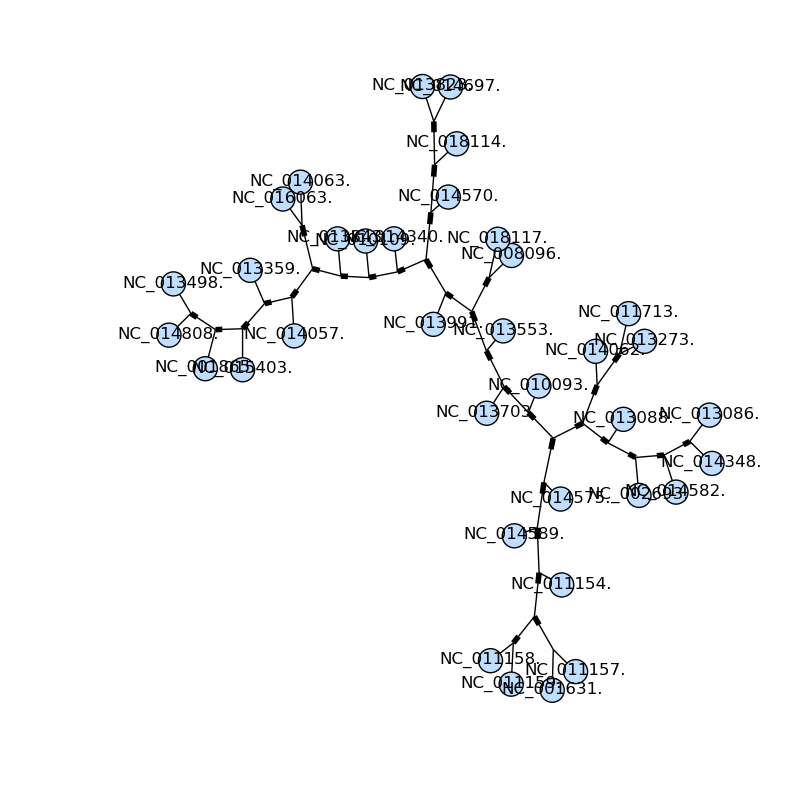{kind=link}
Contributor Information
Jinkui Cheng, Email: jkcheng@implad.ac.cn.
Xu Zeng, Email: zengxu1986@gmail.com.
Guomin Ren, Email: renguomin@implad.ac.cn.
Zhihua Liu, Email: zhliu@implad.ac.cn.
Acknowledgements
This work was supported by National Natural Science Foundation of China (81102746, 81100077, 31201768), Beijing Natural Science Foundation (5113033, 6133033), Special Foundation of China Postdoctoral Science (2012T50067), China Postdoctoral Science Foundation funded project (2011M500014, 2012M510011), Scientific Research Foundation of the State Human Resource Ministry and the Education Ministry for Returned Chinese Scholars, PUMC Youth Fund, the Fundamental Research Funds for the Central Universities, the Research Fund for the Doctoral Program of Higher Education (20111106120028), “Major Drug Discovery” major science and technology research “12nd Five-Year Plan” (2012ZX09301-002-001-025), Basic Scientific Research Operation Cost of State-Leveled Public Welfare Scientific Research Courtyard (YZ-12-15), China Medical Board of New York (A2009001), Program for New Century Excellent Talents in University (NCET-12-0074) granted to Zhihua Liu.
References
- Margulis L. Symbiotic theory of the origin of eukaryotic organelles; criteria for proof. Symp Soc Exp Biol. 1975;29:21–38. [PubMed] [Google Scholar]
- Wicke S, Schneeweiss GM, dePamphilis CW, Muller KF, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol. 2011;76(3–5):273–297. doi: 10.1007/s11103-011-9762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Liang D, Gao T, Pang X, Song J, Yao H, Han J, Liu Z, Guan X, Jiang K. PTIGS-IdIt, a system for species identification by DNA sequences of the psbA-trnH intergenic spacer region. BMC Bioinforma. 2012;12(Suppl 13):S4. doi: 10.1186/1471-2105-12-S13-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J, Lickey EB, Schilling EE, Small RL. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am J Bot. 2007;94(3):275–288. doi: 10.3732/ajb.94.3.275. [DOI] [PubMed] [Google Scholar]
- Asifullah K, Ishtiaq AK, Huma AM, Kamran A. Current trends in chloroplast genome research. Afr J Biotechnol. 2009;9(24):3494–3500. [Google Scholar]
- Lin CP, Huang JP, Wu CS, Hsu CY, Chaw SM. Comparative chloroplast genomics reveals the evolution of Pinaceae genera and subfamilies. Genome Biol Evol. 2010;2:504–517. doi: 10.1093/gbe/evq036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan IC, Liao DC, Wu FH, Daniell H, Singh ND, Chang C, Shih MC, Chan MT, Lin CS. Complete chloroplast genome sequence of an orchid model plant candidate: erycina pusilla apply in tropical oncidium breeding. PLoS One. 2012;7(4):e34738. doi: 10.1371/journal.pone.0034738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien EA, Badidi E, Barbasiewicz A, deSousa C, Lang BF, Burger G. GOBASE--a database of mitochondrial and chloroplast information. Nucleic Acids Res. 2003;31(1):176–178. doi: 10.1093/nar/gkg090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Veeraraghavan N, Richter A, Wall K, Jansen RK, Leebens-Mack J, Makalowska I, dePamphilis CW. ChloroplastDB: the chloroplast genome database. Nucleic Acids Res. 2006;34(Database issue):D692–D696. doi: 10.1093/nar/gkj055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20(17):3252–3255. doi: 10.1093/bioinformatics/bth352. [DOI] [PubMed] [Google Scholar]
- Mazumder R, Kolaskar A, Seto D. GeneOrder: comparing the order of genes in small genomes. Bioinformatics. 2001;17(2):162–166. doi: 10.1093/bioinformatics/17.2.162. [DOI] [PubMed] [Google Scholar]
- Moret BME, Wyman S, Bader DA, Warnow T, Yan M. A new implementation and detailed study of breakpoint analysis. Proc 6th Pacific Symp Biocomput (PSB 2001) 2001. pp. 583–594. [DOI] [PubMed]
- Bourque G, Pevzner PA. Genome-scale evolution: reconstructing gene orders in the ancestral species. Genome Res. 2002;12(1):26–36. [PMC free article] [PubMed] [Google Scholar]
- Yue F, Cui L, dePamphilis CW, Moret BM, Tang J. Gene rearrangement analysis and ancestral order inference from chloroplast genomes with inverted repeat. BMC Genomics. 2008;9(Suppl 1):S25. doi: 10.1186/1471-2164-9-S1-S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Stothard P, Forsythe IJ, Wishart DS. PlasMapper: a web server for drawing and auto-annotating plasmid maps. Nucleic Acids Res. 2004;32(Web Server issue):W660–W664. doi: 10.1093/nar/gkh410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stothard P, Wishart DS. Circular genome visualization and exploration using CGView. Bioinformatics. 2005;21(4):537–539. doi: 10.1093/bioinformatics/bti054. [DOI] [PubMed] [Google Scholar]
- Lohse M, Drechsel O, Bock R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 2007;52(5–6):267–274. doi: 10.1007/s00294-007-0161-y. [DOI] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun SR, Sims GE, Wu GA, Kim SH. Whole-proteome phylogeny of prokaryotes by feature frequency profiles: An alignment-free method with optimal feature resolution. Proc Natl Acad Sci USA. 2009;107(1):133–138. doi: 10.1073/pnas.0913033107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Xu Z, Hao B. Composition vector approach to whole-genome-based prokaryotic phylogeny: success and foundations. J Biotechnol. 2009;149(3):115–119. doi: 10.1016/j.jbiotec.2009.12.015. [DOI] [PubMed] [Google Scholar]
- Liu ZH, Meng JH, Sun X. A novel feature-based method for whole genome phylogenetic analysis without alignment: application to HEV genotyping and subtyping. Biochem Biophys Res Commun. 2008;368(2):223–230. doi: 10.1016/j.bbrc.2008.01.070. [DOI] [PubMed] [Google Scholar]
- Liu ZH, Sun X. Coronavirus phylogeny based on base-base correlation. Int J Bioinform Res Appl. 2008;4(2):211–220. doi: 10.1504/IJBRA.2008.018347. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP - phylogeny inference package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Liu ZH, Jiao D, Sun X. Classifying genomic sequences by sequence feature analysis. Genomics Proteomics Bioinform. 2005;3(4):201–205. doi: 10.1016/S1672-0229(05)03027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZH, Zeng X, Yang D, Ren GM, Chu GY, Yuan ZR, Luo K, Xiao PG, Chen SL. Identification of medicinal vines by ITS2 using complementary discrimination methods. J Ethnopharmacol. 2012;141:242–249. doi: 10.1016/j.jep.2012.01.057. [DOI] [PubMed] [Google Scholar]
- Liu ZH, Zeng X, Yang D, Chu GY, Yuan ZR, Chen SL. Applying DNA barcodes for identification of plant species in the family Araliaceae. Gene. 2012;499:76–80. doi: 10.1016/j.gene.2012.02.016. [DOI] [PubMed] [Google Scholar]
- Zeng X, Yuan ZR, Tong X, Li QS, Gao WW, Qin MJ, Liu ZH. Phylogenetic study of Oryzoideae species and related taxa of the Poaceae based on atpB-rbcL and ndhF DNA sequences. Mol Biol Rep. 2012;39(5):5737–5744. doi: 10.1007/s11033-011-1383-0. [DOI] [PubMed] [Google Scholar]
- Cheng JK, Cao FL, Liu ZH, AGP. A Multimethods Web Server for Alignment-Free Genome Phylogeny. Mol Biol Evol. 2013. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
One by one regional comparison results of genomes.
One by more regional comparison results of genomes.
Overview of the phylogenetic tree constructed in phylogeny analysis.