

Abstract
Mutations in PKD1 and PKD2, the genes encoding the proteins polycystin-1 (PC1) and polycystin-2 (PC2), cause autosomal dominant polycystic kidney disease (ADPKD). Although the leading cause of mortality in ADPKD is cardiovascular disease, the relationship between these conditions remains poorly understood. PC2 is an intracellular calcium channel expressed in renal epithelial cells and in cardiomyocytes, and is thus hypothesized to modulate intracellular calcium signaling and affect cardiac function. Our first aim was to study cardiac function in a zebrafish model lacking PC2 (pkd2 mutants). Next, we aimed to explore the relevance of this zebrafish model to human ADPKD by examining the Mayo Clinic’s ADPKD database for an association between ADPKD and idiopathic dilated cardiomyopathy (IDCM). Pkd2 mutant zebrafish showed low cardiac output and atrioventricular block. Isolated pkd2 mutant hearts displayed impaired intracellular calcium cycling and calcium alternans. These results indicate heart failure in the pkd2 mutants. In human ADPKD patients, we found IDCM to coexist frequently with ADPKD. This association was strongest in patients with PKD2 mutations. Our results demonstrate that PC2 modulates intracellular calcium cycling, contributing to the development of heart failure. In human subjects we found an association between ADPKD and IDCM and suggest that PKD mutations contribute to the development of heart failure.
Keywords: Zebrafish, polycystin-2, ADPKD, calcium, heart, cardiomyopathy
1. Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a common genetic disease affecting approximately 1 in 500 people. It is characterized by renal cysts and numerous extra-renal manifestations [1]. Cardiovascular problems are a major cause of morbidity and a leading cause of mortality in patients with ADPKD [2,3]. Hypertension [4], left ventricular hypertrophy [5], arterial aneurysms [6], and cardiac valve abnormalities [7,8] are associated with the disease. Interestingly, young ADPKD patients with normal blood pressure and renal function exhibit early vascular changes and bi-ventricular diastolic dysfunction [9,10]. The cellular and molecular etiologies of these early changes remain elusive.
Idiopathic dilated cardiomyopathy (IDCM) is a relatively common heart disease characterized by dilated ventricles and weakened systolic function [11]. It is the most frequent form of non-ischemic cardiomyopathy, affecting approximately 1 in 2,500 people [12,13]. An estimated 25–50% of the cases are hereditary [14–16]. The genetic background is varied; to date over two dozen chromosomal loci and disease genes have been linked to IDCM [17]. However, no direct evidence links ADPKD to IDCM.
Mutations in two genes are known to account for ADPKD [18, 19]. PKD1 mutations account for ~85% and PKD2 mutations for ~15% of cases [20]. PKD1 and PKD2 encode the proteins polycystin-1 (PC1) and polycystin-2 (PC2), respectively. PC1 is a transmembrane protein that interacts with PC2, which is a non-selective calcium-regulated cation channel [21, 22]. PC2 is a member of the TRP family (TRPP2) [23] and is primarily found on the endoplasmic/sarcoplasmic reticulum (E/SR) membrane and in primary cilia [24, 25], where it colocalizes with PC1. Most evidence points to PC2 as an intracellular calcium channel that participates in the regulation of intracellular calcium concentration [26–28]. However, a PC2-like protein appears to function as a channel on the plasma membrane in rat ventricular cardiomyocytes [29]. In addition, PC2 is a modulator of the cardiac ryanodine receptor (RyR2) [30]. RyR2, which is a calcium release channel found on the SR membrane, is crucial for calcium-induced calcium release (CICR), which is a prerequisite for excitation-contraction coupling. Altered RyR2 function is seen in heart failure [31], and in patients carrying mutations in RyR2 [32]. PC2 interacts with RyR2, stabilizing the closed state and inhibiting the release of calcium [30]. Loss of inhibition of RyR2 by PC2 in PC2-deficient cardiomyocytes results in a higher frequency of spontaneous calcium oscillations, reduced SR calcium stores, and reduced calcium transient amplitude compared with wildtype (WT) cells [30].
In recent years, zebrafish (Danio rerio) have emerged as a powerful model for studying genetic mechanisms of human cardiovascular diseases [33–35]. We studied cardiac function in a zebrafish model of ADPKD that lacks PC2 [36]. PC2 is ubiquitously expressed in the zebrafish [37, 38] including in muscles where it is expressed in a sarcomeric pattern, strongly suggesting localization on the SR [37]. Our first goal was to determine whether cardiac function is altered in PC2-deficient fish by in vivo monitoring of cardiac performance. Next, we aimed to examine potential underlying causes by studying intracellular calcium cycling and action potentials. Importantly, to determine whether the results of our zebrafish studies are relevant to human ADPKD, we examined the Mayo Clinic ADPKD database. The observation that ADPKD and IDCM coexisted with high frequency had previously suggested a possible association between these two conditions [39]. However, this connection remains unexplored. We hypothesized that the PC2-deficient fish would exhibit altered calcium handling and cardiac dysfunction. We also hypothesized that PKD2 patients would have an increased risk of heart failure compared to non-PKD patients, due to the direct interaction of PC2 with intracellular calcium cycling proteins.
2. Materials and methods
2.1. Zebrafish and morpholino injections
The zebrafish line pkd2/hi4166, which lacks expression of the protein PC2, has been described previously [36]. Morphant embryos were obtained through antisense morpholino oligonucleotide injection into wild-type (WT) eggs at one-cell stage. Fish homozygous for the pkd2 mutation (referred to as pkd2 mutants) were compared to unrelated WT fish in all experiments, unless otherwise stated.
2.2. Zebrafish cardiac physiology
For cardiac output measurements, heart rates were counted and images of blood flow in the dorsal aorta were captured at 125 frames per second. Tracking of red blood cells and measurement of aorta diameter allowed stroke volume to be calculated. To study cardiac function, we found determining cardiac output based on measurements of aortic erythrocyte flow to be the most repeatable method. For measurements such as ventricular ejection fraction and fractional shortening, which are based on determining end-systolic and end-diastolic diameters of the long and short axes of the ventricle, positioning the heart in a standardized way proved difficult in vivo, complicating reliable measurement of these indices. Standardized positioning of the heart was partly complicated by edema in the pkd2 mutant fish.
2.3. Intracellular calcium and action potential imaging, electrical pacing, and SR calcium measurement
For calcium imaging experiments, isolated hearts were loaded with fluo-4 AM, followed by de-esterification before imaging. For optical action potential recordings, isolated hearts were loaded with di-4-ANEPPS. After a baseline recording of spontaneous calcium transients, hearts were electrically stimulated by field pacing. The SR calcium content was determined in calcium-free Tyrode solution by provoking calcium release with caffeine and thapsigargin.
2.4. ADPKD database and mutation screening
Use of the clinical Mayo Clinic ADPKD database and genotyping of research subjects was approved as part of a larger study of genotype-phenotype correlations in Polycystic Kidney Disease by the Mayo Institutional Research Board. The diagnosis of ADPKD was based on Ravine’s criteria in the presence of a positive family history. In the absence of a family history, the criteria for diagnosing ADPKD required at least 20 bilateral renal cysts and absence of clinical findings suggesting the presence of a different cystic disease. A diagnosis of IDCM was made in patients with a LVEF <40%, exclusion of ≥50% obstruction of one or more coronary arteries, exclusion of active myocarditis or a primary or secondary form of heart muscle disease, and exclusion of advanced renal insufficiency (stage 4 or 5 chronic kidney disease). The entire coding and flanking intronic regions of PKD1 and PKD2 were screened for mutations by direct sequencing as previously described [20, 40].
2.5. Statistical Analysis
All values are presented as mean ± s.e.m., determined by Student’s t test. A P value of <0.05 was considered statistically significant.
3. Results
3.1. Pkd2 mutant zebrafish lack cardiac expression of PC2
Pkd2 mutants show dorsal body curvature, making them easy to distinguish from fish lacking or carrying only one copy of the mutant allele (Figure 1A and B). We found PC2 expression throughout the heart of WT fish (Figure 1C), but not in the hearts of pkd2 mutants (Figure 1D) or fish injected with a morpholino silencing pkd2 (Figure 1E). These data verify that the pkd2/hi4166 line lacks expression of the protein PC2.
Fig. 1.

Phenotype of Pkd2 mutant zebrafish embryos and expression of PC2 in zebrafish heart. Images of zebrafish embryos taken at 9 days post fertilization (dpf) show a wildtype (WT) fish (A) and a pkd2 mutant fish with typical dorsal body curvature and edema (B). Immunofluorescent images taken at 48 hours post fertilization (hpf) showing DAPI counterstained nuclei (blue) and PC2 (green) in WT (C), pkd2 mutant (D), and pkd2 morphant fish (E). PC2 is broadly expressed in the WT heart, whereas no PC2 is detectable in the pkd2 mutant and morphant hearts.
3.2. In wildtype hearts PC2 localizes to the sarcoplasmic reticulum
In WT zebrafish hearts we found that PC2 localizes predominantly to the sarcoplasmic reticulum, as shown by the perinuclear co-localization with known SR markers Serca2 and BiP (Supplementary Figure S1).
3.3. Pkd2 mutant zebrafish display impaired cardiac function
Pkd2 mutants had lower heart rates than their siblings with normal phenotype (Figure 2A). Furthermore, this difference in heart rates became more pronounced with increasing age (Figure 2B). This increasing difference in heart rates led us to study cardiac function in more detail. We determined cardiac output based on measurement of aortic erythrocyte flow (Figure 2C). Heart rate (Figure 2D) and stroke volume (Figure 2E) in pkd2 mutants were notably lower than in WT fish. As a result, cardiac output in WT fish was nearly double compared to that in pkd2 mutants (16.7±1.1 nl/min vs. 9.8±0.7 nl/min, P = 0.0002) (Figure 2F). During systole, peak velocities of erythrocytes in the aorta were similar in WTs and pkd2 mutants, 1.94±0.05 mm/s vs. 1.71±0.09 mm/s, P > 0.05.
Fig. 2.

Pkd2 mutant zebrafish show impaired cardiac function. (A) Heart rate in zebrafish embryos at 3, 4, and 5 dpf. The WT/het group consists of siblings of pkd2 mutants with normal wildtype phenotype (no dorsal body curvature). Therefore 2/3 of the WT/het group is heterozygous for the pkd2 mutation, and 1/3 is WT. WT/het 3 dpf n = 144, 4 dpf n = 149, 5 dpf n = 113. Pkd2 mutant (mut.) 3 dpf n = 144, 4 dpf n = 150, 5 dpf n = 113. (B) Difference between the mean heart rates of the WT/het and the pkd2 mutant zebrafish. The pkd2 mutant mean heart rate for each time point was subtracted from the WT/het mean heart rate for the same time point. Propagation of error was used to determine the error bars. (C) Blood flow was imaged in the dorsal aorta of 3 dpf zebrafish. Comparison of heart rate (D), stroke volume (E), and cardiac output (F). WT n = 6, pkd2 mut. n = 7. Pkd2 mutant zebrafish develop edema and arrhythmias (G). Zebrafish embryos (n/group = 27–41) were imaged and cardiac videos recorded at 3, 6, and 9 dpf. All observed arrhythmias were AV blocks, where part of the atrial impulses failed to induce ventricular contraction. Data shown as mean ± s.e.m.
3.4. Pkd2 mutant zebrafish exhibit arrhythmia and edema
We observed an increase in pericardial and abdominal edema in the pkd2 mutants. Analyzed data from videos demonstrate an increase in arrhythmias in the pkd2 mutant hearts in vivo (Figure 2G). In all 17 mutant hearts with arrhythmia, ventricular contraction failed to follow atrial contraction in approximately 30% of the beats (Figure 3 and Supplementary Videos S1 and S2). At 6 dpf, 8/33 of the pkd2 mutants had edema and 6/33 showed atrioventricular (AV) block; of these six arrhythmic fish, one had edema. At 9 dpf, 30/39 of the pkd2 mutants had edema and 11/39 showed AV block; of these 11 arrhythmic fish, nine had edema. This latter finding indicates that the arrhythmia did not develop secondary to the edema in the pkd2 mutants.
Fig. 3.
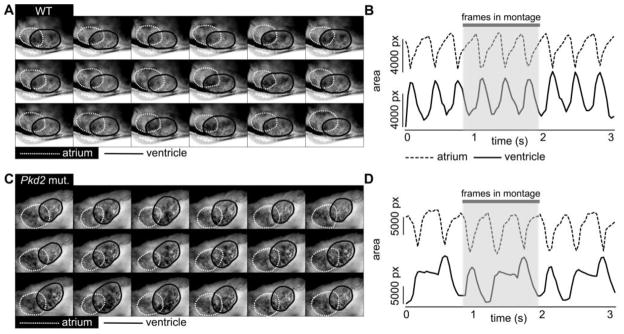
Pkd2 mutant zebrafish show atrioventricular block. Montage of images taken from in vivo video recordings of 6 dpf WT (A) and pkd2 mutant (C) zebrafish hearts. The interval between images in the montage is 1/15s. Outlines of atrium are shown with white dashed line and outlines of ventricle with black line. Panels on the right show corresponding changes in atrial (dashed line) and ventricular (continuous line) area in the WT (B) and the pkd2 mutant (D). The traces in (D) show three beats where the ventricle fails to contract after the atrial contraction, followed by further filling of the ventricle during the next atrial contraction, as shown by the concomitant increase in ventricular area.
AV delay was similar in 3 dpf isolated WT and pkd2 mutant hearts, as measured by the time needed for propagation of the calcium wave from atrium to ventricle. This measurement excludes altered AV delay as a cause for the AV block observed in vivo in the pkd2 mutant hearts. We also excluded AV valve regurgitation in the pkd2 mutant hearts as a cause for the heart failure, as none of the hearts showed significant regurgitation or valvular defects.
The edema seen in pkd2 mutant fish is mainly due to pronephric dysfunction [41]. Additionally, the failing heart, as indicated by reduced stroke volume and heart rate, may contribute to the edema. However, by 9 dpf we failed to observe a significant difference between the WT fish and pkd2 mutants in heart size.
3.5. Pkd2 mutant zebrafish show aberrant intracellular calcium signaling
To investigate potential underlying mechanisms of impaired cardiac function observed in vivo in pkd2 mutant fish, we next examined intracellular calcium cycling at the organ level. Hearts from 3 dpf zebrafish were microdissected and imaged with fluo-4 to record calcium transients (Supplementary Video S3). The shape of spontaneous calcium transients differed between pkd2 mutants and WT fish, especially in the ventricles (Figure 4A). Whereas WT ventricles showed a fast rise of the calcium transient followed by a plateau phase, pkd2 mutants displayed a slow rise and lack of a plateau phase. Pkd2 mutants exhibited increased transient rise times, durations, and decay times, with the differences compared to WT hearts being more pronounced in the ventricle (Figure 4B–D). The temporal characteristics of calcium transients between different regions of the cardiac chambers were similar, indicating that spatio-temporal heterogeneity of calcium release/reuptake did not significantly affect the calcium transient results in non-alternating hearts (Supplementary Figure S2).
Fig. 4.
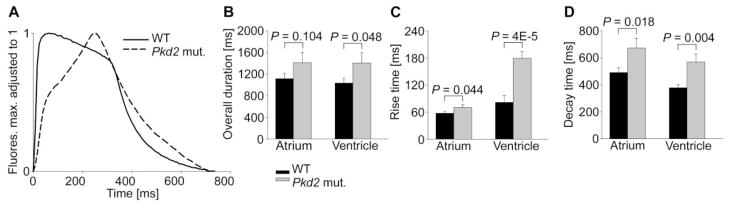
Pkd2 mutant zebrafish hearts show aberrant calcium cycling. (A) Representative spontaneous ventricular calcium transients. Comparison of calcium transient duration (B), rise time from 10% to 90% of peak amplitude (C), and decay time from 90% to 10% of peak amplitude (D). WT n = 14, pkd2 mut. n = 15. Data shown as mean ± s.e.m.
While pacing the isolated hearts in a field stimulation chamber, the pkd2 mutant hearts occasionally displayed calcium transient amplitude alternans, an every other beat variation in the transient amplitude (Figure 5A and B, Supplementary Figure S3). In total, 8/55 (15%) pkd2 mutant hearts showed alternans when paced at 120 beats per minute (bpm), whereas none of WT hearts (0/55) showed alternans at this physiological heart rate. One WT heart showed alternans when pacing was increased to 200 bpm, a rate that is above the physiological heart rate. The pkd2 mutant hearts failed to sustain calcium transients at this high rate of pacing.
Fig. 5.

Pkd2 mutant zebrafish hearts show calcium alternans. When paced at 120 bpm, pkd2 mutant hearts show occasional calcium alternans. (A) An overlay of representative ventricular calcium transients during pacing. (B) Percentage of hearts showing calcium alternans when paced at 120 bpm. WT n= 55, pkd2 mut. n = 55.
The difference in heart rate between WT and pkd2 mutants observed in vivo persisted in the isolated spontaneously beating hearts (Figure 6A). Hearts were paced at 120 bpm and transient amplitudes (F/F0) (Figure 6B), areas (Figure 6C), and baseline fluorescence levels (Figure 6D) were compared to values obtained during spontaneous beating for each heart. Pacing decreased transient amplitude and increased baseline fluorescence significantly more in pkd2 mutant compared to WT ventricles. Changes in atria and in transient areas followed the same trend. Representative raw traces of WT (Figure 6E) and pkd2 mutant (Figure 6F) calcium transients from atria and ventricles demonstrate characteristic transient shapes, relative amplitude of paced vs. spontaneous transients, and rise in baseline fluorescence in response to pacing. The calcium transient area under the curve in response to caffeine and thapsigargin was significantly smaller in pkd2 mutant ventricles (Figure 6G). These results show that PC2 is needed to maintain normal calcium transients and SR calcium stores in the heart.
Fig. 6.

Pacing increases resting Ca2+ levels and decreases Ca2+ cycling in pkd2 mutant hearts. (A) Spontaneous ventricular beating rate of isolated 3 dpf zebrafish hearts, WT n = 27, pkd2 mut. n = 26. Values of normalized calcium transient amplitude (F/F0) (B), area (C), and baseline fluorescence levels (D) during pacing at 120 bpm divided by values obtained from the same hearts during spontaneous beating. WT n = 14, pkd2 mut. n = 17. Representative raw traces of WT (E) and pkd2 mutant (F) calcium transients from atria and ventricles during spontaneous beating and pacing at 120 bpm, arbitrary units. (G) Calcium transient area in response to stimulation with caffeine and thapsigargin, revealing reduced SR calcium stores in pkd2 mutant ventricles. WT n = 4, pkd2 mut. n = 9. Data shown as mean ± s.e.m.
To confirm that our results are not due to differences in the expression of cardiac calcium cycling proteins, we determined mRNA levels of SERCA2a, RyR2a, RyR2b, InsP3R1, InsP3R2, and InsP3R3 in pkd2 mutant fish and their siblings with normal phenotype. There were no differences in mRNA levels to account for the functional differences observed in the fish (Supplementary Figure S4).
3.6. Pkd2 mutant zebrafish have shortened ventricular action potential duration
We recorded action potentials optically from 4–6 dpf isolated zebrafish hearts by staining them with the voltage-sensitive dye di-4-ANEPPS. We found that compared to WT fish, pkd2 mutants show shorter ventricular action potential duration (APD), faster AV conduction, and persistence of action potentials during pacing at higher frequencies (Supplementary Figure S5)
3.7. ADPKD is associated with an increased prevalence of IDCM
To explore whether the impaired calcium cycling and cardiac dysfunction found in pkd2 mutant zebrafish hearts could be relevant to human ADPKD, we examined the ADPKD database at the Mayo Clinic, Rochester, MN for an association between ADPKD and IDCM. The database contained information on 2 620 ADPKD patients seen between 1984 and 2010, including 374 genotyped patients with known PKD mutations (307 with PKD1 and 67 with PKD2 mutations). Of the 307 patients with PKD1 mutations, seven patients from seven different families had a diagnosis of IDCM. Of the 67 patients with PKD2 mutations, six patients from four different families had a diagnosis of IDCM. All identified PKD2 mutations in these four families are predicted to truncate the PC2 protein.
The pedigrees of the four families with coexisting PKD2 and IDCM are shown in Figure 7 and cardiac and renal images in four of these patients in Figure 8. In addition to six patients with IDCM, two additional family members had subclinical dilated cardiomyopathy or stress-induced cardiomyopathy. The first, in family M46 (II-1), had mild left ventricular enlargement and reduction in left ventricular ejection fraction (LVEF) on echocardiographic screening, and an exercise sestamibi scan was negative for ischemia. The second, in family M403 (II-2), had dilated cardiomyopathy with persistent T-wave inversion diagnosed following a motor vehicle accident; LVEF in this patient returned to normal after one month. Another patient in family M26 (III-6) had IDCM in the absence of ADPKD, as confirmed by DNA analysis. Demographic and genetic information on these nine patients, and a summary of the renal and cardiac findings including information on serum creatinine levels and hypertension, are presented in Table 1.
Fig. 7.
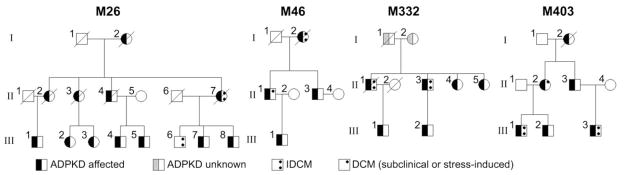
Pedigrees of four families with coexisting PKD2 and IDCM. For simplicity, individuals without ADPKD and/or IDCM or with unknown status are not shown in the figure, but are listed here: M26 Gen II: 1 ADPKD2 female, 3 unaffected brothers and 3 unaffected sisters, all deceased, Gen III 2,3 sibship: 2 unaffected brothers, Gen III 4,5 sibship: 2 unaffected brothers, 3 unaffected sisters, Gen III 6,7,8 sibship: 1 unaffected sister. M46 Gen III: pair of unaffected female twins. M332 Gen II: 2 unaffected brothers, Gen III 1 sibship: 3 unaffected brothers. M403 Gen III 1,2 sibship: 1 unaffected sister, Gen III 3 sibship: one unaffected brother and sister.
Fig. 8.

Cardiac and renal images of four PKD2 patients. Heart outlined with white dotted line and arrows. (A, B) Coronal gadolinium enhanced MR images from a 44-year-old male (M26 III-6) who three years earlier underwent cardiac transplantation for IDCM. A 3.5 cm cyst was present in the right kidney, but he did not carry the PKD2 mutation present in his family. His mother (M26 II-7) had ADPKD and IDCM and died at age 67 from a ruptured intracranial aneurysm. (C, D) Axial contrast enhanced CT images of the heart and kidneys from a 51-year-old male (M46 II-1) with subclinical cardiomyopathy and ADPKD caused by a PKD2 mutation. His mother (M46 I-2) had IDCM and ADPKD and died at age 82 from heart failure. (E, F) Axial unenhanced CT images of the heart and kidneys from a 78-year-old male (M332 II-3) with IDCM and ADPKD caused by a PKD2 mutation. His brother (M332 II-1) also had IDCM and ADPKD and died at age 62 from heart failure. (G, H) Axial unenhanced CT images of the heart and kidneys from a 35-year-old male (M403 III-3) with IDCM and ADPKD caused by a PKD2 mutation. He underwent cardiac transplantation two years later, and started dialysis 10 years after that. (I, J) Coronal and axial unenhanced CT images of the liver and kidneys at age 47 illustrate the progression of the polycystic kidney and liver disease. His cousin (M403 III-1) also has IDCM and ADPKD and his aunt (M403 II-2) has a history of stress-induced cardiomyopathy.
Table 1.
Details of IDMC in PKD2 patients
| Family, Pedigree ID, Gender | PKD2 mutation | Age at Dx of PKD (yrs) | Age at Dx of HT (yrs) | Age at Dx of IDCM (yrs) | Age at evaluation (yrs) | Serum creatinine (μmol/L) | LV Diameter (mm) | LV Ejection Fraction (%) | LVMI (g/m2) | EKG abnormalities | EKG /nuclear scan Exercise | Coronary angiogram | Heart TX |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M26, II-7, F | R361X | 57 | --- | 66 | 66 | 115 | Enlarged | 25 | NA | Sinus tachycardia, LBBB | No ischemia | 40% LAD | No |
| M26, III-6, M | NMD | ---- | ---- | 41 | 41 | 115 | Enlarged | 10 | NA | sinus tachycardia, LBBB | No ischemia | 35% LAD | Yes |
| 41 | 97 | 82/77 | 11 | NA | |||||||||
| M46, I-2, F | R807X | 68 | 68 | 69 | 69 | 97 | 60/NA | 45 | PVCs, LBBB,1st degree AV block, AF | NA | Normal | No | |
| 70 | 115 | 63/56 | 20–25 | ||||||||||
| 71 | 133 | 63/52 | 35–40 | ||||||||||
| 72 | 133 | 61/44 | 48 | ||||||||||
| 74 | 133 | 53/37 | 55 | ||||||||||
| 76 | 124 | 66/48 | 30–35 | 146 | |||||||||
| 81 | 203 | 75/65 | 20–25 | 203 | |||||||||
| 82 | 318 | 75/67 | 20 | 227 | |||||||||
| M46, II-1, M | R807X | 37 | 32 | 56a | 51 | 159 | 54/33 | 66 | 120 | PVCs | No ischemia | ND | No |
| 56 | 177 | 62/38 | 60 | NA | |||||||||
| 57 | 203 | 55/36 | 50 | 122 | |||||||||
| 58 | 239 | 56/38 | 59 | 125 | |||||||||
| M332, II-3, M | 423_430del8 | 63 | 63 | 78 | 55 | 97 | 54/35 | NA | NA | PVCs, multi-focal atrial tachycardia, VT runs, variable AV block | No ischemia | ND | No |
| 78 | 460 | 64/NA | 45 | 133 | |||||||||
| 81 | PD | 71/64 | 18 | 173 | |||||||||
| M332, II-1, M | 423_430del8 | 50 | 50 | 48 | 48 | 88 | 70/50 | NA | NA | PVCs | NA | Normal | No |
| 62 | NA | NA | 11–13 | NA | |||||||||
| M403, III-3, M | IVS4-5A>G | 35 | ---- | 35 | 35 | 115 | 85/74 | 20 | 255 | PVCs, SVT, LBBB | No ischemia | Normal | Yes |
| 35 | 133 | 86/75 | 15–20 | 251 | |||||||||
| 36 | 133 | 86/70 | 20 | 247 | |||||||||
| 36 | 133 | 95/90 | 15 | 246 | |||||||||
| M403, III-1, M | IVS4-5A>G | 40 | --- | 41 | 25 | 60 | NA | NA | Multifocal PVCs | No ischemia | Normal | No | |
| 36 | 80 | 62/49 | 50 | NA | |||||||||
| 41 | 80 | 72/64 | 35–40 | NA | |||||||||
| M403, II-2, F | IVS4-5A>G | 51 | 53 | 51b | 51 | 80 | 47/29 | 35 | NA | T-wave inversion | No ischemia | ND | ND |
| 51 | 80 | 50/31 | 60 | NA |
Abbreviations: NA, not available; NMD, no mutation detected; ND, not done; Dx, diagnosis; HT, hypertension; LV, left ventricular; LVMI, left ventricular mass index; TX, transplant; LBBB, left bundle branch block; LAD, left anterior descending artery; PVC, premature ventricular contraction; AV, atrioventricular; AF, atrial fibrillation; PD, peritoneal dialysis; VT, ventricular tachycardia; SVT, supraventricular tachycardia.
Subclinical cardiomyopathy
Stress-induced cardiomyopathy
4. Discussion
We found cardiac dysfunction and impaired cardiac intracellular calcium cycling in pkd2 mutant zebrafish. Supporting the clinical relevance of these findings, we found that PKD2 mutations and IDCM coexist in ADPKD patients significantly more frequently than expected by chance.
Young normotensive ADPKD patients show bi-ventricular diastolic dysfunction [10], indicating that cardiac dysfunction in ADPKD patients does not develop solely in response to hypertension and renal failure. Our findings support this, as four out of the six PKD2 patients were normotensive at the time of diagnosis of IDCM. Interestingly, pkd2−/− mice die before parturition with edema and other signs of functional heart failure. The mechanisms are unclear, however, two hypotheses are intracranial aneurysms and diastolic dysfunction [42]. Reduced cardiac output and prolonged relaxation are the hallmarks of diastolic dysfunction [43]. Altered intracellular calcium homeostasis as well as reduced sensitivity of myofilaments to calcium is thought to underlie diastolic dysfunction and IDCM.
Our results support a diastolic dysfunction mechanism for heart failure. During systole, peak velocities of erythrocytes in the aorta were similar in WT and pkd2 mutant fish, although cardiac output in the pkd2 mutants was nearly half of that in WT fish. Additionally, preservation of heart size, but poor stress tolerance to pacing in the pkd2 mutants further supports diastolic dysfunction.
Recently, reduced termination threshold of SR calcium release was reported to underlie IDCM associated with RyR2 mutations [44]. Our previous findings using isolated murine cardiomyocytes showed that PC2 binds to RyR2 and functionally maintains the RyR2 in the closed state during the diastolic phase [30]. Similarly, we suggest that loss-of-function mutations in PC2 will lead to prolonged RyR2-dependent calcium release, which may predispose to IDCM. This suggestion is supported by the impaired calcium cycling, including prolonged calcium transients, already observed in the pkd2 mutant hearts at early stages of development and further impaired by stressing with pacing. Pacing also increased the diastolic calcium levels in the pkd2 mutant ventricles, a typical finding in failing hearts. In addition to abnormal SR calcium release [45], also abnormal SR calcium reuptake and prolonged calcium transient decay have been linked to heart failure and hereditary IDCM [46]. Our data support this, as both rise and decay time of calcium transients were significantly prolonged, especially in the ventricles. As suggested by the results of the pacing experiments, the SR calcium stores were reduced in pkd2 mutant ventricles. Our results in the heart are consistent with previous findings in murine vascular smooth muscle cells [27, 47, 48].
Two out of the six PKD2-IDCM patients showed AV block. The AV block observed in pkd2 mutant zebrafish hearts fits our observations on impaired calcium cycling in the ventricles. As heart rate or pacing is increased beyond a threshold, the ventricle fails to contract due to slowed calcium cycling. The development of calcium alternans at physiological rates of pacing in the pkd2 mutant hearts, but not in the WT hearts, supports this interpretation. The fast AV conduction in the pkd2 mutants combined with slow calcium cycling may have contributed to the occasional inability of the action potential to induce an intracellular calcium transient and contraction in the ventricle. In the pkd2 mutant hearts sarcolemmal ion fluxes (ie: action potentials) kept up with pacing at 200 bpm, whereas calcium transients could not be sustained at this high a frequency, and occasionally showed calcium transient alternans already at 120 bpm. These results support impaired calcium cycling as the mechanism behind the AV block observed in vivo. Furthermore, fast AV conduction may have limited ventricular filling and in this way contributed to the lower cardiac output seen in the pkd2 mutants.
We failed to see afterdepolarizations, suggesting that the sodium-calcium-exchanger (NCX) activity remained below the threshold for triggering observable inward currents during diastole [49]. L-type calcium channels (LTCC) are relevant to the plateau phase of the AP in zebrafish [50, 51] and calcium-dependent inactivation of LTCCs is likely playing an important role here [52]. Shorter APD and persistence of action potentials at higher pacing frequencies in the pkd2 mutants may be explained by faster inactivation of LTCC by high intracellular calcium concentrations. The contribution of LTCC and NCX on APD depends, among other issues, on the length of the AP. In species with long APs, including zebrafish, the increase in calcium decreases APD via calcium-dependent inactivation of LTCC [53]. Additionally, sarcolemmal calcium flux may also have contributed to the longer APD in WT fish.
Calcium alternans is thought to be the main cause of action potential alternans, which is seen on the electrocardiogram (EKG) as T-wave alternans TWA [54]. TWA is frequently seen in patients with IDCM and heart failure, and is linked to an increased risk of sudden cardiac death due to ventricular tachyarrhythmias [55]. The subcellular mechanisms of calcium alternans are unclear, but the primary hypothesis is that alternans develops when intracellular calcium cycling is not in balance due to abnormal release and/or reuptake of calcium [56]. One well established molecular candidate is RyR2, as RyR2 dysfunction has been shown to cause calcium alternans [57, 58]. As changes associated with abnormal RyR2 function, such as leaky RyR2s, may disrupt normal calcium release and lead to calcium alternans [59], we propose that PC2 deficiency may promote calcium alternans in a similar manner [30].
RyR2 is the predominant isoform of RyR in the zebrafish heart [60]. Therefore, it would seem likely that the altered calcium release observed in fish lacking PC2 could also be explained by decreased RyR2 and that this would play a role in the development of IDCM and heart failure. However, we did not observe a compensatory mRNA change in RyR2 levels or other calcium signaling proteins (Supplementary Figure S1). Therefore, the changes in calcium signaling and cardiac function are more likely to be explained by alterations to the functional interaction of PC2 with RyR2 or other proteins involved in calcium signaling. Therefore, further studies are needed to decipher the detailed pathomechanisms of PC2 deficiency in the heart.
PC1 has a possible indirect effect on calcium cycling through its interaction with PC2 [21]. Based on this line of thought, we expected to see a greater prevalence of IDCM in PKD2 patients when compared to PKD1 patients. Confirming our hypothesis, IDCM was most common in patients with PKD2 mutations, with six out of 67 (~9%) patients having both conditions. Additionally, two more PKD2 patients had subclinical or stress-induced cardiomyopathy. In contrast, seven out of 307 (~2%) patients with PKD1 mutations also had IDCM. The frequent coexistence of ADPKD and IDCM in the Mayo Clinic ADPKD database is unlikely to be explained by chance association alone, as IDCM prevalence in the general population is 0.04%. Furthermore, it is likely that cases of early or mild IDCM might have gone undiagnosed, especially in ADPKD patients with PKD2 mutations. On the other hand, we must acknowledge a possible selection bias in the ADPKD database. Symptomatic patients with a more severe disease phenotype are probably more likely to undergo cardiac diagnostics. This selection bias could favor PKD1 patients, who tend to present with earlier onset and a more severe phenotype of ADPKD. However, ADPKD mutations cannot entirely account for the development of IDCM, because IDCM did not segregate with ADPKD in one patient in these families. It seems more likely that ADPKD represents a risk factor for the development of cardiomyopathy that requires additional genetic or environmental factors, as observed in our case of stress-induced cardiomyopathy.
Interestingly, 4 of 11 (36%) of the families with mutations identified had a PKD2 mutation, a higher frequency than the 15% usually observed in large clinical series of ADPKD patients. This may be due to an under-representation of PKD2 associated disease in clinical series with a selection bias determined by the severity of the renal disease [61].
In summary, we found a novel association between ADPKD and IDCM and suggest altered calcium signaling as a possible mechanism. We found low cardiac output, impaired intracellular calcium cycling, calcium alternans, and atrioventricular block in pkd2 mutant zebrafish embryo hearts. Together, these results indicate heart failure in the pkd2 mutant fish. In human ADPKD patients, we found IDCM to coexist frequently with PKD2 mutations as PKD2 patients displayed an over 200-fold greater prevalence of IDCM than the general population. We suggest modulation of calcium cycling by PC2 as a potential mechanism. Further studies are warranted to study the relationship between ADPKD and IDCM in more detail, as well as the mechanisms of cardiomyopathy in ADPKD patients with PKD1 and PKD2 mutations. Meanwhile, we propose PKD mutations to be considered a possible contributor to the development of IDCM in the clinical setting.
Supplementary Material
Supplementary Figure S1. Pkd2 localizes to the sarcoplasmic reticulum in zebrafish cardiac cells.
Supplementary Figure S2. Spatial homogeneity of calcium transients within atria and ventricles of isolated zebrafish hearts.
Supplementary Figure S3. Discordant calcium alternans in an isolated pkd2 mutant zebrafish heart.
Supplementary Figure S4. Expression mRNA level for proteins important in cardiac calcium cycling in pkd2 mutants and their siblings with normal phenotype.
Supplementary Figure S5. Optical action potential recordings in isolated zebrafish hearts.
Video S1, in vivo recording of a WT zebrafish heart showing normal rhythm and AV conduction.
Video S2, in vivo recording of a pkd2 mutant zebrafish heart showing normal rhythm and AV block.
Video S3, recording of intracellular calcium transients in an isolated WT zebrafish heart.
Highlights.
Pkd2 mutant zebrafish display reduced cardiac output, atrioventricular block, and edema
Pkd2 mutant hearts show prolonged Ca2+ transients and Ca2+ alternans
Pacing increases resting Ca2+ levels and decreases Ca2+ cycling in pkd2 mutant hearts
ADPKD is associated with an increased prevalence of IDCM
Patients with PKD2 mutations are more likely than PKD1 patients to suffer from IDCM
Acknowledgments
Funding sources
This work was supported by a grant from the Finnish Foundation for Cardiovascular Research to J.P., a scholarship from the German National Academic Foundation to S.S., a Postdoctoral Fellowship from the American Heart Association (R10682) to I.Y.T.K., and grants from the NIH (DK57328 and DK61747) to B.E.E., (DK058816) to P.C.H., and (DK90728) to V.E.T.
We thank Dr Iain Drummond (Department of Medicine, Massachusetts General Hospital, Charlestown, MA, USA) for kindly providing the PC2 antibody. We are grateful to the members of the laboratory of Dr Zhaoxia Sun for assistance with zebrafish. We thank Dr Lawrence Young (Department of Medicine/Cardiology, Yale University, New Haven, CT), Dr Matti Viitasalo (Department of Cardiology, Helsinki University Central Hospital, Helsinki, Finland), and Dr Pasi Tavi (A.I. Virtanen Institute, University of Eastern Finland, Kuopio, Finland) for valuable comments.
Abbreviations
- ADPKD
autosomal dominant polycystic kidney disease
- APD
action potential duration
- AV
atrioventricular
- bpm
beats per minute
- dpf
days post fertilization
- CICR
calcium-induced calcium release
- ER
endoplasmic reticulum
- hpf
hours post fertilization
- IDCM
idiopathic dilated cardiomyopathy
- InsP3R
inositol 1,4,5-trisphosphate receptor
- LTCC
L-type calcium channel
- LVEF
left ventricular ejection fraction
- NCX
sodium-calcium-exchanger
- PC1
polycystin-1
- PC2
polycystin-2
- PKD1
polycystic kidney disease 1
- PKD2
polycystic kidney disease 2
- RyR2
cardiac ryanodine receptor
- SR
sarcoplasmic reticulum
- TRPP2
polycystin-2
- TWA
T-wave alternans
- WT
wildtype
Footnotes
Disclosures
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 2.Fick GM, Johnson AM, Hammond WS, Gabow PA. Causes of death in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1995;5:2048–2056. doi: 10.1681/ASN.V5122048. [DOI] [PubMed] [Google Scholar]
- 3.Perrone RD, Ruthazer R, Terrin NC. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: contribution of extrarenal complications to mortality. Am J Kidney Dis. 2001;38:777–784. doi: 10.1053/ajkd.2001.27720. [DOI] [PubMed] [Google Scholar]
- 4.Chapman AB, Stepniakowski K, Rahbari-Oskoui F. Hypertension in autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17:153–163. doi: 10.1053/j.ackd.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chapman AB, Johnson AM, Rainguet S, Hossack K, Gabow P, Schrier RW. Left ventricular hypertrophy in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1997;8:1292–7. doi: 10.1681/ASN.V881292. [DOI] [PubMed] [Google Scholar]
- 6.Pirson Y, Chauveau D, Torres V. Management of cerebral aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2002;13:269–276. doi: 10.1681/ASN.V131269. [DOI] [PubMed] [Google Scholar]
- 7.Hossack KF, Leddy CL, Johnson AM, Schrier RW, Gabow PA. Echocardiographic findings in autosomal dominant polycystic kidney disease. N Engl J Med. 1988;319:907–12. doi: 10.1056/NEJM198810063191404. [DOI] [PubMed] [Google Scholar]
- 8.Lumiaho A, Ikaheimo R, Miettinen R, Niemitukia L, Laitinen T, Rantala A, et al. Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1. Am J Kidney Dis. 2001;38:1208–16. doi: 10.1053/ajkd.2001.29216. [DOI] [PubMed] [Google Scholar]
- 9.Bardaji A, Vea AM, Gutierrez C, Ridao C, Richart C, Oliver JA. Left ventricular mass and diastolic function in normotensive young adults with autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1998;32:970–5. doi: 10.1016/s0272-6386(98)70071-x. [DOI] [PubMed] [Google Scholar]
- 10.Oflaz H, Alisir S, Buyukaydin B, Kocaman O, Turgut F, Namli S, et al. Biventricular diastolic dysfunction in patients with autosomal-dominant polycystic kidney disease. Kidney Int. 2005;68:2244–9. doi: 10.1111/j.1523-1755.2005.00682.x. [DOI] [PubMed] [Google Scholar]
- 11.Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O’Connell J, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation. 1996;93:841–2. doi: 10.1161/01.cir.93.5.841. [DOI] [PubMed] [Google Scholar]
- 12.Codd MB, Sugrue DD, Gersh BJ, Melton LJ., 3rd Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975–1984. Circulation. 1989;80:564–572. doi: 10.1161/01.cir.80.3.564. [DOI] [PubMed] [Google Scholar]
- 13.Dec GW, Fuster V. Idiopathic dilated cardiomyopathy. N Engl J Med. 1994;331:1564–1575. doi: 10.1056/NEJM199412083312307. [DOI] [PubMed] [Google Scholar]
- 14.Keeling PJ, Gang Y, Smith G, Seo H, Bent SE, Murday V, et al. Familial dilated cardiomyopathy in the United Kingdom. Br Heart J. 1995;73:417–21. doi: 10.1136/hrt.73.5.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baig MK, Goldman JH, Caforio AL, Coonar AS, Keeling PJ, McKenna WJ. Familial dilated cardiomyopathy: cardiac abnormalities are common in asymptomatic relatives and may represent early disease. J Am Coll Cardiol. 1998;31:195–201. doi: 10.1016/s0735-1097(97)00433-6. [DOI] [PubMed] [Google Scholar]
- 16.Grunig E, Tasman JA, Kucherer H, Franz W, Kubler W, Katus HA. Frequency and phenotypes of familial dilated cardiomyopathy. J Am Coll Cardiol. 1998;31:186–94. doi: 10.1016/s0735-1097(97)00434-8. [DOI] [PubMed] [Google Scholar]
- 17.Karkkainen S, Peuhkurinen K. Genetics of dilated cardiomyopathy. Ann Med. 2007;39:91–107. doi: 10.1080/07853890601145821. [DOI] [PubMed] [Google Scholar]
- 18.Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995;10:151–60. doi: 10.1038/ng0695-151. [DOI] [PubMed] [Google Scholar]
- 19.Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–42. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 20.Rossetti S, Consugar MB, Chapman AB, Torres VE, Guay-Woodford LM, Grantham JJ, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007;18:2143–60. doi: 10.1681/ASN.2006121387. [DOI] [PubMed] [Google Scholar]
- 21.Casuscelli J, Schmidt S, DeGray B, Petri ET, Celic A, Folta-Stogniew E, et al. Analysis of the cytoplasmic interaction between polycystin-1 and polycystin-2. Am J Physiol Renal Physiol. 2009;297:F1310–5. doi: 10.1152/ajprenal.00412.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vassilev PM, Guo L, Chen XZ, Segal Y, Peng JB, Basora N, et al. Polycystin-2 is a novel cation channel implicated in defective intracellular Ca(2+) homeostasis in polycystic kidney disease. Biochem Biophys Res Commun. 2001;282:341–50. doi: 10.1006/bbrc.2001.4554. [DOI] [PubMed] [Google Scholar]
- 23.Anyatonwu GI, Ehrlich BE. Calcium signaling and polycystin-2. Biochem Biophys Res Commun. 2004;322:1364–1373. doi: 10.1016/j.bbrc.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 24.Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, et al. Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol. 2002;4:191–7. doi: 10.1038/ncb754. [DOI] [PubMed] [Google Scholar]
- 25.Geng L, Okuhara D, Yu Z, Tian X, Cai Y, Shibazaki S, et al. Polycystin-2 traffics to cilia independently of polycystin-1 by using an N-terminal RVxP motif. J Cell Sci. 2006;119:1383–95. doi: 10.1242/jcs.02818. [DOI] [PubMed] [Google Scholar]
- 26.Tsiokas L, Kim S, Ong EC. Cell biology of polycystin-2. Cell Signal. 2007;19:444–53. doi: 10.1016/j.cellsig.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qian Q, Li M, Cai Y, Ward CJ, Somlo S, Harris PC, et al. Analysis of the polycystins in aortic vascular smooth muscle cells. J Am Soc Nephrol. 2003;14:2280–7. doi: 10.1097/01.asn.0000080185.38113.a3. [DOI] [PubMed] [Google Scholar]
- 28.Li Q, Dai Y, Guo L, Liu Y, Hao C, Wu G, et al. Polycystin-2 associates with tropomyosin-1, an actin microfilament component. J Mol Biol. 2003;325:949–62. doi: 10.1016/s0022-2836(02)01333-5. [DOI] [PubMed] [Google Scholar]
- 29.Volk T, Schwoerer AP, Thiessen S, Schultz JH, Ehmke H. A polycystin-2-like large conductance cation channel in rat left ventricular myocytes. Cardiovasc Res. 2003;58:76–88. doi: 10.1016/s0008-6363(02)00858-1. [DOI] [PubMed] [Google Scholar]
- 30.Anyatonwu GI, Estrada M, Tian X, Somlo S, Ehrlich BE. Regulation of ryanodine receptor-dependent calcium signaling by polycystin-2. Proc Natl Acad Sci U S A. 2007;104:6454–9. doi: 10.1073/pnas.0610324104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marks AR. Cardiac intracellular calcium release channels: role in heart failure. Circ Res. 2000;87:8–11. doi: 10.1161/01.res.87.1.8. [DOI] [PubMed] [Google Scholar]
- 32.Marks AR, Priori S, Memmi M, Kontula K, Laitinen PJ. Involvement of the cardiac ryanodine receptor/calcium release channel in catecholaminergic polymorphic ventricular tachycardia. J Cell Physiol. 2002;190:1–6. doi: 10.1002/jcp.10031. [DOI] [PubMed] [Google Scholar]
- 33.Sehnert AJ, Stainier DY. A window to the heart: can zebrafish mutants help us understand heart disease in humans? Trends Genet. 2002;18:491–494. doi: 10.1016/s0168-9525(02)02766-x. [DOI] [PubMed] [Google Scholar]
- 34.Dahme T, Katus HA, Rottbauer W. Fishing for the genetic basis of cardiovascular disease. Dis Model Mech. 2009;2:18–22. doi: 10.1242/dmm.000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leong IU, Skinner JR, Shelling AN, Love DR. Zebrafish as a model for long QT syndrome: the evidence and the means of manipulating zebrafish gene expression. Acta Physiol (Oxf) 2010;199:257–276. doi: 10.1111/j.1748-1716.2010.02111.x. [DOI] [PubMed] [Google Scholar]
- 36.Sun Z, Amsterdam A, Pazour GJ, Cole DG, Miller MS, Hopkins N. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development. 2004;131:4085–93. doi: 10.1242/dev.01240. [DOI] [PubMed] [Google Scholar]
- 37.Obara T, Mangos S, Liu Y, Zhao J, Wiessner S, Kramer-Zucker AG, et al. Polycystin-2 immunolocalization and function in zebrafish. J Am Soc Nephrol. 2006;17:2706–18. doi: 10.1681/ASN.2006040412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fu X, Wang Y, Schetle N, Gao H, Putz M, von Gersdorff G, et al. The subcellular localization of TRPP2 modulates its function. J Am Soc Nephrol. 2008;19:1342–51. doi: 10.1681/ASN.2007070730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Sullivan DA, Torres VE, Edwards WD, Edwards BS, Griffin MD, Cai Y, et al. Cardiac expression of polycystin 1 and polycystin 2 and idiopathic dilated cardiomyopathy in autosomal dominant polycystic kidney disease [abstract] J Am Soc Nephrol. 1997;8(376A) [Google Scholar]
- 40.Rossetti S, Kubly VJ, Consugar MB, Hopp K, Roy S, Horsley SW, et al. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 2009;75:848–55. doi: 10.1038/ki.2008.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drummond IA. Kidney development and disease in the zebrafish. J Am Soc Nephrol. 2005;16:299–304. doi: 10.1681/ASN.2004090754. [DOI] [PubMed] [Google Scholar]
- 42.Wu G, Markowitz GS, Li L, D’Agati VD, Factor SM, Geng L, et al. Cardiac defects and renal failure in mice with targeted mutations in Pkd2. Nat Genet. 2000;24:75–8. doi: 10.1038/71724. [DOI] [PubMed] [Google Scholar]
- 43.Periasamy M, Janssen PM. Molecular basis of diastolic dysfunction. Heart Fail Clin. 2008;4:13–21. doi: 10.1016/j.hfc.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang Y, Tian X, Wang R, Fill M, Chen SR. Abnormal Termination of Ca2+ Release Is a Common Defect of RyR2 Mutations Associated With Cardiomyopathies. Circ Res. 2012;110:968–77. doi: 10.1161/CIRCRESAHA.111.256560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sen L, Cui G, Fonarow GC, Laks H. Differences in mechanisms of SR dysfunction in ischemic vs. idiopathic dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. 2000;279:H709–18. doi: 10.1152/ajpheart.2000.279.2.H709. [DOI] [PubMed] [Google Scholar]
- 46.Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003;299:1410–3. doi: 10.1126/science.1081578. [DOI] [PubMed] [Google Scholar]
- 47.Torres VE, Cai Y, Chen X, Wu GQ, Geng L, Cleghorn KA, et al. Vascular expression of polycystin-2. J Am Soc Nephrol. 2001;12:1–9. doi: 10.1681/ASN.V1211. [DOI] [PubMed] [Google Scholar]
- 48.Qian Q, Hunter LW, Li M, Marin-Padilla M, Prakash YS, Somlo S, et al. Pkd2 haploinsufficiency alters intracellular calcium regulation in vascular smooth muscle cells. Hum Mol Genet. 2003;12:1875–80. doi: 10.1093/hmg/ddg190. [DOI] [PubMed] [Google Scholar]
- 49.Langenbacher AD, Dong Y, Shu X, Choi J, Nicoll DA, Goldhaber JI, et al. Mutation in sodium-calcium exchanger 1 (NCX1) causes cardiac fibrillation in zebrafish. Proc Natl Acad Sci U S A. 2005;102:17699–704. doi: 10.1073/pnas.0502679102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nemtsas P, Wettwer E, Christ T, Weidinger G, Ravens U. Adult zebrafish heart as a model for human heart? An electrophysiological study. J Mol Cell Cardiol. 2010;48:161–71. doi: 10.1016/j.yjmcc.2009.08.034. [DOI] [PubMed] [Google Scholar]
- 51.Brette F, Luxan G, Cros C, Dixey H, Wilson C, Shiels HA. Characterization of isolated ventricular myocytes from adult zebrafish (Danio rerio) Biochem Biophys Res Commun. 2008;374:143–6. doi: 10.1016/j.bbrc.2008.06.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang PC, Llach A, Sheng XY, Hove-Madsen L, Tibbits GF. Calcium handling in zebrafish ventricular myocytes. Am J Physiol Regul Integr Comp Physiol. 2011;300:R56–66. doi: 10.1152/ajpregu.00377.2010. [DOI] [PubMed] [Google Scholar]
- 53.Han C, Tavi P, Weckstrom M. Modulation of action potential by [Ca2+]i in modeled rat atrial and guinea pig ventricular myocytes. Am J Physiol Heart Circ Physiol. 2002;282:H1047–54. doi: 10.1152/ajpheart.00573.2001. [DOI] [PubMed] [Google Scholar]
- 54.Pruvot EJ, Katra RP, Rosenbaum DS, Laurita KR. Role of calcium cycling versus restitution in the mechanism of repolarization alternans. Circ Res. 2004;94:1083–1090. doi: 10.1161/01.RES.0000125629.72053.95. [DOI] [PubMed] [Google Scholar]
- 55.Narayan SM. T-wave alternans and the susceptibility to ventricular arrhythmias. J Am Coll Cardiol. 2006;47:269–281. doi: 10.1016/j.jacc.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 56.Salerno-Uriarte JA, De Ferrari GM, Klersy C, Pedretti RF, Tritto M, Sallusti L, et al. Prognostic value of T-wave alternans in patients with heart failure due to nonischemic cardiomyopathy: results of the ALPHA Study. J Am Coll Cardiol. 2007;50:1896–904. doi: 10.1016/j.jacc.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 57.Laurita KR, Rosenbaum DS. Cellular mechanisms of arrhythmogenic cardiac alternans. Prog Biophys Mol Biol. 2008;97:332–347. doi: 10.1016/j.pbiomolbio.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pieske B, Kockskamper J. Alternans goes subcellular: a “disease” of the ryanodine receptor? Circ Res. 2002;91:553–555. doi: 10.1161/01.res.0000036862.37203.f4. [DOI] [PubMed] [Google Scholar]
- 59.Lehnart SE, Terrenoire C, Reiken S, Wehrens XH, Song LS, Tillman EJ, et al. Stabilization of cardiac ryanodine receptor prevents intracellular calcium leak and arrhythmias. Proc Natl Acad Sci U S A. 2006;103:7906–10. doi: 10.1073/pnas.0602133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Darbandi S, Franck JP. A comparative study of ryanodine receptor (RyR) gene expression levels in a basal ray-finned fish, bichir (Polypterus ornatipinnis) and the derived euteleost zebrafish (Danio rerio) Comp Biochem Physiol B Biochem Mol Biol. 2009;154:443–448. doi: 10.1016/j.cbpb.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 61.Rossetti S, Adeva M, Kubly V, Consugar MB, Torres VE, Harris PC. An Olmsted County population-based study indicates that PKD2 is more common than previously described. J Am Soc Nephrol. 2007:18. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Pkd2 localizes to the sarcoplasmic reticulum in zebrafish cardiac cells.
Supplementary Figure S2. Spatial homogeneity of calcium transients within atria and ventricles of isolated zebrafish hearts.
Supplementary Figure S3. Discordant calcium alternans in an isolated pkd2 mutant zebrafish heart.
Supplementary Figure S4. Expression mRNA level for proteins important in cardiac calcium cycling in pkd2 mutants and their siblings with normal phenotype.
Supplementary Figure S5. Optical action potential recordings in isolated zebrafish hearts.
Video S1, in vivo recording of a WT zebrafish heart showing normal rhythm and AV conduction.
Video S2, in vivo recording of a pkd2 mutant zebrafish heart showing normal rhythm and AV block.
Video S3, recording of intracellular calcium transients in an isolated WT zebrafish heart.