

Abstract
Objective
Through its location on nociceptors, acid sensing ion channel 3 (ASIC3) is activated by decreases in pH and plays a significant role in musculoskeletal pain. We recently showed that decreases in pH activate ASIC3 located on fibroblast-like synoviocytes (FLS). Since FLS are key cells in the inflammatory process we tested if ASIC3-deficient mice with arthritis have altered inflammation and pain relative to controls.
Methods
Arthritis was induced by injection of a cocktail of anti-type II collagen antibodies induced collagen antibodyarthritis (CAIA). Inflammation and pain parameters in ASIC3−/− and ASIC3+/+ mice were assessed. Disease severity was measured with clinical arthritis scores, joint diameters, histological analysis of joints, and qPCR for synovial gene expression. Pain behaviors were measured by examining withdrawal thresholds of the joint and paw and by measuring physical activity levels in mice. Cell death was assessed with a Live/Dead assay in FLS in response to decreases in pH.
Results
Surprisingly, ASIC3−/− mice with CAIA demonstrated significantly increased joint inflammation, joint destruction and expression of IL-6, MMP-3 and MMP-13 in joint tissue compared to ASIC3+/+ mice. ASIC3+/+ FLS show enhanced cell death when exposed to pH 6.0 in the presence of interleukin-1β that is abolished in ASIC3−/− FLS. Despite enhanced disease severity, ASIC3−/− mice do not develop mechanical hypersensitivity of the paw and show greater levels of physical activity.
Conclusion
These data are consistent with the hypothesis that ASIC3 plays a protective role in inflammatory arthritis conditions by limiting inflammation through enhanced synoviocyte cell death to reduce disease severity and produce pain to reduce joint use.
Acid sensing ion channels (ASICs) are activated by decreased extracellular pH and are the primary acid-sensors in the nervous system. Four genes within mammalian genomes encode several splice variants including ASIC1a, ASIC1b, ASIC1b2, ASIC2a, ASIC2b, ASIC3, and ASIC4, which combine as heteromers to form functional ion channels. Of these, ASIC3 plays a significant role in musculoskeletal pain [20;41;43], including inflammatory pain [21;43;50]. Furthermore, ASIC3 is found in sensory neurons including nociceptors innervating synovial joints [20] and inflammatory pain models show an increased ASIC expression and function in sensory neurons [7;12;20;49;50]. Thus, ASICs are critical players in pain associated with inflammation.
Rheumatoid arthritis (RA) is a chronic inflammatory disease that primarily affects synovial joints. Innate and adaptive immune responses contribute to the initiation and perpetuation of disease, and murine models can provide insight into disease mechanisms [11]. Collagen-antibody induced arthritis (CAIA) in mice bypasses adaptive immunity and permits evaluation of how innate immune responses contribute to synovitis. We recently identified ASIC3 in fibroblast-like synoviocytes (FLS) [20;26], which are key cells that produce inflammatory mediators and contribute to joint damage in RA [18;22;24;34;46]. Cultured FLS respond to decreased pH by increasing intracellular calcium and this effect is blunted in FLS from ASIC3−/− mice [26]. Further, neuronal cells show enhanced cell death in response to decreases in pH that depends on activation of ASICs [23;31;53]. These data suggest that ASIC3 deficiency might suppress synoviocyte activation in inflammatory arthritis through increasing synoviocyte cell death.
Based on these data, we hypothesized that decreases in pH that occur in inflammatory arthritis could produce pain responses and reduce arthritis severity. Low pH could potentially protect the joint by 1) activating ASIC3 on nociceptors to produce pain and limit joint movement, and 2) activating ASIC3 in FLS to cause cell death and inhibit the inflammation.
MATERIALS AND METHODS
All experiments were approved by the Animal Care and Use Committee at the University of Iowa and the University of San Diego.
Mice
These experiments used C57Bl/6 mice (ASIC3+/+) and congenic ASIC3−/− mice on a C57Bl/6 background. ASIC3+/+ and ASIC3−/− mice were bred in-house, housed under the same conditions, but were not littermates. The construction of the ASIC3−/− mice has been previously described [37]. We have previously compared F2 generation mice to congenic ASIC3−/− mice for changes in behavior and show no differences between the groups in animals with inflammation [43].
Induction of arthritis
The CAIA model was induced by injection of 2 mg of anti-CII (collagen) antibody cocktail intravenously on Day 0 via the tail. On Day 3, mice were boosted with 25 μg lipopolysaccharide intraperitoneally (LPS 0111:B4) as previously described [17]. We specifically used a lower dose of the anti-CII antibody cocktail to enhance the ability to observe increased inflammation. Animals were then followed for 12–14 days. Inflammation and pain-behaviors were measured as outlined below. All inflammation and pain behavior assessments were done with the experimenter blinded to group.
Measures of Inflammation
Clinical arthritis
Arthritis scores were determined using a semiquantitative scale of 0 to 4 for each paw (for a total of 16) and by measuring paw diameter in inches [4;5;27;33]. Arthritis scores were taken every other day throughout the 12 days of testing in ASIC3+/+ and ASIC3−/− mice. Two separate experiments were performed and pooled for a total of n=20 for each strain.
Histologic analysis
Ankle joints were removed on day 12, decalcified, paraffin embedded, cut sagittally, and stained with Safranin-O or H&E. Histological changes quantified in a blinded manner on a 0 to 4 scale for changes in joint destruction, synovial thickening, bone erosion or extra-articular inflammation. ‘0’ represents normal and 4 represents severe destruction, thickening, erosion, or inflammation as we previously published [45]. We compared differences between ASIC3+/+ (n=10) and ASIC3−/− (n=10) mice.
Inflammatory cytokines and MMPs
Ankles were harvested and skin, muscle and excess bone was removed by dissection. Joint extracts that included bone, cartilage and symposium were snap frozen and pulverized. RNA isolation and cDNA synthesis was followed by quantitative real-time PCR were performed using Assays on Demand (Applied Biosystems Foster City CA). We applied a standard curve-based method of real-time PCR in order to control for inter-amplicon amplification efficiency and to normalize for differences in sample sizes as previously described. [1]. To control for variation in sample cellularity, expression of the housekeeping gene HPRT1 was measured in separate PCR reactions. Resulting cycle threshold, Ct, data were normalized to standard curves constructed from cDNA of stimulated mouse PBMC assayed simultaneously, yielding cell equivalents, CE, of expression. The ratio between the gene of interest and HPRT1 cell equivalents is reported as Relative Expression Units, REU.
Pain Behavior Measures
Pain-like behaviors were measured by assessing: 1) withdrawal thresholds of the ankle, 2) withdrawal thresholds of the paw, and 3) responsiveness of the paw to repeated mechanical stimuli. Pain-like behaviors were assessed before, 3 days after collagen-antibody injection, Day 6 or 7, Day 10 (paw only) and Day 12 from ASIC3+/+ (n=8–12) and ASIC3−/− (n=9–13) mice.
Mechanical sensitivity of the ankle was tested by squeezing the gastrocnemius muscle of the mice with a calibrated pair of tweezers until the mouse withdrew from the stimulus as previously described for the knee joint [20;40;50]. The force at which the mouse withdrew was measured in mN and called the muscle withdrawal threshold. Each hindlimb was tested 3 times, and the 3 trials were averaged. A decrease in force was considered deep tissue mechanical hyperalgesia.
Withdrawal thresholds of the paw were done using with von Frey filaments as previously described [2]. The 50% withdrawal threshold was determined by using the Dixon up-down method. Testing was initiated with a 0.35 g filament in the middle of the series, and the range of filaments was 0.03 – 1.7 g. Thresholds were calculated noting the stimulus of the first withdrawal. A decrease in force was interpreted as cutaneous mechanical hyperalgesia.
The number of responses to repeated stimulation of the paw to a standard von Frey filament, 0.4mN was assessed as previously described [30]. The filament was applied 5 times, and 10 trials of 5 were averaged. Animals were given 5 trials and this was repeated 10 times. An increase in the number of withdrawals was interpreted as secondary cutaneous hyperalgesia.
Function was assessed by examining physical activity. We measured the amount of time the animal remains active in a standard mouse cage during 60 min using the Activmeter (Bioseb, Inc., Vitrolles, Cedex, France). We measured total activity time which includes low and high speed activities and grooming behavior. We also were able to measure time spent in low speed activities (1–2 cm/s), high speed activities (≥2 cm/s), inactivity, and grooming behavior, and distance traveled. This allowed us to see if animals voluntarily reduce the forces applied to their joints by reducing physical activity levels. Measures of function were assessed before, at peak inflammation on Day 7 and on Day 12 from ASIC3+/+ (n=8) and ASIC3−/− (n=8) mice.
Cell death analysis in cultured fibroblast-like synoviocytes (FLS)
FLS were isolated from 3 WT and 3 ASIC3−/− mice to generate cell lines according to previously published methods [38]. FLS were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% Fetal Bovine Serum (FBS) at 37°C. 20,000 cells/ml were plated onto coverslips coated with poly-L-lysine and incubated at 37° 5% CO2 in DMEM complete media for 24 hours. Media was then replaced with 1% FBS DMEM for 24 hours and then incubated at pH 7.4 or pH 6.0 with or without 1ng/ml IL-1β for 24 h.
LIVE/DEAD Assay
Using the LIVE/DEAD Viability/Cytotoxicity kit (Invitrogen, L-3224, Grand Island, New York), 0.5 mM calcein AM and 0.5 mM ethydium homodimer-1 were used as described by the manufacturer. Stained cells were then mounted on slides with aqueous CMF1+ mounting media and immediately viewed under a fluorescent Olympus BX51 microscope with a SPOT camera at the Central Microscopy Core Facility. To ensure impartial selection, regions with sufficient cells were identified without viewing ethydium staining. Images of the same region were taken at 400x magnification and merged with Image J. Each merged image was quantified by counting at least 500 cells and determining the number of positively stained Live and Dead cells. Data are expressed as the percentage of dead cells.
Statistical Analysis
Pain-behaviors, physical activity scores, daily arthritis scores, and ankle thickness were assessed with a repeated measures ANOVA followed by Tukey’s post-hoc test for differences between groups at each time period. Histology scores and qPCR were analyzed with a t-test for differences between groups. Cell death assay was analyzed with a one-way ANOVA followed by a Tukey’s post-hoc for differences between groups. P< 0.05 was considered significant. Data are expressed as the mean ± SEM.
RESULTS
Increased arthritis severity in ASIC3−/− mice
The CAIA model was performed in ASIC3+/+ (wild type) and ASIC3−/− mice to determine the effect of ASIC3 on the development of disease. As shown in Figures 1A and B, arthritis severity was surprisingly increased in ASIC3−/− mice compared to ASIC3+/+ mice. Histologic analysis of the joints on Day 12 confirmed increased synovitis and demonstrated enhanced proteoglycan destruction, synovial lining thickening, bone erosion and extra-articular inflammation in the ASIC3−/− mice (Figure 2A and B).
Figure 1.

A. Clinical arthritis score from animals with collagen antibody-induced arthritis (CAIA) are enhanced in ASIC3−/− mice (n=20; black squares) when compared to ASIC3+/+ mice (n=20; white circles), *, p<0.05 B. Ankle joint thickness is greater in ASIC3−/− mice (n=20; black squares) when compared to ASIC3+/+ mice (n=20; white circles), *, p<0.05. Data are mean ± S.E.M.
Figure 2.
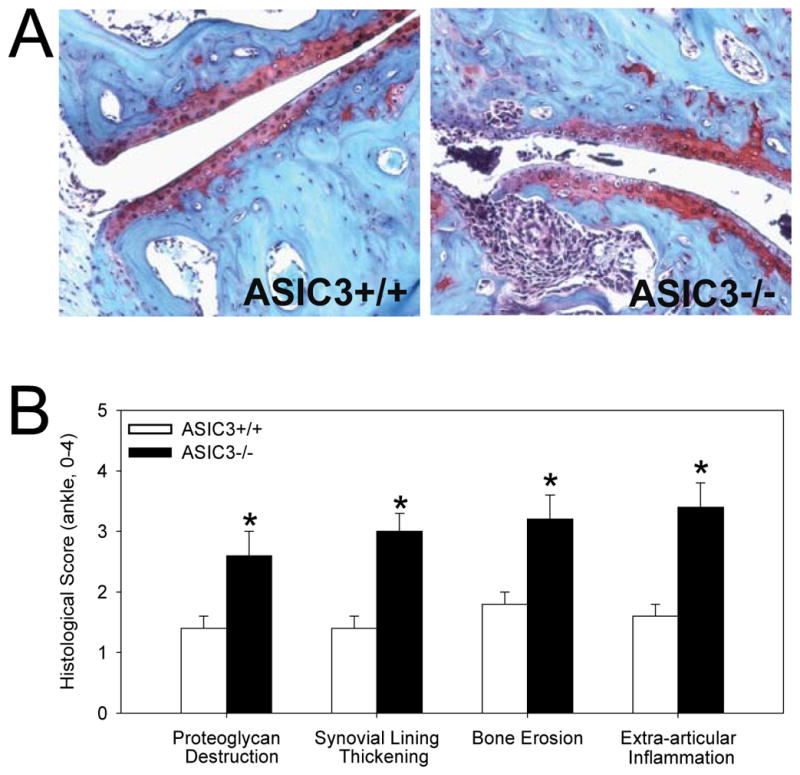
ASIC3−/− mice with collagen antibody-induced arthritis (CAIA) show enhanced histological changes when compared to ASIC3+/+ mice. A. Representative sections from the ankle joint of animals 12 days after induction of CAIA. Sections are stained with Safronin-0. B. Quantification of histological changes on a 0–4 scale where 0 is normal and 4 represents severe destruction, synovial hyperplasia, erosion, or inflammation of the ankle. Greater changes were observed in ASIC3−/− mice compared with ASIC3+/+ mice, *, p<0.01 Data are mean ± S.E.M.
To evaluate whether articular gene expression of inflammatory mediators parallels clinical synovitis, qPCR was performed on joint tissue. As shown in Figure 3, IL-6, MMP-3 and MMP-13 mRNA levels were significantly increased in ASIC3−/− mice compared with ASIC3+/+ mice with arthritis.
Figure 3.
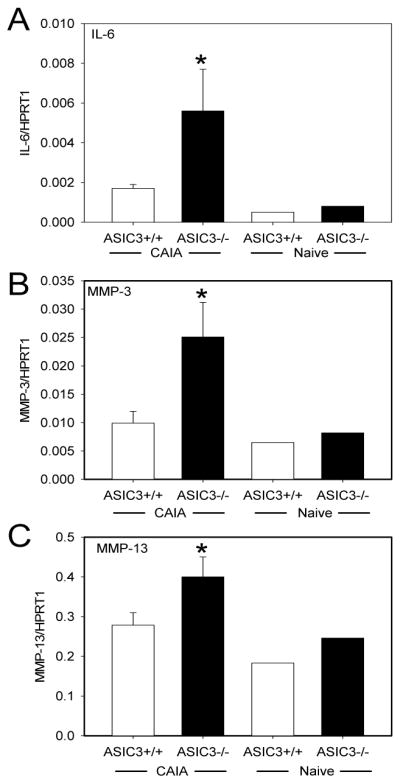
ASIC3−/− mice with collagen antibody-induced arthritis (CAIA) have higher synovial mRNA levels of IL-6, MMP-3 and MMP-13 than ASIC3+/+ mice. mRNA levels for IL-6 (A), MMP-3 (B), and MMP-13(C) were measured in ankle joint extracts normalized to the internal standard HPRT1 from ASIC3+/+ and ASIC3−/− mice with CAIA. (n=20/group). For comparison IL-6 mRNA levels are shown for ASIC3+/+ and ASIC3−/− mice without CAIA (naïve). *p<0.05. Data are mean ± S.E.M.
Enhanced cell death with decreased pH depends on ASIC3
Since prior work shows that decreases in pH cause cell death in neurons through activation of ASICs [23;31;53], we tested if decreases in pH would cause cell death in cultured FLS. ASIC3+/+ FLS treated with pH 6.0 showed no increased cell death when compared to those treated with neutral pH 7.4 (Figure 4). We then tested if cells exposed to IL-1β, a critical inflammatory mediator in arthritis, altered cell death. In ASIC3+/+ FLS, there is a significant increase in the number of dead FLS (30% ± 4.5) when exposed to IL-1β in combination with pH 6.0 when compared to ASIC3+/+ FLS exposed to pH 6.0 alone or ASIC3+/+ exposed to pH 7.4 with or without IL-1β(Figure 4). This increased cell death does not occur in ASC3−/− FLS exposed to pH 6.0 with IL-1β indicating that it is ASIC3-dependent (Figure 4).
Figure 4.

A. Representative examples of the Live/Dead assay from cultured FLS treated with pH 7.4 or pH 6.0 with or without IL-1β taken from ASIC3+/+ or ASIC3−/− mice. Green staining represents live cells, and red staining represents dead cells. Notice the increased number of red nuclei in the panel from ASIC3+/+ cells treated with pH 6.0 and IL-1β. B. The average percent of cells that were dead from the different groups. Three different cell lines were used for each condition. *, significantly different from all other conditions. +, significantly less than ASIC3+/+ treated with pH 6.0 and with IL-1β. Data are mean ± S.E.M.
Pain-like behaviors
To determine if pain-like behaviors in ASIC3−/− mice decrease in inflammatory arthritis as observed for models of musculoskeletal pain [20;43] we examined withdrawal threshold of the paw and ankle, and responsiveness of the paw to repeated mechanical stimulation in mice with CAIA. This allowed us to identify differences in primary hyperalgesia of the inflamed ankle as well as secondary hyperalgesia of the skin. Repeated stimulation of the paw with a 0.4 mN von Frey filament increased the number of withdrawals to the stimulus in ASIC3+/+ mice that peaked on day 10 after induction of CAIA. ASIC3−/− mice showed significant reductions in the responsiveness to repeated stimulation (Figure 5A). Mechanical withdrawal thresholds of the paw also significantly decreased and peaked on day 10 in ASIC3+/+ mice. ASIC3−/− mice did not show this reduction in withdrawal thresholds of the paw (Figure 5B). In contrast, withdrawal thresholds of the ankle significantly decreased and were similar in ASIC3+/+ and ASIC3−/− mice after induction of arthritis (Figure 5C).
Figure 5.

Mechanical pain behaviors are reduced in ASIC3−/− mice compared with ASIC3+/+ mice with passive CIA. A. Responsiveness of the paw to repeated application of mechanical stimulation increases after induction of passive CIA in ASIC3+/+ mice. The increased responsiveness to repeated stimuli is reduced in ASIC3−/− mice at days 6, 10 and 12. B. Withdrawal thresholds of the paw are decreased in ASIC3+/+ mice with passive CIA. These decreases in withdrawal thresholds do not occur in ASIC3−/− mice. C. Withdrawal threshold of the ankle joint are decreased in ASIC3+/+ mice. Similar decreases are observed in ASIC3−/− mice, *significantly different from ASIC3+/+ mice, p<0.05 Data are mean ± S.E.M.
To determine if there were differences in mobility between ASIC3+/+ and ASIC3−/− mice with CAIA we examined physical activity levels. In general, 7 days after induction of CAIA ASIC3+/+ mice showed significant decreases in physical activity and increased inactivity. The decreases in physical activity occurred at low speed walking and high speed walking activities, but not in grooming behavior. The decrease in physical activity resulted in a significant decrease in the total distance walked during the 60 minute time period (Figure 6). By 13 days after induction of arthritis, activity levels returned to baseline. A similar pattern of decreased physical activity and increased inactivity was observed in ASIC3−/− mice; however the changes in behaviors were less pronounced (Figure 6). Specifically, the ASIC3−/− mice spent more time in low and high speed activities and less time being inactive than the ASIC3+/+ mice. This translated into the ASIC3−/− mice walking a greater distance.
Figure 6.

ASIC3−/− mice have greater physical activity than ASIC3+/+ mice with CAIA. Low and high speed walking activity, total time active, inactive time and grooming behavior were measured in a 60 minute test period. A. Both groups of mice showed a decrease in the total time active on Day 7 when compared to baseline. There was no significant difference between groups. B. Both groups showed decreases in time spent in low speed activities on Day 7. ASIC3−/− mice spent significantly more time in low speed activities than ASIC3+/+ mice. C. Both groups showed significantly less time spent in high speed activities on Day 7. ASIC3−/− mice spent significantly more time in high speed activities than ASIC3+/+ mice. D. ASIC3+/+ and ASIC3−/− mice showed no changes in grooming behavior after induction of CAIA and were similar. E. Both ASIC3+/+ and ASIC3−/− mice showed more time being inactive on Day 7. The ASIC3−/− mice however showed less time being inactive than the ASIC3+/+ mice. F. By 7 days both groups showed a significant decrease in the total distance traveled. The ASIC3−/− mice however traveled significantly less distance than the ASIC3+/+ mice. Data are mean ± S.E.M.
DISCUSSION
This study demonstrates the contrasting role of ASIC3 in inflammation and pain responses in a chronic model of arthritis. In particular, this is the first report showing that ASIC3 deficiency paradoxically increases synovial inflammation, joint destruction and inflammatory cytokines and yet is associated with reduced pain-like behaviors. This was associated with increased expression of mediators that are primarily produced by synoviocytes, namely IL-6 and MMPs. We have previously localized ASIC3 not only to nociceptors innervating articular joints but also to FLS [20;21;26], and therefore determined whether altered synoviocyte survival in ASIC3−/− might contribute to increased inflammation. The data show for the first time that exposure of FLS to decreases in pH in combination with inflammatory mediators produces cell death and that this is dependent on ASIC3. Together, our data suggest that ASIC3 in sensory nerves activates nociceptors to produce pain while in synoviocytes ASIC3 prevents excessive inflammation by reducing synoviocyte proliferation.
The observation that ASIC3 deficiency increases synovial inflammation suggests that ASIC3 plays a protective role by suppressing synovitis. The arthritis data presented in the current study contrast with other murine inflammation models. In ASIC3−/− mice with cutaneous, subcutaneous or muscle inflammation there is similar neutrophilic infiltration [43], similar or less severe tissue swelling [41;52], and histological evidence of reduced granuloma formation and vasculitis [52] when compared to ASIC3+/+ mice. Enhanced inflammation in ASIC3 deficient mice with CAIA might be unique to arthritis based on the expression of ASIC3 in synoviocytes.
We previously showed that decreased pH, as occurs in inflammatory arthritis [13;14], activates signaling in FLS [26]. Cultured synoviocytes exposed to an acid environment have increased intracellular calcium and release of hyaluronan that are dependent on ASIC3 [26]. In FLS, IL-1β and TNF increase calcium and activate calcineurin, and stimulate release if IL-6 and MMP [54]. Further, release of inflammatory cytokines from mononuclear cells is prevented by calcium channel blockers [32]. Alternatively, increases in intracellular calcium can activate second messenger systems that reduce activity of pathways involved in cytokine production. For example, pH 6.0 decreased LPS-induced TNF expression [15] that subsequently reduced the phosphorylation of IkB and nuclear translocation of the transcription factor NF-kB [15]. Increased intracellular calcium can enhance activity of protein phosphatase 2a (PP2a) and decrease extracellular signal regulated kinase (ERK)[29]. Our data showing increases in expression of IL-6 and MMPs in joint tissue from ASIC3−/− mice could thus result the loss of a pH-induced decrease in gene expression that normally occurs through pH-activation of ASIC3.
Alternatively, increases in intracellular calcium with acidic pH could enhance synoviocyte cell death to limit synoviocyte proliferation. Indeed our data show that exposure of FLS to decreases in pH in combination with IL-1β results in cell death, and this cell death does not occur in ASIC3−/− FLS. A recent study shows that decreases in pH (pH 5.5) resulted in cell death of cultured synoviocytes through activation of TRPV1 [19]. In neurons activation of ASICs with acidic pH can produce cell death through increased intracellular calcium in central nervous system models of disease [16;23;31;39;51;53]. Thus, we suggest that the decreases in inflammatory mediators observed in ASIC3+/+ mice compared to ASIC3−/− mice are secondary to the reduction in synoviocyte proliferation.
The current study shows decreases in pain behaviors and increases in physical activity in ASIC3−/− mice with CAIA compared to ASIC3+/+ mice. It is, therefore, possible that the increased inflammation in ASIC3−/− mice is a result of the increased physical activity levels and decreases in pain in these animals. However we believe this is unlikely as the activity increases although significant was relatively small (see Figure 6) in comparison to the differences in inflammation (see Figure 1), and decreases in pH activate and kill FLS through ASIC3 (see Figure 4).
ASIC3 plays a critical role in the development and maintenance of musculoskeletal pain, particularly that of inflammatory origin [7;20;20;25;41;43;52]. The current study supports these data and shows a reduction in mechanical hyperalgesia of the paw despite greater intensity of synovitis. However, we also show that ASIC3−/− mice still develop enhanced mechanical hyperalgesia of the ankle similar to ASIC3+/+ mice. These data are consistent with prior work showing ASIC3−/− mice lose injury-induced hyperalgesia outside the site of insult in the skin but still show hyperalgesia at the site of insult [20;43;49]. Hyperalgesia at the site of insult, termed primary hyperalgesia, reflects enhanced nociceptor activity while that outside the site of insult, termed secondary hyperalgesia, is thought to reflect enhanced central neuron activity [6]. The secondary hyperalgesia in ASIC3−/− mice with muscle inflammation is restored by re-expression of ASIC3 into neurons innervating the inflamed muscle and in ASIC3+/+ mice is reduced by downregulation of ASIC3 in neurons innervating the inflamed muscle [43;49]. Further, the enhanced central neuron activity does not occur in ASIC3−/− mice suggesting ASIC3 is critical for induction of enhanced central neuron sensitivity [42] and supporting its role in development of secondary hyperalgesia. Co-administration of the ASIC3 antagonist, APETx2 with CFA into the paw similarly prevents the development of cutaneous hyperalgesia 4h later in rats [7]. In parallel, there is also an upregulation of the expression of ASICs in DRGs innervating inflamed musculoskeletal tissue [21;48;50]. We have further shown that the increase is associated with enhanced ASIC activity in the DRG innervating the inflamed tissue and that blockade of ASICs after onset of inflammation reduces the pain-behaviors [12;50]. The above data further support that localization of ASIC3 to nociceptors innervating the inflamed joint is required for development of hyperalgesia, as well as the accompanying central neuron sensitization.
We also showed that ASIC3 and CAIA affect joint function by measuring physical activity. Importantly, people with inflammatory joint pain show decreased joint function that is manifested as diminished physical activity [28]. In general people with chronic pain conditions such as osteoarthritis and fibromyalgia show a decrease not only in overall activity, but the greatest decreases in activity occur at more vigorous levels of activity [9;10;47]. Indeed in the current study we show that ASIC3+/+ mice with CAIA have decreases in physical activity with the greatest changes occurring at the highest levels of activity; these decreases in physical activity are attenuated in ASIC3−/− mice. This increase in physical activity or function in ASIC3−/− surprisingly occurs despite greater increases in inflammation and joint destruction.
Current pharmacological agents against ASICs have been designed to antagonize the channel to ameliorate pain. Indeed the current data support the use of ASIC3 antagonists to modulate pain behaviors as does a large body of literature [3;7;8;20;25;35–37;41;44;49;50]. However, the role of ASICs in synovitis has previously not been investigated and our data suggests that the use of pharmacological blockers of ASIC3 might enhance the disease severity. An alternative strategy might be to activate ASIC3 pharmacologically in non-neuronal tissues to reduce the inflammatory process while using other strategies to modulate pain.
Acknowledgments
This study was supported by the National Institutes of Health grants (AR053509) and (AR 053509S1)
The authors would like to acknowledge the funding support they have received from the National Institutes of Health, AR053509 and AR053509S1. The authors thank Ms. Ann Lawler for editorial assistance.
Footnotes
Financial disclosures: The authors have nothing to disclose.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for content.
Study design: Sluka, Firestein, Boyle, Walder, Kolker
Acquisition of data: Boyle, Edgar, Rasmussen, O’Donnell
Analysis and interpretation of data: Sluka, Firestein, Boyle, Kolker, O’Donnell
Statistical analysis: Sluka, Boyle, Rasmussen
Project initiation, organization and funding: Sluka, Firestein
Reference List
- 1.Boyle DL, Rosengren S, Bugbee W, Kavanaugh A, Firestein GS. Quantitative biomarker analysis of synovial gene expression by real-time PCR. Arthritis Res Ther. 2003;5:R352–R360. doi: 10.1186/ar1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaplan SR, Pogrel JW, Yaksh TL. Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. J Pharmacol Exp Ther. 1995;269:1117–1123. [PubMed] [Google Scholar]
- 3.Chen CC, Zimmer A, Sun WH, Hall J, Brownstein MJ, Zimmer A. A role for ASIC3 in the modulation of high-intensity pain stimuli. Proc Natl Acad Sci U S A. 2002;99:8992–8997. doi: 10.1073/pnas.122245999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corr M, Crain B. The role of FcgammaR signaling in the K/B x N serum transfer model of arthritis. J Immunol. 2002;169:6604–6609. doi: 10.4049/jimmunol.169.11.6604. [DOI] [PubMed] [Google Scholar]
- 5.Corr M, Firestein GS. Innate immunity as a hired gun: but is it rheumatoid arthritis? J Exp Med. 2002;195:F33–F35. doi: 10.1084/jem.20020389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeSantana JM, Sluka KA. Central mechanisms in the maintenance of chronic widespread noninflammatory muscle pain. Curr Pain Headache Rep. 2008;12:338–343. doi: 10.1007/s11916-008-0057-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deval E, Noel J, Lay N, Alloui A, Diochot S, Friend V, Jodar M, Lazdunski M, Lingueglia E. ASIC3, a sensor of acidic and primary inflammatory pain. EMBO J. 2008;27:3047–3055. doi: 10.1038/emboj.2008.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diochot S, Baron A, Rash LD, Deval E, Escoubas P, Scarzello S, Salinas M, Lazdunski M. A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. EMBO Journal. 2004;23:1516–1525. doi: 10.1038/sj.emboj.7600177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunlop DD, Song J, Semanik PA, Chang RW, Sharma L, Bathon JM, Eaton CB, Hochberg MC, Jackson RD, Kwoh CK, Mysiw WJ, Nevitt MC, Hootman JM. Objective physical activity measurement in the osteoarthritis initiative: Are guidelines being met? Arthritis Rheum. 2011;63:3372–3382. doi: 10.1002/art.30562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ellingson LD, Shields MR, Stegner AJ, Cook DB. Physical activity, sustained sedentary behavior, and pain modulation in women with fibromyalgia. J Pain. 2012;13:195–206. doi: 10.1016/j.jpain.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 12.Gautam M, Benson CJ, Sluka KA. Increased response of muscle sensory neurons to decreases in pH after muscle inflammation. N S. 2010;170:893–900. doi: 10.1016/j.neuroscience.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldie I, Nachemson A. Synovial pH in rheumatoid knee-joints. I. The effect of synovectomy. Acta Orthop Scand. 1969;40:634–641. doi: 10.3109/17453676908989529. [DOI] [PubMed] [Google Scholar]
- 14.Goldie I, Nachemson A. Synovial pH in rheumatoid knee joints. II. The effect of local corticosteroid treatment. Acta Orthop Scand. 1970;41:354–362. doi: 10.3109/17453677008991521. [DOI] [PubMed] [Google Scholar]
- 15.Grabowski J, Vazquez DE, Costantini T, Cauvi DM, Charles W, Bickler S, Talamini MA, Vega VL, Coimbra R, De MA. Tumor necrosis factor expression is ameliorated after exposure to an acidic environment. J Surg Res. 2012;173:127–134. doi: 10.1016/j.jss.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gu L, Yang Y, Sun Y, Zheng X. Puerarin inhibits acid-sensing ion channels and protects against neuron death induced by acidosis. Planta Med. 2010;76:583–588. doi: 10.1055/s-0029-1240583. [DOI] [PubMed] [Google Scholar]
- 17.Han Z, Chang L, Yamanishi Y, Karin M, Firestein GS. Joint damage and inflammation in c-Jun N-terminal kinase 2 knockout mice with passive murine collagen-induced arthritis. Arthritis Rheum. 2002;46:818–823. doi: 10.1002/art.10104. [DOI] [PubMed] [Google Scholar]
- 18.Houssiau FA, Devogelaer JP, Van DJ, de Deuxchaisnes CN, Van SJ. Interleukin-6 in synovial fluid and serum of patients with rheumatoid arthritis and other inflammatory arthritides. Arthritis Rheum. 1988;31:784–788. doi: 10.1002/art.1780310614. [DOI] [PubMed] [Google Scholar]
- 19.Hu F, Sun WW, Zhao XT, Cui ZJ, Yang WX. TRPV1 mediates cell death in rat synovial fibroblasts through calcium entry-dependent ROS production and mitochondrial depolarization. Biochem Biophys Res Commun. 2008;369:989–993. doi: 10.1016/j.bbrc.2008.02.155. [DOI] [PubMed] [Google Scholar]
- 20.Ikeuchi M, Kolker SJ, Burnes LA, Walder RY, Sluka KA. Role of ASIC3 in the primary and secondary hyperalgesia produced by joint inflammation in mice. Pain. 2008;137:662–669. doi: 10.1016/j.pain.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ikeuchi M, Kolker SJ, Sluka KA. Acid-sensing ion channel 3 expression in mouse knee joint afferents and effects of carrageenan-induced arthritis. J Pain. 2009;10:336–342. doi: 10.1016/j.jpain.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inoue T, Boyle DL, Corr M, Hammaker D, Davis RJ, Flavell RA, Firestein GS. Mitogen-activated protein kinase kinase 3 is a pivotal pathway regulating p38 activation in inflammatory arthritis. Proc Natl Acad Sci U S A. 2006;103:5484–5489. doi: 10.1073/pnas.0509188103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jetti SK, Swain SM, Majumder S, Chatterjee S, Poornima V, Bera AK. Evaluation of the role of nitric oxide in acid sensing ion channel mediated cell death. Nitric Oxide. 2010;22:213–219. doi: 10.1016/j.niox.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 24.Kaneko S, Satoh T, Chiba J, Ju C, Inoue K, Kagawa J. Interleukin-6 and interleukin-8 levels in serum and synovial fluid of patients with osteoarthritis. Cytokines CellMol Ther. 2000;6:71–79. doi: 10.1080/13684730050515796. [DOI] [PubMed] [Google Scholar]
- 25.Karczewski J, Spencer RH, Garsky VM, Liang A, Leitl MD, Cato MJ, Cook SP, Kane S, Urban MO. Reversal of acid-induced and inflammatory pain by the selective ASIC3 inhibitor, APETx2. Br J Pharmacol. 2010;161:950–960. doi: 10.1111/j.1476-5381.2010.00918.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kolker SJ, Walder RY, Usachev Y, Hillman J, Boyle DL, Firestein GS, Sluka KA. ASIC3 expressed in Type B synoviocytes and chondrocytes modulates hyaluronan expression and release. Ann Rheum Dis. 2010;69:903–909. doi: 10.1136/ard.2009.117168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–822. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 28.Lee J, Dunlop D, Ehrlich-Jones L, Semanik P, Song J, Manheim L, Chang RW. Public health impact of risk factors for physical inactivity in adults with rheumatoid arthritis. Arthritis Care Res (Hoboken ) 2012;64:488–493. doi: 10.1002/acr.21582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu WH, Chang LS. Caffeine induces matrix metalloproteinase-2 (MMP-2) and MMP-9 down-regulation in human leukemia U937 cells via Ca2+/ROS-mediated suppression of ERK/c-fos pathway and activation of p38 MAPK/c-jun pathway. J Cell Physiol. 2010;224:775–785. doi: 10.1002/jcp.22180. [DOI] [PubMed] [Google Scholar]
- 30.Malmberg AB, Yaksh TL. Voltage-sensitive calcium channels in spinal nociceptive processing: blockade of N-and P-type channels inhibits formalin-ionduced nociception. J Neurosci. 1994;14:4882–4890. doi: 10.1523/JNEUROSCI.14-08-04882.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mari Y, Katnik C, Cuevas J. ASIC1a channels are activated by endogenous protons during ischemia and contribute to synergistic potentiation of intracellular Ca(2+) overload during ischemia and acidosis. Cell Calcium. 2010;48:70–82. doi: 10.1016/j.ceca.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 32.Matsumori A, Nishio R, Nose Y. Calcium channel blockers differentially modulate cytokine production by peripheral blood mononuclear cells. Circ J. 2010;74:567–571. doi: 10.1253/circj.cj-09-0467. [DOI] [PubMed] [Google Scholar]
- 33.Matsumoto I, Staub A, Benoist C, Mathis D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 1999;286:1732–1735. doi: 10.1126/science.286.5445.1732. [DOI] [PubMed] [Google Scholar]
- 34.McNearney T, Baethge BA, Cao S, Alam R, Lisse JR, Westlund KN. Excitatory amino acids, TNF-alpha, and chemokine levels in synovial fluids of patients with active arthropathies. Clinical and Experimental Immunology. 2004;137:621–627. doi: 10.1111/j.1365-2249.2004.02563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naves LA, McCleskey EW. An acid-sensing ion channel that detects ischemic pain. Braz J Med Biol Res. 2005;38:1561–1569. doi: 10.1590/s0100-879x2005001100001. [DOI] [PubMed] [Google Scholar]
- 36.Ohtori S, Inoue G, Koshi T, Ito T, Doya H, Saito T, Moriya H, Takahashi K. Up-regulation of acid-sensing ion channel 3 in dorsal root ganglion neurons following application of nucleus pulposus on nerve root in rats. Spine. 2006;31:2048–2052. doi: 10.1097/01.brs.0000231756.56230.13. [DOI] [PubMed] [Google Scholar]
- 37.Price MP, McIlwrath SL, Xie J, Cheng C, Qiao J, Tarr DE, Sluka KA, Brennan TJ, Lewin GR, Welsh MJ. The DRASIC Cation Channel Contributes to the Detection of Cutaneous Touch and Acid Stimuli in Mice. Neuron. 2001;32:1071–1083. doi: 10.1016/s0896-6273(01)00547-5. [DOI] [PubMed] [Google Scholar]
- 38.Rosengren S, Boyle DL, Firestein GS. Acquisition, culture, and phenotyping of synovial fibroblasts. Methods Mol Med. 2007;135:365–375. doi: 10.1007/978-1-59745-401-8_24. [DOI] [PubMed] [Google Scholar]
- 39.Sherwood T, Franke R, Conneely S, Joyner J, Arumugan P, Askwith C. Identification of protein domains that control proton and calcium sensitivity of ASIC1a. J Biol Chem. 2009;284:27899–27907. doi: 10.1074/jbc.M109.029009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skyba DA, Radhakrishnan R, Sluka KA. Characterization of a method for measuring primary hyperalgesia of deep somatic tissue. Journal of Pain. 2005;6:41–47. doi: 10.1016/j.jpain.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 41.Sluka KA, Price MP, Breese NM, Stucky CL, Wemmie JA, Welsh MJ. Chronic hyperalgesia induced by repeated acid injections in muscle is abolished by the loss of ASIC3, but not ASIC1. Pain. 2003;106:229–239. doi: 10.1016/S0304-3959(03)00269-0. [DOI] [PubMed] [Google Scholar]
- 42.Sluka KA, Price MP, Wemmie JA, Welsh MJ. ASIC3, but not ASIC1, channels are involved in the development of chronic muscle pain. In: Dostrovsky JO, Carr DB, Koltzenburg M, editors. Proceedings of the 10th World Congress on Pain. Seattle: IASP Press; 2003. pp. 71–79. [Google Scholar]
- 43.Sluka KA, Radhakrishnan R, Benson CJ, Eshcol JO, Price MP, Babinski K, Audette KM, Yeomans DC, Wilson SP. ASIC3 in muscle mediates mechanical, but not heat, hyperalgesia associated with muscle inflammation. Pain. 2007;129:102–112. doi: 10.1016/j.pain.2006.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sluka KA, Winter OC, Wemmie JA. Acid-sensing ion channels: A new target for pain and CNS diseases. Curr Opin Drug Discov Devel. 2009;12:693–704. [PMC free article] [PubMed] [Google Scholar]
- 45.Svensson CI, Inoue T, Hammaker D, Fukushima A, Papa S, Franzoso G, Schett G, Corr M, Boyle DL, Firestein GS. Gadd45beta deficiency in rheumatoid arthritis: enhanced synovitis through JNK signaling. Arthritis Rheum. 2009;60:3229–3240. doi: 10.1002/art.24887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sweeney SE, Firestein GS. Rheumatoid arthritis: regulation of synovial inflammation. Int J Biochem Cell Biol. 2004;36:372–378. doi: 10.1016/s1357-2725(03)00259-0. [DOI] [PubMed] [Google Scholar]
- 47.Tonelli SM, Rakel BA, Cooper NA, Angstom WL, Sluka KA. Women with knee osteoarthritis have more pain and poorer function than men, but similar physical activity prior to total knee replacement. Biol Sex Differ. 2011;2:12. doi: 10.1186/2042-6410-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Voilley N, de Weille J, Mamet J, Lazdunski M. Nonsteroid anti-inflammatory drugs inhibit both the activity and the inflammation-induced expression of acid-sensing ion channels in nociceptors. J Neurosci. 2001;21:8026–8033. doi: 10.1523/JNEUROSCI.21-20-08026.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walder RY, Gautam M, Wilson SP, Benson CJ, Sluka KA. Selective Targeting of ASIC3 using miRNAs inhibits primary and secondary hyperalgesia following muscle inflammation. Pain. 2011 doi: 10.1016/j.pain.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walder RY, Rasmussen LA, Rainier JD, Light AR, Wemmie JA, Sluka KA. ASIC1 and ASIC3 Play Different Roles in the Development of Hyperalgesia After Inflammatory Muscle Injury. J Pain. 2010;11:218. doi: 10.1016/j.jpain.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weng XC, Zheng JQ, Jin QE, Ma XY. Inhibition of acid-induced apoptosis by targeting ASIC1a mRNA with short hairpin RNA. Acta Pharmacol Sin. 2007;28:1621–1627. doi: 10.1111/j.1745-7254.2007.00627.x. [DOI] [PubMed] [Google Scholar]
- 52.Yen YT, Tu PH, Chen CJ, Lin YW, Hsieh ST, Chen CC. Role of acid-sensing ion channel 3 in sub- acute-phase inflammation. Mol Pain. 2009;5:1. doi: 10.1186/1744-8069-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yermolaieva O, Leonard AS, Schnizler MK, Abboud FM, Welsh MJ. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci U S A. 2004;101:6752–6757. doi: 10.1073/pnas.0308636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoo SA, Park BH, Park GS, Koh HS, Lee MS, Ryu SH, Miyazawa K, Park SH, Cho CS, Kim WU. Calcineurin is expressed and plays a critical role in inflammatory arthritis. J Immunol. 2006;177:2681–2690. doi: 10.4049/jimmunol.177.4.2681. [DOI] [PubMed] [Google Scholar]