

Abstract
Survival after acute myocardial infarction is decreased in elderly patients. The enhanced rates of apoptosis in the aging heart exacerbate myocardial ischemia/reperfusion (MI/R) injury. We have recently demonstrated that the X-linked inhibitor of apoptosis protein (XIAP), the most potent endogenous inhibitor of apoptosis, was decreased in aging rats’ hearts. XIAP was balanced by two mitochondria proteins, Omi/HtrA2 and Smac/DIABLO. However, the implicative role of XIAP, Omi/HtrA2, and Smac/DIABLO to aging-related MI/R injury has not been previously investigated. In our study, male aging rats (20–24 months) or young adult rats (4–6 months) were subjected to 30 min of myocardial ischemia followed by reperfusion. MI/R-induced cardiac injury was enhanced in aging rats, as evidenced by aggravated cardiac dysfunction, enlarged infarct size, and increased myocardial apoptosis (TUNEL and caspase-3 activity). Then, the XIAP, Omi/HtrA2, and Smac/DIABLO protein and mRNA expression was detected. XIAP protein and mRNA expression was decreased in both aging hearts and aging hearts subjected to MI/R. Meanwhile, myocardial XIAP protein expression was correlated to cardiac function after MI/R. However, Omi/HtrA2, but not Smac/DIABLO, expression was increased in aging hearts. Moreover, the translocation of Omi/HtrA2 from mitochondria to cytosol was increased in both aging hearts and aging hearts subjected to MI/R. Treatment with ucf-101 (a novel and specific Omi/HtrA2 inhibitor) attenuated XIAP degradation and caspase-3 activity and exerted cardioprotective effects. Taken together, these results demonstrated that increased expression and leakage of Omi/HtrA2 enhanced MI/R injury in aging hearts via degrading XIAP and promoting myocardial apoptosis.
Keywords: Aging, Myocardial ischemia/reperfusion, Apoptosis, XIAP, Omi/HtrA2
Introduction
Age is a major independent risk factor for ischemic heart disease (Lakatta and Levy 2003). The progressive structural changes and functional declines make the aging heart more susceptible to ischemia/reperfusion injury and contribute to increased cardiovascular mortality and morbidity in the elderly (Boengler et al. 2009). Considerable evidence from animal studies and clinical observations indicates that aging hearts suffer from greater infarct expansion, more cell death, and poorer clinical outcome (White et al. 1996; Gould et al. 2002; Ananthakrishnan et al. 2011). However, the mechanisms responsible for this age-related pathologic change remain elusive.
Accumulating evidence indicates that myocardial apoptosis, a gene-regulated programmed cell death, markedly increases with aging (Kajstura et al. 1996; Kakarla et al. 2010; Boyle et al. 2011). Recently, data demonstrated the enhanced rates of apoptosis in the aging heart exacerbated myocardial ischemia/reperfusion (MI/R) injury in not only animals but also humans (Liu et al. 2011). However, the molecular mechanisms and signaling transduction pathways responsible for increased susceptibility of cardiomyocytes to apoptosis with aging remain largely unidentified.
Apoptosis is a programmed cell death controlled by the death receptor, the mitochondrial pathways through caspase activation, and also endogenous regulators of apoptotic cell death. X-linked inhibitor of apoptosis protein (XIAP) is the best characterized and most potent endogenous regulators of apoptotic cell death in mammals (Holcik et al. 2001). Many results revealed that it was a major inhibitor of apoptosis in cardiomyocytes (Potts et al. 2005; Souktani et al. 2009). Our primary results found decreased cardiac expression of XIAP in aging rats, and Li et al. found a downregulated XIAP expression in aging transgenic mice hearts (Li and Ren 2007). These results suggested that XIAP was involved in aging-related cardiomyocyte apoptosis. However, why XIAP was reduced in the heart with age and whether this alteration may contribute to increased cardiomyocyte apoptosis in the aging heart need to be investigated further.
Inhibition of apoptosis by XIAP is balanced by mitochondrial proteins, acting as XIAP antagonists, released during the mitochondrial membrane permeabilization. Mitochondrial proteins are considered to be potential therapeutic targets of many apoptosis-related diseases (Soustiel and Larisch 2010). Two main mitochondria proteins have been identified in mammalian cells, Smac/DIABLO and Omi/HtrA2 (Wilkinson et al. 2004). Both of them are located in the mitochondria and translocated into the cytosol following apoptotic insult, where they interact with XIAP and promote caspase-dependent apoptosis. Our group firstly provides a direct evidence that ischemia/reperfusion results in Omi/HtrA2 translocation from the mitochondria to the cytosol, where it promotes cardiomyocyte apoptosis through degradation of XIAP and hence activation of caspases leading to apoptosis (Liu et al. 2005). However, there is very little information available regarding age-related changes in the expression of Omi/HtrA2 and Smac/DIABLO. Whether both were altered with age and promote cardiomyocyte apoptosis by degradation of XIAP and contribute to enhanced MI/R injury in aging has not been previously investigated. Ucf-101 is a highly selective inhibitor for the serine protease activity of Omi/HtrA2. Our and others’ research showed that ucf-101 reduced infarct size and cardiomyocyte apoptosis and attenuated cardiac dysfunction through XIAP degradation in the young adult heart subjected to MI/R (Liu et al. 2005; Bhuiyan and Fukunaga 2008; Li et al. 2009). However, whether ucf-101 exerted cardioprotection and whether Omi/HtrA2 promoted apoptotic cell death in aging hearts subjected to MI/R by a similar mechanism remain unclear.
Therefore, the aims of the present study were (1) to identify the possible mechanism responsible for aging-induced XIAP alteration, Omi/HtrA2, or Smac/DIABLO; (2) to determine whether Omi/HtrA2 or Smac/DIABLO was involved in enhanced postischemia injury in the aging heart; and (3) to investigate whether treatment with ucf-101 (a novel and specific Omi/HtrA2 inhibitor) inhibits XIAP degradation, attenuates caspase-3 activity, and ameliorates cardiac function in MI/R injury of the aging heart.
Materials and methods
Animals
The investigations conformed to the “Guiding Principles in the Use and Care of Animals” published by the National Institutes of Health (NIH publication no. 85-23, revised 1996), and approved by the Institutional Animal Care and Use Committee of Capital Medical University. The 6- to 8-month-old (young rats = Y) and 20- to 24-month-old (aging rats = A) male Sprague-Dawley rats were obtained.
Experimental protocol
Male Sprague-Dawley rats were anesthetized with sodium pentobarbital. Myocardial ischemia was produced by exteriorizing the heart via a left thoracic incision and occluding the left coronary artery (LCA) with a 6-0 silk slipknot. After 30 min of ischemia, the slipknot was released, and the myocardium was reperfused for 1–24 h. The sham-operated control rats (sham) underwent the same surgical procedures except that the LCA was not occluded. Ten minutes before reperfusion, animals were randomized to receive vehicle (DMSO) or ucf-101 (1.5 mol/kg; estimated plasma concentrations, 20 mol/L) via intraperitoneal injection. Ucf-101 has been shown to inhibit 50 to 90 % of Omi/HtrA2 activity but has no effect on other proteases at these concentrations (Cilenti et al. 2003).
Male Sprague-Dawley rats were randomized to receive either sham or MI/R and were divided into the following groups: (1) young/aging (n = 10 each); (2) young/aging sham (n = 10 each); (3) young/aging R0h, R1h, R2h, R3h, R12h, and R24h (n = 6 each, animals were subjected to 30 min of coronary occlusion followed by 0, 1, 2, 3, 12, and 24 h of reperfusion); and (4) vehicle/ucf-101 (treatment with DMSO/ucf-101 via intraperitoneal injection).
Determination of ventricular function
Left ventricular (LV) function was continuously monitored via a Millar Mikro-Tip catheter pressure transducer inserted into the LV via the left carotid artery (Powerlab Hardware; AD Instruments, Charlotte, USA). Left ventricular end-diastolic pressure (LVEDP), left ventricular systolic pressure (LVSP), and maximal rate of rise/decrease of left ventricular pressure (±dp/dtmax) were derived by computer algorithms.
Determination of Myocardial infarct size
At the end of the 24-h reperfusion period, the ligature around the coronary artery was reoccluded through the previous ligation. The I/R area (area at risk (AAR)) was identified by negative Evans blue staining, and the infarct area was identified by negative triphenyltetrazolium chloride (TTC) staining. The Evans blue-stained area (area not at risk), TTC-stained area (ischemic but viable tissue), and TTC-negative staining area (infarcted myocardium) were digitally measured. Myocardial infarct size was expressed as a percentage of infarct area divided by total AAR.
Preparation of mitochondria and cytosolic extracts
Mitochondrial and cytosolic fractions were prepared as previously described (Liu et al. 2005). In brief, 25 mg of myocardial tissue was minced on ice, resuspended in 500 ml of MSH buffer (210 mM mannitol/70 mM sucrose/5 mM HEPES, pH 7.5) supplemented with 1 mM EDTA, and homogenized with a glass Dounce homogenizer and Teflon. Cytosolic and mitochondrial fractions were separated by differential centrifugation (5 min at 1,000 × g, 30 min at 17,530 × g). The mitochondrial pellet was resuspended in MSH buffer, sonicated with Dismembrator for 20 s on ice, and centrifuged at 17,530 × g for 30 min at 4 °C. The resulting supernatant containing mitochondrial extract from this last centrifuge or cytosolic extract from the first centrifugation (5 min at 1,000 × g) was separately mixed with Laemmli’s loading buffer.
Determination of myocardial apoptotic death and caspase-3 protease activity
An in situ apoptosis detection kit (Roche, Switzerland) was used to examine myocardial apoptosis as we reported previously (Zhao et al. 2010). Briefly, the transmural myocardial tissue blocks were fixed using 4 % paraformaldehyde in PBS and embedded in paraffin. The TUNEL staining was then performed on the paraffin slides based on the manufacturer’s protocol. Apoptosis index was assessed by measuring the percentage of apoptotic nuclei from total nuclei in each slide.
The substrate Ac-DEVD-pNA was used to determine caspase-3 activity according to the manufacturer’s instructions (BIOMOL, PA, USA), and the same is true for the substrates Ac-IETD-pNA for caspase-8 and Ac-LEHD-pNA for caspase-9. Briefly, transmural tissue blocks were homogenized in ice-cold lysis buffer and centrifuged at 12,000 rpm for 10 min at 4 °C; 50 μl of supernatant was then incubated with buffer containing 10 mM dithiothreitol and 5 μl Ac-DEVD/IETD/LEHD-pNA (the final concentration was 200 μM) at 37 °C for 1.5 h. Activity of caspase-3, caspase-8, and caspase-9 was determined using a spectrophotometer at 405 nm (Molecular Devices, Sunnyvale, CA), and the results were expressed as -fold of the sham group.
Detection of XIAP, Omi/HtrA2, and Smac/DIABLO proteins by Western blot
The area at risk myocardium was saved for detecting XIAP, Omi/HtrA2, and Smac/DIABLO proteins. Tissue block (50 mg) was lysed and homogenized. The protein concentration in the supernatant was detected using BCA kit; 30 μg of protein was loaded on a 10 % sodium dodecyl sulfate–polyacrylamide gel, electrophoresed (80 V for 30 min followed by 120 V for 90 min), transferred onto a nitrocellulose membrane (100 V, 350 mA for 15–35 min), blocked with 5 % bovine serum albumin for 2 h at room temperature, and then incubated using the primary antibody at 4 °C overnight. The membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG at room temperature for 1 h. The primary antibodies used were rabbit anti-HtrA2/Omi (cell signaling, #2176), rabbit anti-XIAP (cell signaling, #2042), rabbit anti-Smac/DLABLO (cell signaling, #2954), rabbit anti-GAPDH (cell signaling, #2118), and rabbit anti-CoxIV (cell signaling, #4844). Specific antibody binding was detected using electrochemiluminescence. The density of the scanned protein bands was measured by image analysis software, and the results were presented as percentage change of the loading control.
Detection of XIAP, Omi/HtrA2, and Smac/DIABLO mRNAs by real-time PCR
Analysis of gene expression was studied using real-time quantitative PCR with SYBR Green (Sigma) detection in the Mx3005 real-time PCR system (Stratagene, CA). Total RNA was extracted from the area at risk myocardium with Trizol reagent (Invitrogen, USA); 3 μg of total RNA was reversely transcribed into cDNA (TaKaRa Perfect Real Time #DRR063S). The thermal profile for SYBR Green real-time PCR was 94 °C for 2 min, followed by 55 cycles of 94 °C for 15 s, 60 °C for 1 min, and 72 °C for 1 min. The primer sequences were as follows: XIAP (forward primer 5′-GAG TTA GCT AGT GCT GGA CTC TAC TAC A-3′ and reverse primer 5′-CGT AAT CCA CTT CCA CTA TTT CAC TTC-3′ (GenBank accession number AF183429), Omi/HtrA2 (forward primer 5′-CCT TGC TTT TCT AGT CTA GGT TCT GA-3′ and reverse primer 5′-CAA GAA ACA CCA CAC AAG ACA GAT GA-3′), and Smac/DIABLO (forward primer 5′-TGT GAC GAT TGG CTT TGG AGT AAC-3′ and reverse primer 5′-TTC AAT CAA CGC ATA TGT GGT CTG-3′). Samples were normalized against β-actin expression (forward primer 5′-GGC TAC AGC TTC ACC ACC AC-3′ and reverse primer 5′-TCA GGA GGA GCA ATG ATC TTG-3′ GenBankTM accession number NM_031144) to ensure equal loading. The specificity of the amplified product was monitored by its melting curve. The results, expressed as the -fold difference in the number of target gene (XIAP, Omi/HtrA2, or Smac/DIABLO) copies relative to the number of 18 s gene copies, were determined by the relative quantitative 2−ΔΔCt method (Yoshida et al. 2003), ΔΔCt = ΔCt (target gene) − ΔCt (β-actin) and ΔCt (target gene) = Ct (experimental target gene) − Ct (control target gene) and ΔCt (β-actin) = Ct (experimental β-actin) − Ct (control β-actin).
Statistical analysis
All values in the text and figures are presented as mean ± SEM. All data (except Western blot density) were subjected to ANOVA followed by the Bonferroni correction as a post hoc test. Western blot densities were analyzed with the Kruskal–Wallis test followed by Dunn’s post hoc test. Probabilities of 0.05 or less were considered statistically significant.
Results
Two hundred and six rats were instrumented to obtain one hundred and ninety-two successful experiments. Five aging rats were excluded due to intractable ventricular arrhythmias. Two aging rats were excluded due to tumors, and two aging rats were excluded due to bleeding. The ages, body weights, and areas at risk were similar in the aging groups.
Aging enhanced the myocardial ischemia/reperfusion injury
Hemodynamics
Utilizing a Millar Mikro-Tip catheter and pressure transducer, direct cardiac function index, LVSP, LVEDP, and ± dp/dtmax values were obtained. To fully realize the characters of myocardial ischemia/reperfusion injury in aging hearts, time-dependent alteration of hemodynamics among the experimental groups were detected. As shown in Fig. 1, aging rats within 24 h after being subjected to MI/R showed significantly decreased LVSP (Fig. 1a), increased LVEDP (Fig. 1b), and decreased ± dp/dtmax (Fig. 1c, d) in comparison with the younger rats.
Fig. 1.
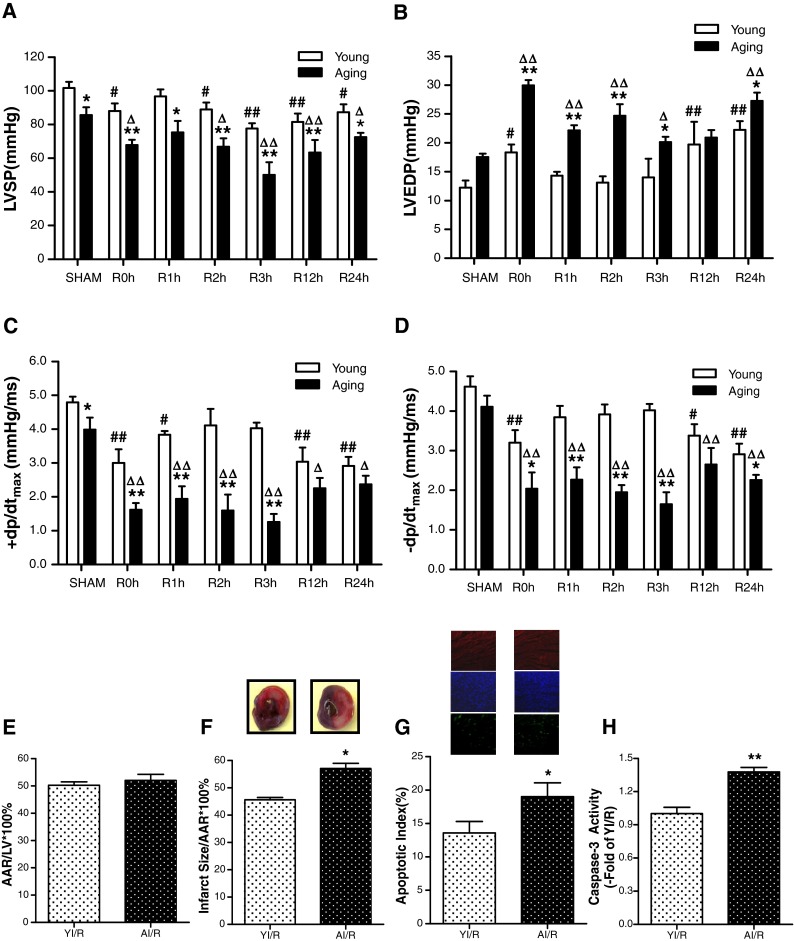
Aging enhanced myocardial ischemia/reperfusion injury. a Time-dependent alteration of left ventricular systolic pressure (LVSP) in both the young group (YI/R) and aging group (AI/R) after myocardial ischemia/reperfusion. b Left ventricular end-diastolic pressure (LVEDP). c, d Maximal rate of rise/decrease of left ventricular pressure (±dp/dtmax). e Percentage of area at risk (AAR), as identified by negative Evans blue staining, divided by total left ventricular (LV) area. f Percentage of infarct area (as identified by negative TTC staining) divided by AAR. g, h MI/R-induced cardiomyocyte apoptosis with aging. Apoptosis was determined by TUNEL labeling, and positive apoptotic nuclei were shown as the bright green fluorescence. Sections were counterstained with anti-α-actin antibody and Hoechst to identify cardiac myocytes (red) and nuclei (blue). Apoptotic index = number of positively stained apoptotic myocytes / total number of myocytes counted × 100 %. Activity of caspase-3 was determined at the end of reperfusion. Data expressed as mean ± SEM (n = 6 each). * P < 0.05, ** P < 0.01 vs. YI/R; # P < 0.05, ## P < 0.01 vs. young sham; ∆ P < 0.05, ∆∆ P < 0.01 vs. a sham
Myocardial infarct size and myocardium apoptosis
Consistent with previously published results by other investigators (Zhang et al. 2007), MI/R-induced cardiac injury was enhanced in aging rats as evidenced by enlarged infarct size (Fig. 1e, f) and increased apoptosis (Fig. 1g, h).
Aging increased myocardial apoptosis in rat heart
In myocardial tissue from the 6-month-old rats, a very low level of TUNEL-positive staining was observed, while a significant number of TUNEL-positive cells were observed in myocardial tissue from the 24-month-old rats (Fig. 2a). Normal aging is associated with the activation of a caspase-3-mediated apoptotic pathway in several tissues. As shown in Fig. 2b, the caspase-3 activity was increased with aging. Caspase-3 can be activated by either caspase-8 or by caspase-9, and then both of them were detected. The data showed that caspase-9 activity in the aging heart was enhanced (Fig. 2c), but caspase-8 activity was not (Fig. 2d).
Fig. 2.

Aging increased cardiomyocyte apoptosis in rat heart. a TUNEL labeling. b Caspase-3, c caspase-8, and d caspase-9 activities in the heart of young and aging rats. Data expressed as mean ± SEM (n = 10 each). * P < 0.05, ** P < 0.01 vs. young
The age-associated XIAP protein expression might be related to enhanced MI/R injury in aging rats
XIAP is the best characterized and the most potent caspase inhibitor. As shown in Fig. 3a, XIAP protein expression was reduced in the heart of aging rats compared with that in the young rats. Then, the mRNA content of XIAP was determined by real-time PCR. Consistent with the protein expression analyses, the amount of XIAP mRNA in aging rat hearts was lower than that in the young rat hearts (Fig. 3a, b).
Fig. 3.

a Age-dependent differences in XIAP protein expression. Densitometry analysis for expression of XIAP protein by Western blot (n = 10 each) b The quantitative analysis for expression of XIAP mRNA by real-time PCR (n = 10 each) c Representative Western blot of XIAP in young and aging myocardial tissue subject to ischemia/reperfusion (I/R). Lane 1, young sham (YSham); lane 2, aging sham (ASham); lane 3, young myocardial tissue subject to I/R (YI/R); and lane 4, aging myocardial tissue subject to I/R (AI/R) (n = 6 each). d A significant linear positive correlation between the + dp/dtmax and XIAP protein expression among all MI/R groups (r = 0.658, n = 42). Bar heights represent mean values, and brackets indicate SEM. * P < 0.05, ** P < 0.01 vs. young; # P < 0.05 vs. young sham; ∆ P < 0.05 vs. YI/R
Moreover, XIAP protein expression in the aging and young rat hearts was determined after myocardial ischemia/reperfusion. XIAP was decreased in aging hearts after MI/R and lower than that in young adult hearts subjected to MI/R (Fig. 3c). Using a statistical logistic regression method, we found a clearly significant correlation (r = 0.658, p < 0.01) between XIAP protein expression and + dp/dtmax in 42 rats subjected to MI/R (Fig. 3d). These results demonstrated that the XIAP protein expression was related to MI/R injury in aging rats.
Increased translocation of Omi/HtrA2 from mitochondria to cytosol induced XIAP downregulation in aging hearts after MI/R
Smac/DIABLO
Smac/DIABLO, a mitochondrial protein and negative regulator of XIAP, enhances apoptosis by antagonizing to XIAP. Total Smac/DIABLO was observed in the heart of young adult and aging rats. As shown in Fig. 4a, b, there is no statistical significance of Smac/DIABLO protein and mRNA expression in the two groups.
Fig. 4.

a, b Age-dependent differences in Smac/DIABLO protein and mRNA expression (n = 10 each). c, d Age-dependent differences in Omi/HtrA2 protein and mRNA expression (n = 10 each). e Representative Western blots showing cytosolic Omi/HtrA2 expression. Cyt. indicates cytosolic extract, and β-actin was used as a cytoplasm loading control; Mit. indicates mitochondria extract, and Cox IV was used as a mitochondrial loading control (n = 6 each). f Effect of ucf-101 on XIAP protein expression in both aging (top) and aging MI/R (AI/R, bottom) group. Bar heights represent mean values, and brackets indicate SEM (n = 6 each). ** P < 0.01 vs. young; # P < 0.05, ## P < 0.01 vs. young sham; ∆ P < 0.05 vs. YI/R
Omi/HtrA2
Another novel protein identified to have similar characteristics in antagonizing XIAP function is Omi/HtrA2. Omi/HtrA2 was detected in myocardial tissue obtained from the young and the aging rats. Interestingly, Omi/HtrA2 protein and mRNA expression was significantly increased in the aging hearts compared with that in the young adult hearts (Fig. 4c, d).
It is reported that Omi/HtrA2 inactivates XIAP irreversibly and facilitates caspase activity in apoptosis. Recent in vitro studies have shown that cytosolic stimulation, such as UV irradiation and pathological conditions such as MI/R in vivo, results in Omi/HtrA2 translocation from the mitochondria to the cytosol. The mitochondrial and cytosolic fraction of myocardium was isolated separately and incubated with Omi/HtrA2 antibody. As show in Fig. 4e, the cytosolic Omi/HtrA2 expression was significantly increased in the aging group compared with that in the young group. Moreover, cytosolic Omi/HtrA2 protein expression was significantly increased in the aging MI/R group compared with that in the young MI/R group, indicating more translocation of Omi/HtrA2 from mitochondria to cytosol in not only the aging heart but also the aging heart subjected to MI/R. Meanwhile, inhibition of Omi/HtrA2 by ucf-101 increased the XIAP protein expression in both the aging heart and the aging heart subjected to MI/R (Fig. 4f).
Increased Omi/HtrA2 expression enhanced MI/R injury in the aging heart
Inhibition of Omi/HtrA2 reduced cardiomyocyte apoptosis in the aging heart
Having demonstrated that Omi/HtrA2 was increased with age, we then sought to determine whether Omi/HtrA2 plays an important role in apoptosis in the aging myocardium. Ucf-101, a novel and specific inhibitor of Omi/HtrA2, was used in vivo. The results showed that inhibition of Omi/HtrA2 reduced cardiomyocyte apoptosis in the aging heart evidenced by decreased caspase-3 activity (Fig. 5a).
Fig. 5.

a Effect of ucf-101 on caspase-3 activity in the aging heart. b Effect of ucf-101 on caspase-3 activity in aging heart subject to myocardial ischemia/reperfusion (MI/R). c Effect of ucf-101 on left ventricular systolic pressure (LVSP) in aging heart subject to MI/R. d Left ventricular end-diastolic pressure (LVEDP). e, f Maximal rate of rise/decrease of left ventricular pressure (±dp/dtmax). g Percentage of area at risk (AAR), as identified by negative Evans blue staining, divided by total left ventricular (LV) area. h Percentage of infarct area (as identified by negative TTC staining) divided by AAR. Ten minutes before reperfusion, animals were randomized to receive vehicle (DMSO) or ucf-101 (1.5 mol/kg) via intraperitoneal injection (IP). Data expressed as mean ± SEM (n = 6 each). * P < 0.05, ** P < 0.01 vs. vehicle
Inhibition of Omi/HtrA2 exerted significant cardiac protective effects against MI/R injury in the aging heart
The data above show that the MI/R caused more Omi/HtrA2 translocation in the aging hearts, so we then sought to determine whether Omi/HtrA2 after translocation induced more serious aging postischemic myocardial injury. As shown in Fig. 5b, ucf-101 treatment significantly attenuated caspase-3 activity in the aging heart subjected to MI/R, which suggested that Omi/HtrA2 plays a critical role in aging postischemic myocardial apoptosis. To determine whether the cardioprotective effects of ucf-101 were sustained, aging rats were subjected to 30 min of ischemia and 3 h of reperfusion, and the effect of ucf-101 on cardiac function was determined. As summarized in Fig. 5c, d, f, ucf-101 treatment (1.50 μmol/kg) restored ± dp/dtmax and LVSP, but not LVEDP (Fig. 5e). As shown in Fig. 5g, h, ucf-101 reduces the infarct size of aging heart subjected to MI/R. All of these results demonstrate that inhibition of Omi/HtrA2 significantly improves heart function after reperfusion.
Discussion
Aging is associated with a reduced functional reserve and altered responsiveness of the heart to I/R injury, but the molecular basis for this deficiency has not been elucidated. This study has defined for the first time that the expression of Omi/HtrA2 is increased in the aging heart, which promotes cardiomyocyte apoptosis through degradation of XIAP. Moreover, we have demonstrated that cytosolic Omi/HtrA2 in the aging myocardium was higher than that in the adult heart subjected to MI/R, suggesting increased translocation of Omi/HtrA2 from mitochondria to cytosol in the aging heart after MI/R. Finally, we have demonstrated that treatment with a novel and specific Omi/HtrA2 inhibitor before ischemia/reperfusion could attenuate XIAP degradation and exerted cardioprotective effects in the aging heart, as evidenced by ameliorated cardiac function and decreased infarct size and caspase-3 activity ratio. Taken together, these results provide strong evidence that increased expression of Omi/HtrA2 plays a causative role in enhanced postischemic injury in the aging heart.
With aging, baseline cardiac function declines. When the aging heart is exposed to various forms of stress, a further deterioration of cardiac function is observed (Boengler et al. 2009). In the present study, a rat model of MI/R was used to investigate the effects of aging on the alteration of myocardial function and possible mechanisms. Numerous reports have demonstrated that aging enhanced MI/R injury (Zhang et al. 2007; Fujita et al. 2009; Vessey et al. 2009). However, only a few reports evaluate cardiac function and were not enough to comprehensively realize the features of MI/R injury. Our results provided time-dependent alteration of ventricular function in both young and aging MI/R rats and showed that the events of MI/R injury to aging rats induced more severe myocardial dysfunction within 24 h after reperfusion compared to young rats. Moreover, the aging heart showed a more serious decrease in postischemic recovery of cardiac function, evidenced by no statistical difference of hemodynamics between the young MI/R heart and the young sham heart but exaggerated cardiac dysfunction in the aging MI/R heart compared with the aging sham heart at 1 h after reperfusion. These results that were consistent with reperfusion therapies confer less benefit in the aging with myocardial infarction than in younger adults.
Clinical studies have shown that elderly patients have a higher morbidity and mortality following acute myocardial infarction than younger patients (Ferdinandy et al. 2007). Attenuation of myocardial protective and repair processes may be reflected in a greater occurrence of cardiomyocyte loss through apoptosis in the aging, when compared to young adults during MI/R. In 1996, Kajstura et al. demonstrated that apoptosis in the left ventricles increased by more than 200 % in 24-month-old rats compared with 16-month-old rats (Kajstura et al. 1996). Our data confirm this conclusion with more TUNEL-positive cell and higher caspase-3 activity in 24-month-old rats. Moreover, caspase-9 activity in the aging heart was enhanced, indicating that the mitochondrial pathway (caspase-9/caspase-3 pathway) may be related to cardiomyocyte apoptosis with aging.
One of the most widely recognized biochemical features of apoptosis is the activation of a class of cysteine proteases known as caspases. Among numerous proteins that are known to regulate caspase activity, XIAP is the most potent inhibitor of apoptosis (Siu et al. 2005) and possesses three baculovirus IAP repeat (BIR1, BIR2, and BIR3) domains and a RING domain. It binds to and inhibits caspase-3, caspase-7, and caspase-9. The BIR2 domain of XIAP inhibits caspase-3 and caspase-7, while BIR3 binds to and inhibits caspase-9. The RING domain utilizes E3 ubiquitin ligase activity and enables IAPs to catalyze ubiquitination of self, caspase-3, or caspase-7 (Dubrez-Daloz et al. 2008). It has been demonstrated that in the failing human heart, XIAP is downregulated, indicating that the cardiomyocytes were more sensitive to apoptosis (Haider et al. 2009). Previous studies showed that XIAP expression in myocardial tissue was reduced after ischemia or hypoxia (Lyn et al. 2002). The administration of pharmacologically mimicking endogenous XIAP, PTD-BIR3/RING, reduces myocardial infarct size when injected during reperfusion through interruption of caspase activation (Souktani et al. 2009). These results suggested that reduced XIAP expression plays an important role in MI/R injury. Interestingly, our current study found that XIAP protein and mRNA expression was decreased in the aging heart and demonstrated that MI/R reduced XIAP protein expression more in the aging than in the young adult heart, which may induce exaggerated apoptosis in both the aging heart and the aging heart subjected to MI/R. In addition, we found a significant correlation between XIAP protein expression and + dp/dtmax in 42 MI/R rats. Cumulatively, these data suggested that the decline of XIAP expression may be a major factor related to increased susceptibility of aging.
XIAP protein was balanced by Smac/DIABLO and Omi/HtrA2, two nuclear-encoded proteins released into the cytoplasm by the mitochondria. Both Smac/DIABLO and Omi/HtrA2 are synthesized as precursor proteins with N-terminal mitochondrial localization signal peptides that are removed during maturation in the mitochondria to expose their N-terminal IAP-binding motif. During apoptosis, both proteins are released from the intermembrane space of the mitochondria into the cytoplasm and promote caspase activation and apoptosis by binding to the BIR domain(s) of XIAP (Vaux and Silke 2003). In our present study, both Smac/DIABLO and Omi/HtrA2 expressions were detected. However, Smac/DIABLO protein expression was not significantly different in the hearts of the aging rat compared with the young adult, while Omi/HtrA2 protein and mRNA expression was significantly increased. Moreover, there was Omi/HtrA2 protein leakage from the mitochondria to the cytoplasm in the aging heart. Although Omi/HtrA2 and Smac/DIABLO both seem to target XIAP, an increasing evidence suggests that Omi/HtrA2 may play a unique role in apoptosis. Several different Smac/DIABLO-deficient cells respond normally to various apoptotic stimuli, suggesting the existence of a redundant molecule or molecules compensating for a loss of Smac/DIABLO function (Okada et al. 2002). However, overexpression of Omi/HtrA2 sensitizes cells to apoptosis, and its removal by RNA interference reduces cell death (Martins et al. 2002). Meanwhile, our results showed that the administration of ucf-101 (Omi/HtrA2-specific protease inhibitor) could attenuate the decreased XIAP protein expression under normal conditions in aging rats. In 2004, Suzuki et al. found that Omi/HtrA2 protease activity could reduce XIAP expression, and the release of HtrA2 from mitochondria might catalytically cleave and inactivate XIAP (Suzuki et al. 2004). Taken together, these data indicated a nonredundant essential function of Omi/HtrA2 in the induction of apoptosis though inactivation of XIAP in the aging heart. As for why XIAP mRNA was reduced, there were no relationships between XIAP mRNA and Omi/HtrA2 as far as we know. There might be other factors related to the reduced XIAP mRNA. The first is mir-23a, which gradually increases with age and was predicted to bind and inhibit XIAP mRNA expression (Lin et al. 2011; Siegel et al. 2011). The second is HuR (human antigen R), which decreases during replicative senescence and regulates XIAP expression by stabilizing its mRNA (Masuda et al. 2009; Zhang et al. 2009). The third is TGF-β. The level of active TGF-β1 is decreased during replicative senescence, and XIAP gene expression and function is positively regulated by exposure to TGF-β (Zeng et al. 1996; Van Themsche et al. 2010). However, these hypotheses have not been tested in this paper and will be investigated in future studies.
In addition, Liu and her colleagues first provided a direct evidence that a normal level of endogenously expressed Omi/HtrA2 contributes to apoptosis under myocardial ischemia followed by reperfusion in vivo (Liu et al. 2005). Our current study showed that the release of Omi/HtrA2 from mitochondria to cytosol was significantly increased in the aging MI/R rats compared with that in the young MI/R group, providing a strong clue that Omi/HtrA2 plays a causative role in increased postischemic cardiomyocyte apoptosis in the aging heart. In order to investigate whether Omi/HtrA2 exactly plays an important role in age-related MI/R injury, we observed the effect of ucf-101, a highly selective Omi/HtrA2 inhibitor, on MI/R injury in aging rats. Treatment with ucf-101 in aging MI/R animal models could reduce the caspase-3 activity and attenuate the cardiac dysfunction and infarct size, strongly suggesting that Omi/HtrA2 plays a causative role in enhanced aging MI/R injury.
Our results demonstrated that increased expression and leakage of Omi/HtrA2 enhanced MI/R injury in aging hearts via degrading XIAP and promoting myocardial apoptosis, suggesting that therapeutic interventions that inhibit Omi/HtrA2 in the aging patient may improve outcomes after myocardial ischemia.
Acknowledgments
This study was supported by the Natural Sciences Foundation of China (NSFC) grants (30973163, to Huirong Liu) and the Funding Project for Academic Human Resources Development in Institutions of Higher Learning Under the Jurisdiction of Beijing Municipality (PHR201106112, to Huirong Liu).
References
- Ananthakrishnan R, Li Q, Gomes T, Schmidt AM, Ramasamy R. Aldose reductase pathway contributes to vulnerability of aging myocardium to ischemic injury. Exp Gerontol. 2011;46(9):762–767. doi: 10.1016/j.exger.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuiyan MS, Fukunaga K. Activation of HtrA2, a mitochondrial serine protease mediates apoptosis: current knowledge on HtrA2 mediated myocardial ischemia/reperfusion injury. Cardiovasc Ther. 2008;26(3):224–232. doi: 10.1111/j.1755-5922.2008.00052.x. [DOI] [PubMed] [Google Scholar]
- Boengler K, Schulz R, Heusch G. Loss of cardioprotection with ageing. Cardiovasc Res. 2009;83(2):247–261. doi: 10.1093/cvr/cvp033. [DOI] [PubMed] [Google Scholar]
- Boyle AJ, Shih H, Hwang J, Ye J, Lee B, Zhang Y, Kwon D, Jun K, Zheng D, Sievers R, Angeli F, Yeghiazarians Y, Lee R. Cardiomyopathy of aging in the mammalian heart is characterized by myocardial hypertrophy, fibrosis and a predisposition towards cardiomyocyte apoptosis and autophagy. Exp Gerontol. 2011;46(7):549–559. doi: 10.1016/j.exger.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cilenti L, Lee Y, Hess S, Srinivasula S, Park KM, Junqueira D, Davis H, Bonventre JV, Alnemri ES, Zervos AS. Characterization of a novel and specific inhibitor for the pro-apoptotic protease Omi/HtrA2. J Biol Chem. 2003;278(13):11489–11494. doi: 10.1074/jbc.M212819200. [DOI] [PubMed] [Google Scholar]
- Dubrez-Daloz L, Dupoux A, Cartier J. IAPs: more than just inhibitors of apoptosis proteins. Cell Cycle. 2008;7(8):1036–1046. doi: 10.4161/cc.7.8.5783. [DOI] [PubMed] [Google Scholar]
- Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007;59(4):418–458. doi: 10.1124/pr.107.06002. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Ishino K, Nakanishi K, Fujii Y, Kawada M, Sano S. Age-dependent vulnerability to ischemia-reperfusion injury of cyanotic myocardium in a chronic hypoxic rat model. Acta Med Okayama. 2009;63(5):237–242. doi: 10.18926/AMO/31840. [DOI] [PubMed] [Google Scholar]
- Gould KE, Taffet GE, Michael LH, Christie RM, Konkol DL, Pocius JS, Zachariah JP, Chaupin DF, Daniel SL, Sandusky GE, Jr, Hartley CJ, Entman ML. Heart failure and greater infarct expansion in middle-aged mice: a relevant model for postinfarction failure. Am J Physiol Heart Circ Physiol. 2002;282(2):H615–621. doi: 10.1152/ajpheart.00206.2001. [DOI] [PubMed] [Google Scholar]
- Haider N, Arbustini E, Gupta S, Liu H, Narula N, Hajjar R, Moorjani N, Westaby S, Semigran MJ, Dec GW, Chandrashekhar Y, Narula J. Concurrent upregulation of endogenous proapoptotic and antiapoptotic factors in failing human hearts. Nat Clin Pract Cardiovasc Med. 2009;6(3):250–261. doi: 10.1038/ncpcardio1452. [DOI] [PubMed] [Google Scholar]
- Holcik M, Gibson H, Korneluk RG. XIAP: apoptotic brake and promising therapeutic target. Apoptosis. 2001;6(4):253–261. doi: 10.1023/A:1011379307472. [DOI] [PubMed] [Google Scholar]
- Kajstura J, Cheng W, Sarangarajan R, Li P, Li B, Nitahara JA, Chapnick S, Reiss K, Olivetti G, Anversa P. Necrotic and apoptotic myocyte cell death in the aging heart of Fischer 344 rats. Am J Physiol. 1996;271(3Pt2):H1215–1228. doi: 10.1152/ajpheart.1996.271.3.H1215. [DOI] [PubMed] [Google Scholar]
- Kakarla SK, Rice KM, Katta A, Paturi S, Wu M, Kolli M, Keshavarzian S, Manzoor K, Wehner PS, Blough ER. Possible molecular mechanisms underlying age-related cardiomyocyte apoptosis in the F344XBN rat heart. J Gerontol A Biol Sci Med Sci. 2010;65(2):147–155. doi: 10.1093/gerona/glp203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107(1):139–146. doi: 10.1161/01.CIR.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- Li Q, Hueckstaedt LK, Ren J. The protease inhibitor UCF-101 ameliorates streptozotocin-induced mouse cardiomyocyte contractile dysfunction in vitro: role of AMP-activated protein kinase. Exp Physiol. 2009;94(9):984–994. doi: 10.1113/expphysiol.2009.049189. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Li Q, Ren J. Influence of cardiac-specific overexpression of insulin-like growth factor 1 on lifespan and aging-associated changes in cardiac intracellular Ca2+ homeostasis, protein damage and apoptotic protein expression. Aging Cell. 2007;6(6):799–806. doi: 10.1111/j.1474-9726.2007.00343.x. [DOI] [PubMed] [Google Scholar]
- Lin ST, Ptácek LJ, Fu YH. Adult-onset autosomal dominant leukodystrophy: linking nuclear envelope to myelin. J Neurosci. 2011;31(4):1163–1166. doi: 10.1523/JNEUROSCI.5994-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HR, Gao E, Hu A, Tao L, Qu Y, Most P, Koch WJ, Christopher TA, Lopez BL, Alnemri ES, Zervos AS, Ma XL. Role of Omi/HtrA2 in apoptotic cell death after myocardial ischemia and reperfusion. Circulation. 2005;111(1):90–96. doi: 10.1161/01.CIR.0000151613.90994.17. [DOI] [PubMed] [Google Scholar]
- Liu M, Zhang P, Chen M, Zhang W, Yu L, Yang XC, Fan Q (2011) Aging might increase myocardial ischemia/reperfusion-induced apoptosis in humans and rats. Age. doi:10.1007/s11357-011-9259-8 [DOI] [PMC free article] [PubMed]
- Lyn D, Bao S, Bennett NA, Liu X, Emmett NL. Ischemia elicits a coordinated expression of pro-survival proteins in mouse myocardium. ScientificWorld Journal. 2002;2:997–1003. doi: 10.1100/tsw.2002.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins LM, Iaccarino I, Tenev T, Gschmeissner S, Totty NF, Lemoine NR, Savopoulos J, Gray CW, Creasy CL, Dingwall C, Downward J. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J Biol Chem. 2002;277(1):439–444. doi: 10.1074/jbc.M109784200. [DOI] [PubMed] [Google Scholar]
- Masuda K, Marasa B, Martindale JL, Halushka MK, Gorospe M. Tissue- and age-dependent expression of RNA-binding proteins that influence mRNA turnover and translation. Aging (Albany NY) 2009;1(8):681–698. doi: 10.18632/aging.100073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada H, Suh WK, Jin J, Woo M, Du C, Elia A, Duncan GS, Wakeham A, Itie A, Lowe SW, Wang X, Mak TW. Generation and characterization of Smac/DIABLO-deficient mice. Mol Cell Biol. 2002;22(10):3509–3517. doi: 10.1128/MCB.22.10.3509-3517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts MB, Vaughn AE, McDonough H, Patterson C, Deshmukh M. Reduced Apaf-1 levels in cardiomyocytes engage strict regulation of apoptosis by endogenous XIAP. J Cell Biol. 2005;171(6):925–930. doi: 10.1083/jcb.200504082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel C, Li J, Liu F, Benashski SE, McCullough LD. miR-23a regulation of X-linked inhibitor of apoptosis (XIAP) contributes to sex differences in the response to cerebral ischemia. Proc Natl Acad Sci U S A. 2011;108(28):11662–11667. doi: 10.1073/pnas.1102635108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu PM, Bryner RW, Murlasits Z, Alway SE. Response of XIAP, ARC, and FLIP apoptotic suppressors to 8 wk of treadmill running in rat heart and skeletal muscle. J Appl Physiol. 2005;99(1):204–209. doi: 10.1152/japplphysiol.00084.2005. [DOI] [PubMed] [Google Scholar]
- Souktani R, Pons S, Guegan C, Bouhidel O, Bruneval P, Zini R, Mandet C, Onteniente B, Berdeaux A, Ghaleh B. Cardioprotection against myocardial infarction with PTD-BIR3/RING, a XIAP mimicking protein. J Mol Cell Cardiol. 2009;46(5):713–718. doi: 10.1016/j.yjmcc.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Soustiel JF, Larisch S. Mitochondrial damage: a target for new therapeutic horizons. Neurotherapeutics. 2010;7(1):13–21. doi: 10.1016/j.nurt.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Takahashi-Niki K, Akagi T, Hashikawa T, Takahashi R. Mitochondrial protease Omi/HtrA2 enhances caspase activation through multiple pathways. Cell Death Differ. 2004;11(2):208–216. doi: 10.1038/sj.cdd.4401343. [DOI] [PubMed] [Google Scholar]
- Van Themsche C, Chaudhry P, Leblanc V, Parent S, Asselin E. XIAP gene expression and function is regulated by autocrine and paracrine TGF-beta signaling. Mol Cancer. 2010;9:216. doi: 10.1186/1476-4598-9-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux DL, Silke J. Mammalian mitochondrial IAP binding proteins. Biochem Biophys Res Commun. 2003;304(3):499–504. doi: 10.1016/S0006-291X(03)00622-3. [DOI] [PubMed] [Google Scholar]
- Vessey DA, Kelley M, Li L, Huang Y. Sphingosine protects aging hearts from ischemia/reperfusion injury: superiority to sphingosine 1-phosphate and ischemic pre- and post-conditioning. Oxid Med Cell Longev. 2009;2(3):146–151. doi: 10.4161/oxim.2.3.8622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White HD, Barbash GI, Califf RM, Simes RJ, Granger CB, Weaver WD, Kleiman NS, Aylward PE, Gore JM, Vahanian A, Lee KL, Ross AM, Topol EJ. Age and outcome with contemporary thrombolytic therapy. Circulation. 1996;94(8):1826–1833. doi: 10.1161/01.CIR.94.8.1826. [DOI] [PubMed] [Google Scholar]
- Wilkinson JC, Cepero E, Boise LH, Duckett CS. Upstream regulatory role for XIAP in receptor-mediated apoptosis. Mol Cell Biol. 2004;24(16):7003–7014. doi: 10.1128/MCB.24.16.7003-7014.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A, Suzuki N, Nakano Y, Oho T, Kawada M, Koga T. Development of a 5′ fluorogenic nuclease-based real-time PCR assay for quantitative detection of Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. J Clin Microbiol. 2003;41(2):863–866. doi: 10.1128/JCM.41.2.863-866.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Tao L, Jiao X, Gao E, Lopez BL, Christopher TA, Koch W, Ma XL. Nitrative thioredoxin inactivation as a cause of enhanced myocardial ischemia/reperfusion injury in the aging heart. Free Radic Biol Med. 2007;43(1):39–47. doi: 10.1016/j.freeradbiomed.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zou T, Rao JN, Liu L, Xiao L, Wang PY, Cui YH, Gorospe M, Wang JY. Stabilization of XIAP mRNA through the RNA binding protein HuR regulated by cellular polyamines. Nucleic Acids Res. 2009;37(22):7623–7637. doi: 10.1093/nar/gkp755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao HX, Wang XL, Wang YH, Wu Y, Li XY, Lv XP, Zhao ZQ, Zhao RR, Liu HR. Attenuation of myocardial injury by postconditioning: role of hypoxia inducible factor-1alpha. Basic Res Cardiol. 2010;105(1):109–118. doi: 10.1007/s00395-009-0044-0. [DOI] [PubMed] [Google Scholar]
- Zeng G, McCue HM, Mastrangelo L, Millis AJ. Endogenous TGF-beta activity is modified during cellular aging: effects on metalloproteinase and TIMP-1 expression. Exp Cell Res. 1996;228(2):271–276. doi: 10.1006/excr.1996.0326. [DOI] [PubMed] [Google Scholar]