

Abstract
Although the positive symptoms of schizophrenia are reasonably well-controlled by current antipsychotics, cognitive impairment remains largely unaddressed. The Matrics initiative lays out a regulatory path forward and a number of targets have been tested in the clinic, so far without much success. To address this translational disconnect, we have developed a mechanism-based humanized computer model of a relevant key cortical brain network with schizophrenia pathology involved with the maintenance aspect of working memory (WM). The model is calibrated using published clinical experiments on N-back WM tests. We further simulate the opposite effect of γ-aminobutyric acid (GABA) modulators lorazepam and flumazenil and of a published augmentation trial of clozapine with risperidone, illustrating the introduction of new targets and the capacity of predicting the effects of polypharmacy. This humanized approach allows for early prospective and quantitative assessment of cognitive outcome in a central nervous system (CNS) research and development project, thereby hopefully increasing the success rate of clinical trials.
Current antipsychotic treatment reasonably addresses the positive symptoms in schizophrenia but not the cognitive and negative symptoms hampering patients to regain their professional status.1 Therefore, the US Food and Drug Administration, industry, and the National Institutes of Health developed the Matrics initiative;2 and a few compounds have been tested in the TURNS initiative,3 such as davunetide4 and CX5165 without much success. Developing new compounds for addressing cognitive impairment in schizophrenia is a challenge given the complexity of the human brain and the limited translationability of preclinical animal models.
Important translational issues for cognitive impairment in schizophrenia include appropriate attention to comedications, genotypes that interfere with the primary pharmacology, and the appropriate patient population. For instance, the catechol-o-methyl transferase Val158Met genotype modulates clearance of dopamine and norepinephrine and significantly affects human cognition.6 This genotype is not present in rodents, although specific knockouts or knockins have been developed.7 A mechanistically based computer model allows for the effect of this genotype on dopamine receptor activation level in human brain to be simulated.8
There is increasing evidence that the excitatory–inhibitory balance in cortical networks is fundamentally different between primates and rodents,9 with primate basket interneuron cells having a higher input resistance and a lower firing threshold, generating more spikes at near-threshold current intensities. Different interneuron subtypes with short spike duration are specific for the primate cortex, but not for rodents.10
To provide better guidance on clinical central nervous system (CNS) candidate selection and development, a more humanized model of cognitive performance such as computer-based disease modeling could be potentially helpful at critical junctures. Indeed, Quantitative Systems Pharmacology has been proposed as a tool for improving pharmaceutical research and development, especially in complex disease situations with many interacting circuits.11 Detailed computer-based biophysically realistic models of cortical and hippocampal networks have been developed and calibrated extensively using single-unit electrophysiology data in preclinical models.12 However, the absence of neuromodulatory drug targets, such as dopamine and serotonin, norepinephrine, and clinical calibration make these computer models less useful for supporting CNS research and development programs.
Any useful computer-based quantitative systems pharmacology model must be able to predict clinical scales. With limited preclinical animal data and the absence of many detailed data for the human neurophysiology situation, we elected to determine the relevant biological coupling parameters—within a biologically constrained range—by simulating clinical experiments of an N-back working memory (WM) task in human subjects13 and optimizing the correlation between model and clinical outcome. Within such a hybrid framework, we limited experimental uncertainty about preclinical data and focused on clinically relevant information. Such an approach allowed us to blindly predict an unexpected clinical outcome in schizophrenia, which was missed by traditional animal models.14
We also report on the effect of γ-aminobutyric acid (GABA) modulators15 in healthy controls and patients with schizophrenia and the effect of risperidone augmentation therapy on clozapine16 as independent validations.
Results
Implementation of schizophrenia pathology
We first studied the sensitivity of the network on the different schizophrenia pathology processes. Outcome variability (range of outcomes divided by average outcome) was greatest for N-methyl-D-aspartate (0.63), followed by GABA (0.43), noise (0.26), and D1 receptor (D1R) activation level (0.16).
A virtual subject trial simulation for “healthy controls,” assuming a Gaussian distribution with a variance(s) of 10% for N-methyl-D-aspartate-R, 5% for the GABA-R conductance, 15% for D1R activation level, and 10% for the background noise, resulted in an average WM span of 9.18 ± 0.66 s.
Implementing schizophrenia pathology by reducing basal N-methyl-D-aspartate by 12.5%, gGABA by 5%, basal D1R activity level by 6.5%, and increasing background noise level by 20% resulted in an average WM span of 8.12 s or a decrease of 1.06/0.66 = 1.61 SD below normal performance. Although it is difficult to derive quantitative values for the observed clinical changes,17,18,19,20 these numbers illustrate the rather modest pathological changes associated with schizophrenia, especially with regard to other degenerative diseases such as Alzheimer's disease,21 in which WM spans are in the range of 4–7 s.
A virtual patient trial with schizophrenia pathology and with values drawn from Gaussian distributions around these pathological values shows a clear distinction between patients with schizophrenia and controls (Figure 1a).
Figure 1.

Virtual subject histogram. (a) Histogram of working memory model outcomes for a virtual patient and a virtual healthy subject population with variances of 10, 5, 15, and 10% around the fixed values for NMDA conductance, GABA conductance, D1R activation level, and noise level. Although the two distributions are statistically different, there is a large overlap; but the variability of the schizophrenia population is larger than the variability of the healthy population. This allows to define the SD for each population. Correlation between model and clinical outcome. (b) Calibration between model outcomes of 17 different interventions (antipsychotics, schizophrenia pathology, COMT genotype, scopolamine, mecamylamine. Tolcapone, see text for more details) and the corresponding clinically reported effects on the 2-Back working memory test. The data suggest a robust correlation between model outcomes and clinical effects. New therapeutic interventions can be simulated and an anticipated clinical response on a 2-Back working memory test can be derived, allowing to estimate the differentiability with existing treatments. COMT, catechol-O-methyltransferase; D1R, D1 receptor; GABA, γ-aminobutyric acid; NMDA, N-Methyl-D-aspartate; SZ, schizophrenia.
Calibration of the cortical model
The remaining free biological coupling parameters were calibrated to optimize the correlation between the model outcome and the actual clinical performance on the 2-Back WM test for 17 different interventions. Note that each value is the average for a group of subjects submitted to the intervention (Supplementary Data online). We first applied a design-of-experiment approach where the slopes in the Pareto plots were proportional to the contribution of the processes that were driving the optimization. After three iterations, the Pareto plots indicated no further improvement (slopes close to zero) and we switched to a local maximum search. Figure 1b shows that we can get a robust correlation between model outcome and clinical observations.
To compare this result with a more traditional regression statistical analysis, we also performed a multivariate regression correlation analysis between the receptor occupancies (ROs) of each drug–dose combination and their respective WM outcomes. ROs were calculated using the formula Dose/(Dose + Ki) with Ki the dose for 50% of RO. Because displacement data for the receptors in the model are unknown, in a first approximation, we set Ki = KD2 (Aff-x/Aff-D2), where KD2 is the dose for 50% of raclopride RO; Aff-x and Aff-D2 are the affinities of the drug for the receptor X (any of the non-D2R) and D2, respectively. The correlation for this multivariate analysis is about 0.12 (P = 0.31), much lower than for our mechanism-based computer model (r2 = 0.71, P < 0.001).
GABA modulators on cognitive performance
As an independent verification, we simulated a clinical trial with GABA modulators,15 where in 11 treated patients and 11 controls the effect of GABA agonist lorazepam and GABA antagonist flumazenil22 in a crossover, double-blind design on WM performance were studied. GABA-Aα1 and GABA-Aα2 receptor subunits are located on pyramidal, but only GABA-Aα1 is on inhibitory interneurons. Postmortem cortical tissue from patients with schizophrenia showed a relative increase in GABA-Aα2 subunits in layer 2 at pyramidal cells.23 Both drugs affect both GABA subunits; therefore, we assume that their modulation is 25% higher on inhibitory–excitatory synapses. Except for lithium, we simulated the individual patients with their full antipsychotic medication to which lorazepam and flumazenil was added, using the receptor competition model; we assumed a 50% N-desmethylclozapine metabolite in the clozapine active moiety.24
In the absence of target engagement values, we first studied the dose–response for both drugs to identify a target engagement value that gave a robust change in performance. Figure 2a shows that control subjects are essentially unaffected, whereas in schizophrenia conditions, flumazenil dose-dependently improves and lorazepam dose-dependently decreases outcome.
Figure 2.

Effect on healthy and Schizophrenia pathology.(a) Dose–response of increasing target engagement for lorazepam and flumazenil in healthy controls and in patients with schizophrenia. Although there is not much effect for the healthy controls with regard to both treatments, lorazepam in patients with schizophrenia dose-dependently worsens, whereas flumazenil dose-dependently improves cognitive outcome. Virtual trial model outcome and clinical outcome. (b,c) Effect of lorazepam and flumazenil on network performance in a healthy control environment and in schizophrenics. In healthy subjects, none of the compounds had a significant effect on the working memory span outcome for the computer model, in line with the clinical observations, whereas in schizophrenia, lorazepam induces a statistically significant decrease and flumazenil induces a statistically significant improvement in working memory performance. This same differential effect is indeed observed in the clinical outcome. Individual patient simulation. (d) Simulation of switching individual patients in a crossover study from placebo to each of the two GABA modulators. The schizophrenia subjects who switched from placebo to lorazepam did 15.6 ± 1.1% worse (P < 0.01), whereas the subjects who switched to flumazenil did 3.9 ± 2.0 % better (P = 0.089). FLU, flumazenil; GABA, γ-aminobutyric acid; LOR, lorazepam; SZ, schizophrenia; WM, working memory.
For this particular trial with lorazepam, we assumed a 5% GABA-Aα1 agonist effect on inhibitory–inhibitory and a 6.25% GABA agonist effect on inhibitory–excitatory synapses and for flumazenil, a 2% GABA decrease on inhibitory–inhibitory and a 2.5% GABA decrease on inhibitory–excitatory synapse. Simulating the 11 healthy subjects, flumazenil improved the WM span from 9.41 ± 0.22 s to 9.42 ± 0.18 s (0.11% improvement) whereas lorazepam decreased WM to 9.32 ± 0.19 s (0.92% decrease). The schizophrenia subjects with their individual comedications who switched from placebo to lorazepam did 15.6 ± 1.1% worse (P < 0.01), whereas the subjects who switched to flumazenil did 3.9 ± 2.0% better (P = 0.089), in line with the clinical observations (Figure 2b,c). The relatively modest improvement with flumazenil is likely due to the limited contribution of the GABA-Aα2 subunit to the inhibitory tone (25% of the GABA-Aα1 contribution). Figure 2d shows the simulated effect of individual patient outcomes. For flumazenil, 8 out of 11 patients improved over their placebo values; in contrast all lorazepam patients deteriorated as compared with placebo.
Risperidone augmentation therapy in clozapine as independent validation
The second independent test was the simulation of a clinical study,16 in which 68 patients with schizophrenia on clozapine were randomized to augmentation therapy with placebo or 3 mg risperidone. Although there was no improvement on positive and negative symptoms in Schizophrenia clinical scales, risperidone-treated patients did significantly worse on cognitive outcomes. Using our receptor competition model, we first calculated functional brain concentration of risperidone and clozapine from cortical D2R radiotracer imaging with 11C-fallypride.25 We then calculated the postsynaptic non-D2R receptor activations for the combination therapy using the competition of neurotransmitter, clozapine, its metabolite N-desmethylclozapine, and the 9-OH risperidone metabolite, which accounts for up to 75% of the active moiety of risperdal,26 assuming no pharmacokinetic (PK) interaction between clozapine and risperidone.
Figure 3a shows the pharmacology of clozapine, N-desmethylclozapine, risperidone, and 9-OH risperidone.
Figure 3.

Pharmacology of clozapine and risperidone. (a) Radar plot of affinities for clozapine, N-desmethyclozapine, risperidone, and 9-OH risperidone in pKA values. High-affinity values are located at the periphery; i.e., 9-OH risperidone has the highest affinity for the D3R. As expected, the affinities of clozapine and its major metabolite are very similar as are the risperidone and 9-OH risperidone pharmacology. Note that N-desmethylclozapine is a partial agonist at the D2R (Emax = 32%), D3R (Emax = 50%), 5-HT1A (Emax = 69%), and various mAChR (Emax ranging between 32% and 100%). Virtual patient outcome. (b) Individual results from a virtual patient trial of 35 subjects taking clozapine and 35 subjects taking clozapine and risperidone. Adding risperidone clearly impairs working memory. The same effect is observed in the actual clinical trial, suggesting that the model captures the complex nonlinear pharmacodynamic interaction between the two compounds. CLO, clozapine; D3R, D3 receptor; Emax, maximum effect; mAChR, muscarinic acetylcholine receptor; pKA, logarithmic value for dissociation constant; RIS, risperidone.
We simulated the clinical trial conditions using a virtual patient trial with the same number of subjects (35) in each group with parameters sampled from Gaussian probability distributions of schizophrenia pathology-related changes, in addition to receptor activation levels changes that are likely due to PK variability and specific genotypes. For the latter, we started from published PK studies26 that suggested ranges of 50 and 30% around the average plasma levels for clozapine and risperidone, respectively. In addition, human genotypes such as catechol-O-methyltransferase (COMT)27 and the 5-HTTLPR promotor isoform28 determine the dynamics of dopamine, norepinephrine, and serotonin. With the receptor competition model, a range of receptor activations from which to sample individual subjects from a Gaussian distribution was defined.
The average WM for the clozapine placebo group was 8.27 ± 1.60 s (n = 35), while adding risperdal resulted in a 19% lower WM of 6.93 ± 1.55 s (P = 0.001). Close inspection of the underlying processes in the model suggest that this is likely due to a greater D1R inhibition (by the 9-OH risperidone metabolite) and greater presynaptic 5-HT1BR inhibition that tend to increase free 5-HT and activate 5-HT4, 5-HT3, and 5-HT6R. Increasing 5-HT4 works procognitively, but increasing 5-HT3 and 5-HT6 activity reduces cognitive performance.
Discussion
This report documents a computer-based mechanistic and biophysically realistic disease model of a cortical network involved in the maintenance of cognitive WM tasks. The output is the time over which a memory trace can be stabilized and introduction of the schizophrenia pathology is shown to reduce this readout. The effect of clinical interventions, such as specific antipsychotics in the presence of specific COMT genotypes in schizophrenics and the effect of anticholinergics and COMT inhibitors in healthy volunteers is calculated based on the corresponding changes in activation levels of all receptors, affected by the drugs. Optimizing the correlation found between the model outcome of the different interventions and the corresponding clinical results on the 2-back WM test allows for the calibration of the remaining biological parameters using a set of 17 published clinical experiments. When more publications with drug effects on the N-back WM test become available, we can use them for additional validation.
The best validation for such a platform would be to blindly and prospectively test the clinical outcome based on the primary pharmacology and target engagement data in humans, as we did successfully for a phase II outcome in schizophrenia14 and for a symptomatic Alzheimer's drug in a Ph I scopolamine-induced deficit.29 This, however, would necessitate a long-term collaboration with a pharmaceutical company over several years and so far we were unable to identify a project on cognitive impairment in schizophrenia. The next best test is to predict the outcome of retrospective data that have not been used for the calibration.
The first such test is the correct prediction of GABA modulators, suggesting that new targets can be introduced in a physiologically realistic way leading to reasonably good predictions. Indeed, flumazenil has been associated with an improvement in cognitive symptoms in humans,30 whereas first-generation and nonselective GABA allosteric potentiators like lorazepam have consistently shown a reduced cognitive performance even at subanesthetic doses.31 We believe our approach is successful because we have embedded these new targets in the model using preclinical and clinical neurophysiology information.
The second example includes the combination therapy of clozapine and risperidone. Many drugs have been tested in combination with clozapine for improving the performance of treatment-resistant schizophrenia. Although not significantly improving clinical efficacy, augmentation therapy leads to the introduction of serious side effects such as extra-pyramidal symptoms and cognitive deficits. Many of the existing antipsychotics do have a complex pharmacology that affects many receptor systems; in the case of risperidone and its active metabolite 9-OH risperidone, the additional effect on 5-HT3 and 5-HT6 receptor activation through its indirect block of the 5-HT1B presynaptic autoreceptor in addition to its higher D1R block can significantly impair the cognitive outcome. In addition, 9-OH risperidone's additional block of the 5-HT2AR is likely to be reduced by the increased free 5-HT level as a consequence of the presynaptic 5-HT1B autoreceptor block. The clinical result16 is recapitulated correctly by the model, suggesting that these anticognitive effects likely more than compensate for the procognitive effect of increased 5-HT1AR and 5-HT4R activation level.
A similar computational biophysical model has been proposed recently,32 in which schizophrenia pathology was introduced as a combination of glutamate and GABA changes. In our model, we have additionally implemented the neurophysiology of diverse neuromodulatory membrane-bound receptors, added the hypodopaminergic pathology and increased background noise, and finally calibrated the model with actual human clinical data on 17 therapeutic interventions so the model output can be related to clinical scales.
Another computational model33 describes the effect of GABA pathology on network activity and γ power oscillations. Although we do not model γ oscillations in this article, our result suggest a similar outcome. Reducing GABA tone by decreased synthesis through a deficit in GAD67, a key finding in schizophrenia,34 has the capacity to reduce network activity because of an asymmetric effect between inhibitory tone on interneurons and pyramidal cells. The conclusion that reducing GABA tone will always increase firing has to be interpreted with caution; using a biophysically realistic computer-based approach enables us to identify the underlying biological rationale.
In general, the WM task consists of sequential encoding, maintenance, and retrieval processes. A major limitation of the actual model is that it accounts only for the maintenance part of WM and therefore, we assume that the cognitive deficit is driven to a substantial degree by the decline in the maintenance process. A recent study35 suggests that cognitive deficits in schizophrenia are driven by dysfunctional context maintenance processes in an N-back test, whereas semantic inhibition was the major driver in a dysfunctional Stroop test. Alternatively, we can see this computational model as a representation of the stability of a memory trace in a brain region, the stability of which is essential for encoding or retrieval or processing. The fact that we can get such a robust correlation between this computational model and clinical data suggests that either the maintenance phase or the representation of the trace stability is fundamental to the different stages of the WM task to the extent that it drives a large part of the functional clinical outcome.
The sensitivity analysis suggests a major role for glutamatergic and GABAergic and to a lesser extent dopamine modulation in driving the schizophrenia pathology. The small changes in these parameters for our model is driven most importantly by the performance reduction of 1.5 SD below normal controls and is determined also by the excitatory–inhibitory balance of the network. Of note, clinical studies with glutamatergic modulators such as mGluR2 agonist or Glycine modulators in schizophrenia36,37 also suggest that this balance is tightly regulated.
More complex mathematical models for cognition, involving the interaction between different cortical regions and basal ganglia have been recently proposed.38 These models (Prefrontal cortex, Basal Ganglia WM, or PGWM model) make it possible to consider multiple cognitive representations to be accounted for and are “calibrated” using actual neuropsychological experiments with well-defined cognitive tasks, such as the AX-Continuous Performance Task. However, the level of detail of the biophysical representations does not allow for the pharmacological effects of drugs to be simulated. We plan, however, to include aspects of this more complete computer model for cognitive control in a future version, where we will explicitly bring in the neurophysiology of the direct and indirect pathway of the basal ganglia in addition to other subcortical brain regions. Another major limitation of our current model is the lack of differentiation between neocortex and hippocampus;39 the model is a generic version of a cortical or hippocampal recurrent network and certainly does not address the complex division of labor between these two brain regions.
Not all receptors supposed to have an effect on cognition are currently implemented—for example β-adrenergic receptors.40 The current model also has only two GABA-A subunit types and no GABA-B subunits41 that can have clinical benefit. Furthermore, as not many clinical trials using the Matrics scale have been published, it is not possible to calibrate the outcome in terms of this Food and Drug Administration-approved clinical scale. However, when these are published, we will be able to provide a better quantitative prediction of actual clinical scales using correlation with these clinical data.
Being aware of the differences in absolute affinities of drugs for human receptors reported in different labs, we derive these numbers mostly from the standardized human Psychoactive Drug Screening Program database. In this way, differences between individual drugs and neurotransmitters are consistent; in addition, we use normalization to calculate the effect of changed receptor activation levels on neurophysiological processes. Another problem is that target engagement of scopolamine, tolcapone, and mecamylamine in the human trials is not reported; usually these are titrated to yield a clinically relevant deficit.
Statistical data analysis or pharmacodynamic modeling of the drug effects on clinical outcomes has been used before,42 especially in those cases in which the outcome is dominated by one process, such as the D2 occupancy in schizophrenia psychosis. To our knowledge, this type of PK/pharmacodynamic modeling for predicting the effect of procognitive drugs in patients with schizophrenia has not been used in cognitive trials.
This Quantitative Systems Pharmacology platform can be used in CNS research and development to assess the quantitative clinical dose–response of multitarget drugs on cognitive outcome in WM paradigms long before a clinical trial is initiated. It also helps define the exclusion criteria for comedication in clinical trials. Alternatively, this approach can be used to estimate the cognitive impairment of off-target pharmacology of other types of medications targeted at positive and negative symptoms in Schizophrenia total.
Alternative approaches include systems biology analyses where large data sets are mined for statistical analysis and pattern recognition. We showed here that studying the correlation between RO of the same drugs and using the same clinical data set, the correlation is more than fivefold lower than with our quantitative systems pharmacology model, even when including the COMT genotype as an independent discrete variable. Although the traditional regression analysis approach assumes independent variables and linear interactions, the computer model takes into account many more nonlinear interactions, embedded in the neurophysiology (the threshold for an action potential) or the indirect effect between two neurotransmitter systems (i.e., one system influencing the release and dynamics of another neurotransmitter system).
In summary, this article presents a computer model that can be useful in supporting drug discovery and development for cognitive impairment in schizophrenia and in the rational management of polypharmacy.
Methods
Pharmacology of psychoactive compounds. The affinity parameters for each drug and neurotransmitter for human receptors were derived from the standardized Psychoactive Drug Screening Program database (http://pdsp.med.unc.edu/indexR.html). We implemented the active moiety of antipsychotics, taking into account the activity of metabolites. For instance N-desmethylclozapine, the major clozapine metabolite, accounts for 50% of the active moiety43 and has partial agonism at different receptors, such as 5-HT1A (maximum effect (Emax) 69%; EC50 = 501 nmol/l), and M1 mAChR (Emax 67%; EC50 = 42 nmol/l).44
The receptor competition model. To calculate the functional free concentration of the drug, we use the receptor competition model (see Supplementary Data online), ordinary differential equations that describe the competition between neurotransmitter, drug, metabolite, and a radiotracer with time-dependent changes in pre- and postsynaptic receptor activations, neurotransmitter, and drug levels in the synaptic cleft.45 Presynaptic neurotransmitter physiology is implemented using a phenomenological approach and calibrated with preclinical experiments using rapid-cyclic fast voltammetry, constrained by human imaging data.
The functional free intrasynaptic concentration of the drugs is determined by simulating the competition between total active moiety and tracer at the postsynaptic D2R in a positron emission tomography radiotracer displacement study with clinical data of various antipsychotics.25 This value for the active moiety is the functional intrasynaptic concentration that is dependent upon the PK properties of the drug (assuming steady-state plasma values), which can be used to calculate the postsynaptic receptor activation in other nondopaminergic D2 synapses (such as 5-HT or Ach). This change in the postsynaptic receptor activation drives the conductance change in voltage-mediated ion channels that drive the excitability of the network.
The effect of the COMTVal158Met genotype on dopamine and norepinephrine half-life in the cortex is simulated8 using human imaging data from the displacement of the D1-specific radiotracer NNC-112 in healthy subjects with different COMT genotypes.27
The cortical network. We extended a biophysically realistic model of a network of 20 four-compartment pyramidal cells and 10 two-compartment GABA interneurons12 with the receptor physiology of 18 different dopaminergic, serotonergic, noradrenergic, and cholinergic receptors21 and adjusted the relative fraction of inhibitory synapses46 (Figure 4a). This model has been calibrated from in vivo single-unit recordings in primates during a WM task and as such reduces some of the species–specific problems associated with a difference in inhibitory tone.
Figure 4.

Receptor neurophysiology in network model. (a) Schematic diagram of the connectivity and receptors in the prefrontal cortex network. The localization of different types of cholinergic, dopaminergic, serotonergic, noadrenergic, glutamate, and GABA receptors is shown according to their preclinical data. The memory stimulus “M” is given at 2,000 ms into the simulation and represents a sensory or conceptual stimulus that is introduced into the network. “B” is a background stimulus that represents the interaction of this particular network with the rest of the cortex and the brain and is described as a Poisson process. State diagram of network. (b) State diagram of the working model output. Each line of dots represents the activity of a single neuron, with each dot indicating an action potential occurring at that time for the neuron. For simplicity we only show 10 excited pyramidal neurons, 10 bystander pyramidal cells, and 10 GABA interneurons. At time 2,000 ms, a current (equivalent to a “memory” stimulus) is injected into the attractor neurons (lowest 10 lines); these neurons then fire in synchrony for a certain period until the stochastic noise starts to deteriorate. From bottom to top: 10 pyramidal neurons that are stimulated with the memory stimulus, 10 nonstimulated pyramidal cells, and 10 interneurons. The time period over which the 10 stimulated pyramidal cells fire action potential independently is defined as the working memory span. GABA, γ-aminobutyric acid.
Synchronous firing of the target pyramidal cells is initiated by injecting a transient current at t = 2,000 ms. The network then fires in a synchronized pattern before it gets degraded by the background noise and the interference of the distractor neurons. This WM span (see Supplementary Data online and Figure 4b), is usually in the range of 4–10 s and corresponds to the time a certain pattern is held in WM.47
Implementation of receptor pharmacology. We assume a linear normalized relationship between receptor activation and biological effect on physiological responses such as 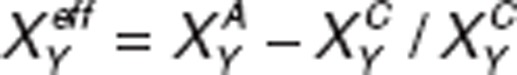 ; where
; where 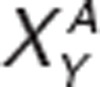 and
and 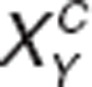 are the actual activation levels of receptor XY (for instance D1) in placebo (C) and after treatment (A). The modified conductances for ion-channel Z downstream of a specific receptor are given by
are the actual activation levels of receptor XY (for instance D1) in placebo (C) and after treatment (A). The modified conductances for ion-channel Z downstream of a specific receptor are given by
 |
where ParamZY is an adjustable parameter (see below).
The model includes the physiology of the dopamine D1, D2, and D4 receptors, the serotonin 5-HT1A, 5-HT2A, 5-HT3, 5-HT4, and 5-HT6 receptors, the adrenergic α2AR and the cholinergic M1, M2 mAChR, the α7 and α4β2 nAChR in a quantitative way from published preclinical studies (see Supplementary Data online).
Calibrating the model. Schizophrenia pathology is implemented using a hypodopaminergic cortical D1R tone,48 N-methyl-D-aspartate hypofunction,18 documented by a hypocortical-hyperstriatal imbalance in metabolic imaging,19 a GABA deficit20 applied here to the network interneurons, and a noisier background signal,17 resulting in a clinical cognitive deficit that is dependent upon the clinical readout, but on average is 1.5 SDs lower than healthy controls.49
We will simulate 100 different virtual “normal subjects” with variable parameters for the four processes, allowing us to determine a value of 1.5 SDs and the corresponding minimal change.
In a second step, all remaining 11 free biological coupling factors ParamYX (two for 5-HT, three for Ach, two for DA, two for glutamate, and two for GABA) are calibrated using clinical data (Supplementary Data online) on the N-back WM test in schizophrenia patients and healthy controls with anticholinergics or COMT inhibitors, and stratified according to the COMT genotype.
The calibration is performed using “design-of-experiment” statistical techniques.50 A good robust approach uses only 2n simulations, where n is the number of free parameters, as compared with 2n for a one-factor-at-a-time design.
The Pareto-effect, Parj (j = 1··n), is the difference Average+j − Average−j and indicates both the strength and the sign of the gradient toward the optimum. Here, Average+j =  where Posij = 1 if eij = “P” and Posij = 0 if eij = “M” and an opposite definition for Average−j, with eij a [2nxn] matrix of “P” and “M.”
where Posij = 1 if eij = “P” and Posij = 0 if eij = “M” and an opposite definition for Average−j, with eij a [2nxn] matrix of “P” and “M.”
Author contributions
H.G. wrote the manuscript and designed the research; A.S. performed the research; H.G., A.S., and P.R. analyzed the data; and A.S. and P.R. contributed new reagents/analytical tools.
Conflict of interest
All authors are employees of In Silico Biosciences, Inc.
Study Highlights

Supplementary Material
References
- Gold J.M., Harvey P.D. Cognitive deficits in schizophrenia. Psychiatr. Clin. North Am. 1993;16:295–312. [PubMed] [Google Scholar]
- Nuechterlein K.H., Barch D.M., Gold J.M., Goldberg T.E., Green M.F., Heaton R.K. Identification of separable cognitive factors in schizophrenia. Schizophr. Res. 2004;72:29–39. doi: 10.1016/j.schres.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Buchanan R.W., Freedman R., Javitt D.C., Abi-Dargham A., Lieberman J.A. Recent advances in the development of novel pharmacological agents for the treatment of cognitive impairments in schizophrenia. Schizophr. Bull. 2007;33:1120–1130. doi: 10.1093/schbul/sbm083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt D.C., et al. Effect of the neuroprotective peptide davunetide (AL-108) on cognition and functional capacity in schizophrenia. Schizophr. Res. 2012;136:25–31. doi: 10.1016/j.schres.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Goff D.C., et al. A placebo-controlled add-on trial of the Ampakine, CX516, for cognitive deficits in schizophrenia. Neuropsychopharmacology. 2008;33:465–472. doi: 10.1038/sj.npp.1301444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weickert T.W., et al. Catechol-O-methyltransferase val108/158met genotype predicts working memory response to antipsychotic medications. Biol. Psychiatry. 2004;56:677–682. doi: 10.1016/j.biopsych.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Tammimäki A., Männistö P.T. Effect of genetic modifications in the synaptic dopamine clearance systems on addiction-like behaviour in mice. Basic Clin. Pharmacol. Toxicol. 2011;108:2–8. doi: 10.1111/j.1742-7843.2010.00647.x. [DOI] [PubMed] [Google Scholar]
- Spiros, A., Geerts H. A quantitative way to estimate clinical off-target effects for human membrane brain targets in CNS research and development. J. Exp Pharmacol. 2012;4:53–61. doi: 10.2147/JEP.S30808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povysheva N.V., Zaitsev A.V., Rotaru D.C., Gonzalez-Burgos G., Lewis D.A., Krimer L.S. Parvalbumin-positive basket interneurons in monkey and rat prefrontal cortex. J. Neurophysiol. 2008;100:2348–2360. doi: 10.1152/jn.90396.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitsev A.V., et al. Interneuron diversity in layers 2-3 of monkey prefrontal cortex. Cereb. Cortex. 2009;19:1597–1615. doi: 10.1093/cercor/bhn198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agoram B.M., Demin O. Integration not isolation: arguing the case for quantitative and systems pharmacology in drug discovery and development. Drug Discov. Today. 2011;16:1031–1036. doi: 10.1016/j.drudis.2011.10.001. [DOI] [PubMed] [Google Scholar]
- Durstewitz D., Seamans J.K., Sejnowski T.J. Dopamine-mediated stabilization of delay-period activity in a network model of prefrontal cortex. J. Neurophysiol. 2000;83:1733–1750. doi: 10.1152/jn.2000.83.3.1733. [DOI] [PubMed] [Google Scholar]
- Carter C.S., Perlstein W., Ganguli R., Brar J., Mintun M., Cohen J.D. Functional hypofrontality and working memory dysfunction in schizophrenia. Am. J. Psychiatry. 1998;155:1285–1287. doi: 10.1176/ajp.155.9.1285. [DOI] [PubMed] [Google Scholar]
- Geerts H., Spiros A., Roberts P., Twyman R., Alphs L., Grace A.A. Blinded prospective evaluation of computer-based mechanistic schizophrenia disease model for predicting drug response. PLoS ONE. 2012;7:e49732. doi: 10.1371/journal.pone.0049732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies L., et al. Effects of gamma-aminobutyric acid-modulating drugs on working memory and brain function in patients with schizophrenia. Arch. Gen. Psychiatry. 2007;64:156–167. doi: 10.1001/archpsyc.64.2.156. [DOI] [PubMed] [Google Scholar]
- Honer W.G., Clozapine and Risperidone Enhancement (CARE) Study Group et al. Clozapine alone versus clozapine and risperidone with refractory schizophrenia. N. Engl. J. Med. 2006;354:472–482. doi: 10.1056/NEJMoa053222. [DOI] [PubMed] [Google Scholar]
- Winterer G., et al. Schizophrenia: reduced signal-to-noise ratio and impaired phase-locking during information processing. Clin. Neurophysiol. 2000;111:837–849. doi: 10.1016/s1388-2457(99)00322-3. [DOI] [PubMed] [Google Scholar]
- Coyle J.T. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell. Mol. Neurobiol. 2006;26:365–384. doi: 10.1007/s10571-006-9062-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Lindenberg A., et al. Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat. Neurosci. 2002;5:267–271. doi: 10.1038/nn804. [DOI] [PubMed] [Google Scholar]
- Volk D.W., Lewis D.A. Impaired prefrontal inhibition in schizophrenia: relevance for cognitive dysfunction. Physiol. Behav. 2002;77:501–505. doi: 10.1016/s0031-9384(02)00936-8. [DOI] [PubMed] [Google Scholar]
- Roberts P.D., Spiros A., Geerts H. Simulations of symptomatic treatments for Alzheimer's disease: computational analysis of pathology and mechanisms of drug action. Alzheimers. Res. Ther. 2012;4:50. doi: 10.1186/alzrt153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- File S.E., Pellow S. Intrinsic actions of the benzodiazepine receptor antagonist Ro 15-1788. Psychopharmacology (Berl.) 1986;88:1–11. doi: 10.1007/BF00310505. [DOI] [PubMed] [Google Scholar]
- Beneyto M., Abbott A., Hashimoto T., Lewis D.A. Lamina-specific alterations in cortical GABA(A) receptor subunit expression in schizophrenia. Cereb. Cortex. 2011;21:999–1011. doi: 10.1093/cercor/bhq169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Esquivel D.F., et al. Plasma levels of clozapine and norclozapine in Mexican schizophrenia patients. Arzneimittelforschung. 2011;61:335–339. doi: 10.1055/s-0031-1296207. [DOI] [PubMed] [Google Scholar]
- Xiberas X., et al. Extrastriatal and striatal D(2) dopamine receptor blockade with haloperidol or new antipsychotic drugs in patients with schizophrenia. Br. J. Psychiatry. 2001;179:503–508. doi: 10.1192/bjp.179.6.503. [DOI] [PubMed] [Google Scholar]
- Mannens G., Huang M.L., Meuldermans W., Hendrickx J., Woestenborghs R., Heykants J. Absorption, metabolism, and excretion of risperidone in humans. Drug Metab. Dispos. 1993;21:1134–1141. [PubMed] [Google Scholar]
- Slifstein M., et al. COMT genotype predicts cortical-limbic D1 receptor availability measured with [11C]NNC112 and PET. Mol. Psychiatry. 2008;13:821–827. doi: 10.1038/mp.2008.19. [DOI] [PubMed] [Google Scholar]
- Fisher P.M., et al. 5-HTTLPR status predictive of neocortical 5-HT4 binding assessed with [(11)C]SB207145 PET in humans. Neuroimage. 2012;62:130–136. doi: 10.1016/j.neuroimage.2012.05.013. [DOI] [PubMed] [Google Scholar]
- Nicholas T., et al. Systems pharmacology modeling in 5HT4: prediction and outcome of a clinical scopolamine impairment trial and further application to Alzheimer's disease pathology. Alzheimer's & Dementia:The Journal of the Alzheimer's Association. 2012;8 (suppl.):400. [Google Scholar]
- Girdler N.M., et al. A randomised crossover trial of post-operative cognitive and psychomotor recovery from benzodiazepine sedation: effects of reversal with flumazenil over a prolonged recovery period. Br. Dent. J. 2002;192:335–9; discussion 331. doi: 10.1038/sj.bdj.4801369. [DOI] [PubMed] [Google Scholar]
- Stonnington C.M., Snyder P.J., Hentz J.G., Reiman E.M., Caselli R.J. Double-blind crossover study of the cognitive effects of lorazepam in healthy apolipoprotein E (APOE)-epsilon4 carriers. J. Clin. Psychiatry. 2009;70:1379–1384. doi: 10.4088/JCP.08m04593. [DOI] [PubMed] [Google Scholar]
- Cano-Colino M., Compte A. A computational model for spatial working memory deficits in schizophrenia. Pharmacopsychiatry. 2012;45 (suppl. 1):S49–S56. doi: 10.1055/s-0032-1306314. [DOI] [PubMed] [Google Scholar]
- Volman V., Behrens M.M., Sejnowski T.J. Downregulation of parvalbumin at cortical GABA synapses reduces network gamma oscillatory activity. J. Neurosci. 2011;31:18137–18148. doi: 10.1523/JNEUROSCI.3041-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa E., et al. A GABAergic cortical deficit dominates schizophrenia pathophysiology. Crit. Rev. Neurobiol. 2004;16:1–23. doi: 10.1615/critrevneurobiol.v16.i12.10. [DOI] [PubMed] [Google Scholar]
- Chan R.C., Huang J., Guo L., Cao X., Hong X., Gao Z. Executive control in schizophrenia in task involving semantic inhibition and working memory. Psychiatry Res. 2010;179:259–266. doi: 10.1016/j.psychres.2009.07.014. [DOI] [PubMed] [Google Scholar]
- Kinon B.J., HBBI Study Group et al. A multicenter, inpatient, phase 2, double-blind, placebo-controlled dose-ranging study of LY2140023 monohydrate in patients with DSM-IV schizophrenia. J. Clin. Psychopharmacol. 2011;31:349–355. doi: 10.1097/JCP.0b013e318218dcd5. [DOI] [PubMed] [Google Scholar]
- Singh S.P., Singh V. Meta-analysis of the efficacy of adjunctive NMDA receptor modulators in chronic schizophrenia. CNS Drugs. 2011;25:859–885. doi: 10.2165/11586650-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Hazy T.E., Frank M.J., O'Reilly R.C. Towards an executive without a homunculus: computational models of the prefrontal cortex/basal ganglia system. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 2007;362:1601–1613. doi: 10.1098/rstb.2007.2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly R.C., Bhattacharyya R., Howard M.D., Ketz N. Complementary Learning Systems. Cogn. Sci. 2011. pp. 1–20. [DOI] [PubMed]
- Yu J.T., Wang N.D., Ma T., Jiang H., Guan J., Tan L. Roles of ß-adrenergic receptors in Alzheimer's disease: implications for novel therapeutics. Brain Res. Bull. 2011;84:111–117. doi: 10.1016/j.brainresbull.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Bowery N.G. GABAB receptor: a site of therapeutic benefit. Curr. Opin. Pharmacol. 2006;6:37–43. doi: 10.1016/j.coph.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Kimko H.C., Reele S.S., Holford N.H., Peck C.C. Prediction of the outcome of a phase 3 clinical trial of an antischizophrenic agent (quetiapine fumarate) by simulation with a population pharmacokinetic and pharmacodynamic model. Clin. Pharmacol. Ther. 2000;68:568–577. doi: 10.1067/mcp.2000.110975. [DOI] [PubMed] [Google Scholar]
- Tadori Y., Forbes R.A., McQuade R.D., Kikuchi T. In vitro pharmacology of aripiprazole, its metabolite and experimental dopamine partial agonists at human dopamine D2 and D3 receptors. Eur. J. Pharmacol. 2011;668:355–365. doi: 10.1016/j.ejphar.2011.07.020. [DOI] [PubMed] [Google Scholar]
- Lameh J., Burstein E.S., Taylor E., Weiner D.M., Vanover K.E., Bonhaus D.W. Pharmacology of N-desmethylclozapine. Pharmacol. Ther. 2007;115:223–231. doi: 10.1016/j.pharmthera.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Spiros A., Carr R., Geerts H. Not all partial dopamine D(2) receptor agonists are the same in treating schizophrenia. Exploring the effects of bifeprunox and aripiprazole using a computer model of a primate striatal dopaminergic synapse. Neuropsychiatr. Dis. Treat. 2010;6:589–603. doi: 10.2147/NDT.S12460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFelipe J. Cortical interneurons: from Cajal to 2001. Prog. Brain Res. 2002;136:215–238. doi: 10.1016/s0079-6123(02)36019-9. [DOI] [PubMed] [Google Scholar]
- Levy R., Goldman-Rakic P.S. Segregation of working memory functions within the dorsolateral prefrontal cortex. Exp. Brain Res. 2000;133:23–32. doi: 10.1007/s002210000397. [DOI] [PubMed] [Google Scholar]
- Durstewitz D., Seamans J.K. The dual-state theory of prefrontal cortex dopamine function with relevance to catechol-o-methyltransferase genotypes and schizophrenia. Biol. Psychiatry. 2008;64:739–749. doi: 10.1016/j.biopsych.2008.05.015. [DOI] [PubMed] [Google Scholar]
- Saykin A.J., et al. Neuropsychological deficits in neuroleptic naive patients with first-episode schizophrenia. Arch. Gen. Psychiatry. 1994;51:124–131. doi: 10.1001/archpsyc.1994.03950020048005. [DOI] [PubMed] [Google Scholar]
- Box H.C., Hunter W., Hunter S.Statistics for Experimenters1st edn.,(Wiley Series in Probability and Mathematical Statistics, Hoboken, NJ,1978
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.