

Abstract
Overactivation of c-Jun N-terminal kinase (JNK)/c-Jun signaling is a central mechanism of hepatocyte injury and death including that from oxidative stress. However, the functions of JNK and c-Jun are still unclear, and this pathway also inhibits hepatocyte death. Previous studies of menadione-induced oxidant stress demonstrated that toxicity resulted from sustained JNK/c-Jun activation as death was blocked by the c-Jun dominant negative TAM67. To further delineate the function of JNK/c-Jun signaling in hepatocyte injury from oxidant stress, the effects of direct JNK inhibition on menadione-induced death were examined. In contrast to the inhibitory effect of TAM67, pharmacological JNK inhibition by SP600125 sensitized the rat hepatocyte cell line RALA255-10G to death from menadione. SP600125 similarly sensitized mouse primary hepatocytes to menadione toxicity. Death from SP600125/menadione was c-Jun dependent as it was blocked by TAM67, but independent of c-Jun phosphorylation. Death occurred by apoptosis and necrosis and activation of the mitochondrial death pathway. Short hairpin RNA knockdowns of total JNK or JNK2 sensitized to death from menadione, whereas a jnk1 knockdown was protective. Jnk2 null mouse primary hepatocytes were also sensitized to menadione death. JNK inhibition magnified decreases in cellular ATP content and β-oxidation induced by menadione. This effect mediated cell death as chemical inhibition of β-oxidation also sensitized cells to death from menadione, and supplementation with the β-oxidation substrate oleate blocked death. Components of the JNK/c-Jun signaling pathway have opposing functions in hepatocyte oxidant stress with JNK2 mediating resistance to cell death and c-Jun promoting death.
Keywords: APOPTOSIS, ATP, FATTY ACID OXIDATION, MENADIONE, NECROSIS
INTRODUCTION
Overactivation of the mitogen-activated protein kinase (MAPK) c-Jun N-terminal kinase (JNK) is a mechanism of hepatocyte death from central mediators of liver injury such as oxidants and tumor necrosis factor α (TNF) [Czaja et al., 2003; Gunawan et al., 2006; Liu et al., 2002; Malhi et al., 2006; Schwabe et al., 2004; Xu et al., 1997]. Two JNK genes, jnk1 and jnk2, each encode for four distinct JNK isoforms expressed in all cells including hepatocytes [Czaja, 2010]. The classic function of JNK is to phosphorylate and activate c-Jun to increase AP-1 gene transcription, but JNK regulates cellular responses through other mechanisms such as altering protein degradation [Chang et al., 2006; Kodama et al., 2009b]. The mechanisms underlying the effects of JNK/c-Jun on hepatocyte death, and how the specific jnk isoforms individually regulate this process, remain unclear.
Recent investigations have yielded varied findings on the functions of the two JNK genes in liver injury. JNK1 was initially reported to promote death from toxins such as galactosamine [Chang et al., 2006]. However, we demonstrated that JNK2 mediates death from galactosamine plus lipopolysaccharide [Wang et al., 2006], a finding confirmed by others in this model [Kodama et al., 2009b], additional hepatotoxic models [Gunawan et al., 2006], and in other forms of liver injury such as ischemia/reperfusion [Devey et al., 2009; Theruvath et al., 2008]. However, JNK function in hepatic injury varies depending on the setting of the injury and the stimulus. For example, in contrast to the findings from liver injury induced by toxins or ischemia/reperfusion, JNK1 promotes liver injury in hepatic steatosis, whereas JNK2 is protective [Schattenberg et al., 2006; Singh et al., 2009c]. Contributions of JNK in different hepatic cell types may explain in part some of these differences [Kluwe et al., 2010; Kodama et al., 2009a]. Distinct JNK functions that promote or inhibit hepatocyte death depending on the injury, may also explain these conflicting findings and others suggesting that JNK does not modulate injury [Hong et al., 2009].
Studies of JNK function have been performed with the pharmacological JNK inhibitor SP600125 [Bennett et al., 2001] or in jnk1 and jnk2 knockout mice or primary hepatocytes. SP600125 cannot differentiate between JNK1 and JNK2 effects and may have off-target effects [Bain et al., 2003]. Studies of jnk knockout mice and cells differentiate JNK1 and JNK2 functions, but these models may have unknown compensatory responses from the loss of these critical genes. Another strategy has been to block the downstream JNK effector c-Jun with the dominant negative protein TAM67 [Bradham et al., 2001]. The present study was predicated on the finding that SP600125 and TAM67 had opposing effects on hepatocyte death from menadione-induced oxidant stress. To resolve this apparent contradiction, we generated JNK isoform-specific knockdowns in a hepatocyte cell line and found that JNK2 was hepatoprotective whereas c-Jun promoted death. These findings demonstrate that JNK and c-Jun have distinct, opposing functions in oxidant-induced hepatocyte death which has important implications for the manipulation of this signaling pathway to prevent liver injury.
MATERIALS AND METHODS
CELL CULTURE AND TREATMENTS
The adult rat hepatocyte line RALA255-10G (RALA hepatocytes) is conditionally immortalized with a mutant SV40 virus expressing a temperature sensitive T antigen [Chou, 1983]. Cells were routinely cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Mediatech, Manassas, VA) supplemented with 4% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA), and antibiotics (Invitrogen, Carlsbad, CA) at the permissive temperature of 33°C. For experiments cells were plated and cultured at 33°C for 24 h, and then cultured in DMEM, 2% fetal bovine serum, antibiotics and 1 μM dexamethasone (Sigma, St. Louis, MO) at the restrictive temperature of 37°C, as previously described [Jones et al., 2000]. Under these culture conditions, T antigen expression is suppressed, the cells are nontransformed, and become differentiated [Chou, 1983]. Cells were then placed in serum-free medium containing dexamethasone for 18 h prior to the start of an experiment.
Mouse primary hepatocytes from C57BL/6, jnk1−/− or jnk2−/− mice (Jackson Labs, Bar Harbor, ME) were obtained by collagenase perfusion [Xu et al., 1998b]. The cells were purified by Percoll (Sigma) density centrifugation and cultured in RPMI 1640 with 5% fetal bovine serum, 1.7 μM insulin, 1 μM dexamethasone and antibiotics on Biocoat plates (Becton Dickinson, Franklin Lakes, NJ). The medium was changed 3 h after plating and again at 18 h at which time experiments were started. All studies were approved by the Animal Care and Use Committee of the Albert Einstein College of Medicine and followed the National Institutes of Health guidelines on the care and use of animals.
Cells were treated with menadione (Sigma) at the concentrations indicated, SP600125 (Calbiochem, San Diego, CA) at a 10 μM concentration unless otherwise noted, 15 ng/ml TNF (R&D Systems, Minneapolis, MN), 50 ng/ml phorbol myristate acetate (Sigma), 10 μM Q-Val-Asp-OPh (Q-VD-OPh; MP Biomedicals, Aurora, OH), 25 μM etomoxir and 2.5 mM 2-deoxyglucose (Sigma). Unless otherwise indicated, cells were pretreated with SP600125, Q-VD-OPh or etomoxir for 1 h prior to menadione administration. Control cells received an equivalent amount of dimethyl sulfoxide vehicle alone for studies involving SP600125 or Q-VD-OPh. Oleic acid (Sigma) was conjugated to bovine serum albumin as previously described [Singh et al., 2009a], and used at a 250 μM concentration.
ADENOVIRUSES
Adenoviruses were grown in 293 cells, purified by banding twice on CsCl gradients, tittered by plaque assay and used to infect RALA hepatocytes at an MOI of 20, as previously described [Jones et al., 2000; Xu et al., 1998a]. As a control, cells were infected with the adenovirus Ad5LacZ which expresses the β-galactosidase gene [Bradham et al., 2001]. Cells were also infected with Ad5TAM which expresses TAM-67, a dominant negative c-Jun [Bradham et al., 2001], or viruses expressing Bcl-2 or Bcl-XL [Liedtke et al., 2002; Shinoura et al., 2000].
JNK KINASE ASSAY
JNK activity was measured by commercial kit according to the manufacturer’s instructions (Cell Signaling, Beverly, MA). JNK was immobilized from cell lysates by binding to an N-terminal c-Jun (1–89) fusion protein bound to glutathione sepharose beads. The kinase reaction was performed in the presence of cold ATP using the c-Jun fusion protein as a substrate. The reaction products were resolved on 10% SDS-polyacrylamide gels, and the amount of phosphorylated c-Jun detected with an antibody specific for c-Jun phosphorylated at serine 63. Total c-Jun levels were determined to assess the equivalency of loading among samples.
3-(4,5-DIMETHYLTHIAZOL-2-YL)-2,5-DIPHENYLTETRAZOLIUM BROMIDE (MTT) ASSAY
Cell death was quantified by MTT assay [Xu et al., 1998a]. At 24 h after menadione treatment, the cell culture medium was replaced by an equal volume of a 1 mg/ml MTT solution, pH 7.4, in DMEM. After incubation at 37°C for 1 h, the MTT solution was discarded, the formazan product solubilized with 1-propanol and the absorbance of this compound measured at 560 nm in a spectrophotometer. The percentage of cell death was determined by dividing the optical density of the treated group by the optical density for untreated, control cells, multiplying by 100, and subtracting that number from 100.
FLUORESCENCE MICROSCOPY
The steady-state levels of apoptotic and necrotic cells were quantified by examining acridine orange and ethidium bromide costained cells under fluorescence microscopy, as previously described [Xu et al., 1996]. Cells with condensed or fragmented nuclei and a shrunken cytoplasm by acridine orange staining were considered apoptotic, and necrotic cells were detected by positive staining with ethidium bromide. A minimum of 400 cells per dish were examined, and the numbers of apoptotic and necrotic cells expressed as a percentage of the total number of cells.
PROTEIN ISOLATION AND WESTERN BLOTTING
Total protein was isolated from cells as described previously [Wang et al., 2004]. Protein concentrations were determined by the Bio-Rad (Hercules, CA) protein assay according to the manufacturer’s instructions. Western blotting was performed, as previously described [Singh et al., 2009b]. Nitrocellulose membranes were exposed to antibodies that recognized phosphorylated and total JNK1 and JNK2, phosphorylated and total c-Jun (Santa Cruz Biotechnology, Santa Cruz, CA and Abcam, Cambridge, MA), phosphorylated and total ERK1/2 (Cell Signaling, Beverly, MA). Membranes were stripped and reprobed with β-actin (Abcam) to assess for equivalent loading among samples.
Mitochondrial and cytosolic protein fractions were isolated as previously described [Wang et al., 2006]. Western blotting was performed as above with antibodies to cytochrome c (BD Biosciences, San Diego, CA), cytochrome oxidase (MitoSciences, Eugene, OR) and tubulin (Cell Signaling).
LUCIGENIN ASSAY
ROS production was determined by lucigenin chemiluminescence. Cells were exposed to 1 mg/ml of lucigenin in Krebs-Ringer solution. Chemiluminescence was measured in a microplate reader and normalized to cellular protein.
LENTIVIRUS CONSTRUCTION AND INFECTION
The nucleotide sequences shown in Table 1 were used for shRNAs to jnk, jnk1 and jnk2. Oligonucleotides were annealed and cloned into the BglII-XhoI site of pSUPER (Ambion). The SmaI-XhoI fragments of the corresponding pSUPER plasmids, which included the H1 promoter-shRNA cassette, were subcloned into the EcoRV-XhoI sites of the lentiviral vector pCCL.sin.PPT.hPGK.GFPWpre [Piva et al., 2006].
Table 1.
shRNA Sequences
| jnk | 5′-GCATGGGCTACAAGGAGAA-3′ |
| jnk1 | 5′-GGAATAGTGTGTGCAGCTT-3′ |
| jnk2 | 5′-GGAATTGTTTGTGCTGCTT-3′ |
High titer lentiviral stocks were produced by calcium phosphate-mediated transfection of the modified transfer vectors and the packaging vectors pMDLg/pRRE, pRSV-Rev and pMD2.VSVG into HEK-293T cells. Supernatants were harvested over 36 to 48 h, titered by plaque assay and used at an MOI of 5 to infect RALA hepatocytes. The efficiency of infection was determined at 72 h by determining the numbers of GFP positive cells under fluorescence microscopy which exceeded 98% for all constructs. All studies were performed in stably-infected cells.
ATP ASSAY
Intracellular ATP concentrations were determined by colorimetric kit (BioVision, Mountain View, CA) using the manufacturer’s instructions. Values were normalized to protein concentration and expressed as values relative to untreated control cells.
LACTATE ASSAY
Levels of lactate in the cell culture medium were assayed by commercial kit (BioVision) using the manufacturer’s instructions. Lactate production was measured over a 2 or 4 h period and levels normalized to total cellular protein.
β-OXIDATION ASSAY
Rates of fatty acid β-oxidation were determined by a modification of the method of Hoppel et al. [Hoppel et al., 1979], as previously described [Singh et al., 2009a]. Rates of β-oxidation were measured from the production of radioactive carbon dioxide from the oxidation of [14C]oleate. Rates are expressed as cpm normalized to total protein.
STATISTICAL ANALYSIS
The numerical results are reported as mean ± S.E. and derived from at least three independent experiments. Groups were compared by the Student’s t-test with statistical significance defined as P<0.05 using Sigma Plot (Jandel Scientific, San Rafael, CA).
RESULTS
PHARMACOLOGICAL JNK INHIBITION SENSITIZES RALA HEPATOCYTES TO MENADIONE-INDUCED CELL DEATH
Previous studies demonstrated that RALA [Czaja et al., 2003; Wang et al., 2004] and primary [Conde de la Rosa et al., 2006] hepatocytes are sensitized to oxidant-induced cell death from the superoxide generator menadione by sustained JNK/c-Jun activation. In RALA hepatocytes death was blocked by the c-Jun dominant negative TAM67, suggesting that JNK-induced activation of c-Jun mediates cell death [Czaja et al., 2003]. However, in light of findings that TAM67 can directly inhibit JNK as well as c-Jun [Reuther-Madrid et al., 2002], we reevaluated the roles of JNK and c-Jun in oxidant-induced hepatocyte death.
Initially we investigated whether TAM67 affected menadione-induced JNK activation. Similar to fibroblasts [Reuther-Madrid et al., 2002], adenoviral TAM67 expression inhibited JNK activation in RALA hepatocytes as determined by in vitro kinase activity assay employing c-Jun as substrate. In cells infected with the control virus Ad5LacZ, JNK activity increased with menadione treatment as indicated by increased c-Jun phosphorylation on immunoblots (Fig. 1A). TAM67 is a c-Jun truncation mutant lacking amino acids 3-122 [Brown et al., 1993]. As expected, Ad5TAM did not alter wild-type 43 kD c-Jun levels on immunoblots probed with an amino-terminal anti-c-Jun antibody (Fig. 1B). TAM67 was detected with a carboxy-terminal anti-c-Jun antibody in Ad5TAM-infected cells as a series of 20–30 kD bands representing post-translationally modified TAM67 proteins (Fig. 1B), as previously reported [Brown et al., 1993]. TAM67 decreased basal JNK activity in untreated cells, and blocked the menadione-induced increase (Fig. 1A). These data suggested that our previous findings with TAM67 may have resulted from direct effects of this construct on JNK rather than c-Jun.
Fig. 1.
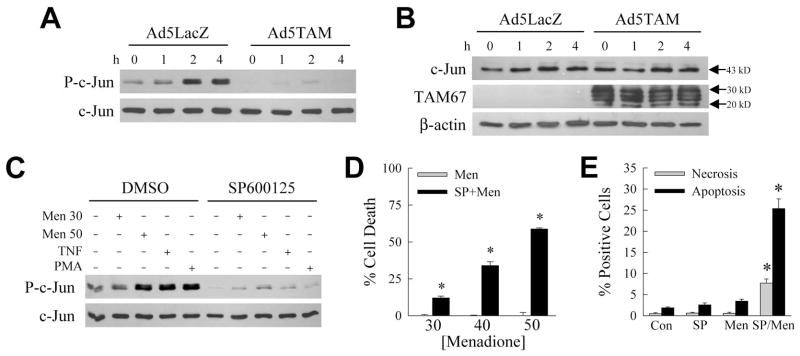
SP600125 sensitizes RALA hepatocytes to menadione-induced death despite inhibiting JNK. A: Cells were infected with control virus Ad5LacZ or Ad5TAM and treated with menadione for the indicated number of hours. JNK activity was determined by an in vitro kinase assay with c-Jun as the substrate. Activity was assayed by immunoblots for phosphorylated c-Jun (P-c-Jun), and membranes were reprobed for total c-Jun to assess loading equivalency. B: Proteins from Ad5LacZ- and Ad5TAM-infected cells treated with menadione for the indicated hours and immunoblotted with an amino-terminal anti-c-Jun antibody for wild-type c-Jun (c-Jun) and a carboxy-terminal anti-c-Jun antibody that detects the truncated c-Jun TAM67 (TAM67). β-actin is a loading control. C: JNK assay of cells pretreated with vehicle dimethyl sulfoxide (DMSO) or SP600125 alone, or in combination with 30 or 50 μM menadione (Men), TNF, or phorbol myristate acetate (PMA). D: Percentage cell death in RALA hepatocytes treated with menadione alone (Men) or with SP600125 (SP+Men) at the indicated μM concentrations of menadione for 24 h (*P<0.0002 as compared to menadione alone; n=5). E: Percentages of untreated control (Con) cells, or cells treated for 12 h with SP600125 (SP) or 50 μM menadione (Men) alone, or in combination (SP/Men), that were positive by acridine orange/ethidium bromide costaining for necrosis or apoptosis under fluorescence microscopy (*P<0.0001 as compared to menadione alone; n=6).
To determine the direct actions of JNK on hepatocyte death from menadione, we examined the effects of the JNK inhibitor SP600125 [Bennett et al., 2001]. Pretreatment with 10 μM SP600125 decreased JNK activity in cells that were untreated or treated with nontoxic (30 μM) and toxic (50 μM) menadione concentrations or the JNK inducers TNF and phorbol myristate acetate (Fig. 1C). Concentrations of 20–100 μM SP600125 were only slightly more effective in blocking JNK, and 50–100 μM concentrations were toxic (data not shown). With the efficacy of SP600125 established, its ability to block death from menadione was examined with the expectation that similar to TAM67 this chemical would prevent death that was mediated by JNK directly, or JNK-dependent c-Jun activation. Surprisingly, rather than protecting against death, SP600125 sensitized RALA hepatocytes to death from 30–50 μM menadione as determined by MTT assay (Fig. 1D). The cytotoxicity of SP600125 and 50 μM menadione was confirmed by fluorescence microscopy of acridine orange/ethidium bromide costained cells to determine steady-state levels of necrosis and apoptosis. SP600125 or 50 μM menadione alone failed to induce necrosis or apoptosis (Fig. 1E). In contrast, SP600125/menadione cotreatment significantly increased the numbers of cells undergoing necrosis and apoptosis (Fig. 1E). The majority of cell death occurred through the typical menadione-induced cell death cascade mediated by the mitochondrial death pathway and caspase activation. The menadione-induced reduction in mitochondrial cytochrome c and release into the cytoplasm were increased by SP600125 cotreatment (Fig. 2A). Adenoviral over expression of Bcl-2 or Bcl-XL, which block the mitochondrial death pathway, inhibited death by 65–80% (Fig. 2B). An equivalent decrease was achieved with the caspase inhibitor Q-VD-OPh (Fig. 2C). In contrast to the protective effect of the c-Jun dominant negative TAM67, JNK inhibition by SP600125 had the diametrically opposed effect of sensitizing to death from oxidant stress in RALA hepatocytes.
Fig. 2.

Death from SP600125/menadione occurs through mitochondrial death pathway-mediated caspase activation. A: RALA hepatocytes were treated with 50 μM menadione and SP600125 alone or in combination for 8 h. Cytosolic and mitochondrial protein fractions were immunoblotted with antibodies for cytochrome c (Cyt c), and cytochrome oxidase (Cyt ox) and tubulin to demonstrate fraction purity. B: Percentage cell death by MTT assay in cells infected with Ad5LacZ (LacZ) or Bcl-2- or Bcl-XL-expressing adenoviruses and treated with SP600125 and 50 or 60 μM menadione for 24 h (*P<0.005 as compared to Ad5LacZ-infected cells; n=6). C: Percentage cell death 24 h after treatment with SP600125 and 40 or 50 μM menadione in the absence (SP) or presence (SP+QVD) of Q-VD-OPh (*P<0.001 as compared to cells without Q-VD-OPh; n=5). D: Percentage cell death in primary hepatocytes treated with the indicated μM concentration of menadione alone (Men) or with SP600125 pretreatment (SP+Men) for 24 h (*P<0.05, **P<0.001 as compared to menadione alone; n=5). E: Relative ROS levels in the absence (SP−) or presence (SP+) of SP600125 and left untreated (No tx) or treated with 50 μM menadione (Men 50) for 1 h (n=4–5).
PRIMARY HEPATOCYTES ARE SENSITIZED TO DEATH FROM MENADIONE BY JNK INHIBITION
To insure that the findings were not an artifact of the RALA cell line, the effect of SP600125 was examined on menadione toxicity in primary hepatocytes. SP600125 cotreatment significantly increased cell death in mouse primary hepatocytes over a range of menadione concentrations (Fig. 2D). JNK signaling was critical for resistance to oxidant stress identical to findings in RALA hepatocytes.
SP600125 DOES NOT INCREASE ROS PRODUCTION FROM MENADIONE
SP600125’s effects are not completely JNK specific [Bain et al., 2003], suggesting that sensitization to menadione death may be through another mechanism. One off-target effect could be to alter levels of oxidant stress. To exclude an effect secondary to altered oxidative stress, menadione-induced ROS production was determined in the absence and presence of SP600125. Previous studies demonstrated that ROS peak at 1 h after menadione [Wang et al., 2010]. ROS were undetectable in untreated cells, and minimally increased in SP600125-treated cells (Fig. 2E). In contrast, ROS levels increased markedly in response to menadione but were equivalent with SP600125 cotreatment (Fig. 2E), indicating that sensitization to menadione toxicity by SP600125 was not due to increased ROS production.
PHARMACOLOGICAL JNK INHIBITION SENSITIZES TO DEATH DESPITE BLOCKING C-JUN PHOSPHORYLATION
Phosphorylated c-Jun levels represent the net effect of JNK phosphorylation and phosphatase-mediated dephosphorylation. Although SP600125 inhibited JNK activity as measured by phosphorylation of c-Jun (Fig. 1A), menadione-induced oxidant stress may decrease phosphatase activity resulting in an overall increase in c-Jun phosphorylation and cell death. To exclude this possibility, SP600125 effects on phospho-c-Jun levels with menadione treatment were examined. By Western blotting, SP600125 blocked both c-Jun and JNK phosphorylation from menadione (Fig. 3A). TAM67 similarly blocked c-Jun and JNK phosphorylation (Fig. 3B). Previously we identified the MAPK ERK1/2 as a JNK inhibitor that promotes resistance to menadione [Czaja et al., 2003]. SP600125 and TAM67 specifically inhibited JNK/c-Jun signaling as neither agent altered ERK1/2 phosphorylation (Fig. 3A and 3B). Thus, SP600125 sensitized to death from menadione despite inhibition of c-Jun phosphorylation.
Fig. 3.

Cell death from SP600125/menadione treatment occurs from a block in early JNK activation and is independent of c-Jun phosphorylation. A: RALA hepatocytes were treated with dimethyl sulfoxide (DMSO) or SP600125 alone or with 50 μM menadione for the indicated hours. Total protein was isolated and immunoblotted for phospho- (P-c-Jun) and total (c-Jun) c-Jun, phospho- (P-JNK) and total (JNK) JNK, phospho- (P-ERK1/2) and total (ERK1/2) ERK1/2 and β-actin as a loading control. B: Cells were infected with Ad5LacZ or Ad5TAM and treated with 50 μM menadione. Total protein from these cells was immunoblotted with the same antibodies as in (A). C: Percentage cell death at 24 h in untreated control (Con) RALA hepatocytes, and cells treated with 50 μM menadione together with SP600125 1 h before (−1h), or 1 (+1h), 2 (+2h) or 4 (+4h) h after menadione treatment (*P<0.01, **P<0.001 as compared to cells treated with SP600125 1 h before menadione; n=4).
EARLY JNK SIGNALING MEDIATES RESISTANCE TO MENADIONE
An immediate, transient JNK activation occurs in RALA or primary hepatocytes from non-toxic menadione concentrations [Conde de la Rosa et al., 2006; Czaja et al., 2003]. With toxic menadione doses JNK activation becomes prolonged, and triggers cell death [Conde de la Rosa et al., 2006; Czaja et al., 2003]. These findings imply that transient JNK activation is beneficial, but prolonged activation initiates cell death. We therefore varied the onset of JNK inhibition to examine whether SP600125 exerts its death-promoting effect by interfering with early, protective JNK activation. Death decreased only slightly when SP600125 was given 1 h after menadione (Fig. 3C). However, death decreased significantly when SP600125 was delayed until 2 h after menadione treatment, and the effect of SP600125 was lost completely with treatment at 4 h after menadione (Fig. 3C). Inhibition of early JNK activation by SP600125 abolishes hepatocyte resistance to death from menadione.
SENSITIZATION TO MENADIONE BY SP600125 IS C-JUN DEPENDENT
The finding that SP600125 sensitized to death from menadione despite blocking c-Jun phosphorylation suggested that death may be c-Jun independent. The effect of direct c-Jun inhibition by TAM67 was therefore investigated in SP600125/menadione-treated cells. TAM67 markedly decreased death from SP600125/menadione (Fig. 4A), demonstrating that death was c-Jun dependent, although independent of c-Jun phosphorylation.
Fig. 4.

SP600125/menadione death is c-Jun dependent. A: RALA hepatocytes were infected with Ad5LacZ or Ad5TAM, pretreated with SP600125 and treated with 40 or 50 μM menadione. Percentage cell death was determined at 24 h by MTT assay (*P<0.00004 as compared to Ad5LacZ-infected cells; n=4–8). B: Immunoblots for total JNK and β-actin in jnk knockdown cells. C: Percentage cell death 24 h after treatment with the indicated menadione concentrations in knockdown cells (*P<0.002, #P<0.01 as compared to VEC cells; n=5–7). D: Percentages of apoptotic and necrotic cells by fluorescence microscopy 12 h after 50 μM menadione treatment (*P<0.01 as compared to menadione-treated VEC cells; n=4). E: Percentage cell death in wild-type (WT), jnk1−/− and jnk2−/− mouse primary hepatocytes by MTT assay after 24 h treatment with the menadione concentrations shown (*P<0.03, **P<0.005 as compared to menadione-treated wild-type cells; n=3–5).
JNK MEDIATES RESISTANCE TO MENADIONE TOXICITY
The divergent effects of the JNK inhibitor SP600125 and the c-Jun dominant negative TAM67 suggested that rather than the two signaling molecules functioning in sequence to mediate a common effect, they have distinct and opposing roles in the menadione death pathway. The ability of TAM67 to prevent death may have resulted from an ability to block phosphorylation-independent c-Jun activity directly, whereas unphosphorylated c-Jun function was unaffected by SP600125. The finding that TAM67 blocked death from SP600125/menadione supports this conclusion. However, this possibility does not completely explain the experimental findings, as SP600125 was not only ineffective in preventing death, but also promoted death from oxidative stress. The data therefore suggested that JNK may have a c-Jun-independent cytoprotective effect that when blocked by SP600125 sensitizes to death from menadione.
To more specifically determine the effect of JNK inhibition, we established stable, lentiviral shRNA-mediated knockdowns of both jnk genes (siJNK cells), jnk1 (siJNK1 cells) and jnk2 (siJNK2 cells) in RALA hepatocytes. An shRNA directed against a common sequence of both genes reduced both p54 and p46 JNK (Fig. 4B). Consistent with findings in jnk1 and jnk2 mouse knockouts [Singh et al., 2009c], and primary hepatocytes from these livers [Kodama et al., 2009b], an shRNA directed against jnk1 predominantly decreased levels of p46 JNK, whereas the jnk2-targeted shRNA reduced p54 JNK (Fig. 4B).
Cells infected with empty lentiviral vector (VEC cells) and the three knockdowns were treated with menadione. The total jnk knockdown sensitized to death from menadione (Fig. 4C). siJNK2 cells were similarly sensitized to death, whereas siJNK1 cells had decreased toxicity (Fig. 4C). These MTT findings were confirmed by fluorescence microscopic quantification of the numbers of apoptotic and necrotic cells (Fig. 4D). These results demonstrate that inhibition of both jnk genes sensitized to death from menadione identical to the JNK inhibitor SP600125. The mechanism of this effect was due in large part to JNK2 as the jnk2 and total jnk knockdown cells were equally sensitized to death. We confirmed this finding by examining the sensitivity of primary hepatocytes from jnk knockout mice to menadione. Jnk2 null mouse primary hepatocytes were significantly more sensitive to death from menadione than wild-type hepatocytes (Fig. 4E). In contrast, jnk1 knockout hepatocytes were protected against cell death from menadione (Fig. 4E). Thus, in both knockdown RALA hepatocytes, and knockout primary hepatocytes, JNK2 mediated survival from menadione whereas JNK1 promoted cell death.
JNK INHIBITION ALTERS ENERGY HOMEOSTASIS DURING MENADIONE-INDUCED INJURY
Our recent studies demonstrated that maintenance of energy homeostasis is critical for RALA hepatocyte resistance to menadione [Wang et al., 2010]. Others have shown that JNK regulates mitochondrial bioenergetics in response to acetaminophen [Hanawa et al., 2008]. We therefore investigated whether JNK inhibition compromised energy levels during menadione-induced stress. ATP levels were moderately decreased 2 h after 50 μM menadione treatment (Fig. 5A), but returned back towards baseline by 4 h (Fig. 5B). In contrast, SP600125/menadione cotreatment led to significantly greater decreases in ATP at 2 and 4 h than menadione alone, and by 4 h levels had decreased further rather than normalizing (Fig. 5A and 5B). Cellular ATP is maintained by the glycolytic breakdown of glucose or the β-oxidation of fatty acids. Glycolysis was assessed by lactate production which decreased significantly with menadione, but to an equivalent extent in the absence or presence of SP600125 (Fig. 5C and 5D). In contrast, the menadione-induced decrease in fatty acid β-oxidation was significantly greater with SP600125 cotreatment (Fig. 5E and 5F). The decreases in ATP and β-oxidation induced by JNK inhibition preceded cell death as apoptotic and necrotic cells were not detected by fluorescence microscopy until 8 h after menadione administration (data not shown).
Fig. 5.

SP600125/menadione leads to ATP depletion and decreased β-oxidation. A: ATP levels in RALA hepatocytes treated with 50 μM menadione for 2 h in the absence (SP−) or presence (SP+) of SP600125 (*P<0.001 as compared to SP-/menadione-treated cells; n=4). B: ATP levels in identically treated cells at 4 h (*P<0.00004; n=3). C, D: Lactate production in similarly treated cells at 2 h (n=3–4) (C), and 4 h (n=4) (D) after menadione. E, F: Rates of β-oxidation at 2 h (E) and 4 h (F) after menadione (*P< 0.02; n=5–6).
A genetic JNK knockdown had similar effects on energy homeostasis. Identical to wild-type cells, a nontoxic menadione dose significantly decreased ATP content in control VEC cells at 2 h (Fig. 6A), but levels almost returned to baseline within 4 h (Fig. 6B). ATP levels in siJNK1 and siJNK2 cells followed a similar pattern as VEC cells in response to menadione (Fig. 6A and 6B). However, siJNK cells had a significantly larger decrease in ATP at 2 and 4 h with a greater depletion by 4 h (Fig. 6A and 6B). Lactate levels decreased equivalently with menadione in VEC cells and all three knockdowns at both 2 and 4 h (Fig. 6C and 6D). In contrast, although β-oxidation rates were decreased by menadione in all cell types at both time points, the decrease was significantly greater in siJNK cells (Fig. 6E and 6F). Thus, chemical or genetic inhibition of total JNK function sensitized to death from menadione in association with a more profound depletion of ATP and decrease inβ-oxidation.
Fig. 6.
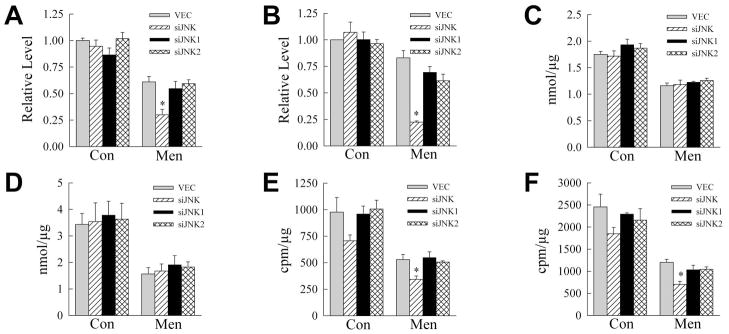
Total jnk knockdown decreases ATP levels and rates of β-oxidation. A, B: ATP levels in VEC and jnk knockdown cells at 2 (A) and 4 (B) h after treatment with 50 μM menadione (*P<0.004 as compared to VEC cells; n=3–5). C, D: Cumulative lactate production over 2 (C) and 4 (D) h (n=3). E, F: Rates of β-oxidation after 2 h (*P<0.03 as compared to VEC cells; n=3) (E), and 4 h (*P<0.002 as compared to VEC cells; n=3) (F) of menadione.
β-OXIDATION MEDIATES RESISTANCE TO MENADIONE TOXICITY
To determine whether the reduction in β-oxidation was causal in menadione-induced death, the effect of an inhibition of β-oxidation on hepatocyte death from menadione was examined. Wild-type RALA hepatocytes were treated with the carnitine palmitoyltransferase-1 inhibitor etomoxir to block β-oxidation [Abdel-aleem et al., 1994]. Etomoxir treatment for 24 h was not toxic, but this agent significantly increased death from menadione (Fig. 7A). In contrast, inhibition of glycolysis with 2-deoxyglucose was mildly toxic but failed to amplify menadione toxicity (data not shown). Menadione treatment in the setting of an inhibition of β-oxidation led to a significant reduction in ATP at 4 h (Fig. 7B).
Fig. 7.

Inhibition of β-oxidation sensitizes to death from menadione. A: Percentage cell death in wild-type cells 24 h after treatment with etomoxir (Eto) or 50 μM menadione (Men) alone, or in combination (Men+Eto) (*P<0.00001 as compared to menadione-treated cells; n=4–6). B: ATP levels 4 h after menadione in the absence (Eto−) or presence (Eto+) of etomoxir (*P<0.001 as compared to menadione-treated cells; n=4). C: Percentage cell death in siJNK cells 24 h after 50 or 60 μM menadione treatment without (OL−) or with (OL+) oleate supplementation (*P<0.0005 as compared to OL- cells; n=3). D: ATP levels in siJNK cells treated with 60 μM menadione for 4 h in the absence (OL−) or presence (OL+) of oleate (*P<0.001 as compared to OL- cells; n=3). E: Percentage cell death at 24 h in wild-type RALA hepatocytes treated with the indicated concentrations of menadione and SP600125 alone (SP) or with oleate (SP+OL) (*P<0.00003 as compared to SP cells; n=5).
Previously we demonstrated that the β-oxidation substrate oleic acid increases ATP levels and blocks death from menadione [Wang et al., 2010]. To determine whether increased β-oxidation could replenish ATP and prevent sensitization to death from menadione by JNK inhibition, siJNK cells were supplemented with oleate. Oleate blocked death from menadione in siJNK cells (Fig. 7C) in parallel with the maintenance of normal levels of ATP (Fig. 7D). Oleate similarly blocked death from SP600125/menadione (Fig. 7E). Thus, a decreased rate of β-oxidation is the mechanism by which JNK inhibition sensitizes cells to death from menadione.
DISCUSSION
JNK/c-Jun overactivation is central to a variety of forms of hepatocyte injury, but the biological outcome from JNK signaling varies depending on the injury [Czaja, 2010; Schwabe, 2006]. This complexity undoubtedly results in part from undelineated differences in function of the JNK isoforms. In addition, potentially opposing, functions of JNK and c-Jun may exist. Although c-Jun is the major downstream effector of JNK signaling, JNK can function independently of c-Jun [Chang et al., 2006; Kodama et al., 2009b], and c-Jun may have JNK-independent functions as well. The present study explains some of the complexity of JNK/c-Jun signaling in hepatocyte injury by identifying distinct JNK and c-Jun functions in death from oxidant stress.
Hepatocyte resistance to oxidant stress is regulated by cell signaling pathways including JNK [Czaja, 2007]. In previous studies, the c-Jun dominant negative TAM67, which inhibits both JNK and c-Jun function, blocked death from menadione in RALA hepatocytes [Czaja et al., 2003]. This finding suggested that JNK/c-Jun signaling promotes death in superoxide-induced injury. In contrast, in the present study pharmacological JNK inhibition with SP600125 sensitizes RALA hepatocytes to death from menadione-induced oxidant stress. An identical effect of SP600125 was seen in mouse primary hepatocytes. This finding differs from a previous report in which SP600125 protected against menadione-induced death in rat primary hepatocytes [Conde de la Rosa et al., 2006]. However, the prior study did not directly examine overall cell death which may have included necrosis, but only caspase activation which is an indirect measure of apoptosis. Although reduced, caspase activation was still significantly elevated with SP600125/menadione [Conde de la Rosa et al., 2006]. Thus, the effects of SP600125 on death in this previous study are unclear. Alternatively, the discordant results may reflect the use of mouse versus rat primary hepatocytes or differences in cell culture conditions.
The present findings demonstrate opposing cell protective JNK effects and death promoting c-Jun effects in oxidant-induced hepatocyte injury. The effect of c-Jun was phosphorylation independent because c-Jun-dependent death from menadione occurred despite SP600125 inhibition of c-Jun phosphorylation. Both SP600125 and TAM67 blocked menadione-induced c-Jun phosphorylation as determined by kinase activity assays and immunoblotting, yet SP600125 promoted death whereas TAM67 prevented it. These findings suggest that a c-Jun independent JNK function mediates hepatocyte resistance to death from menadione. However, this JNK effect becomes irrelevant when c-Jun function is blocked as TAM67 prevented death from SP600125/menadione. These results suggest that although TAM67 inhibited JNK function as measured by c-Jun phosphorylation, this dominant negative protein failed to block other protective functions of JNK. Alternatively, c-Jun may represent the ultimate mediator of death from menadione, thus c-Jun inhibition renders other upstream protective signals irrelevant. Little is known about direct interactions of TAM67 with JNK. It is possible that the JNK inhibitor SP600125 may block JNK2 functions that are unaffected by TAM67. This possibility cannot be tested because of the lack of a JNK2-specific functional assay.
The results with the pharmacological inhibitor SP600125 were confirmed by the finding that a genetic jnk knockdown similarly sensitized RALA hepatocytes to death from menadione. These data confirm that the death-sensitizing effect of SP600125 was not an off target effect of this chemical. The ability of a selective jnk2, but not jnk1, knockdown to sensitize to death from menadione indicated that JNK2 was the protective isoform, a finding confirmed in primary jnk2 null hepatocytes. Early JNK activation mediated resistance to menadione as SP600125 had to be administered within 1 h of menadione treatment to sensitize to death. These findings suggest that conclusions on JNK’s role in liver injury based on findings with SP600125 or other global JNK inhibitors must be interpreted with caution. A negative result from global inhibition is not conclusive that JNK is uninvolved in the liver injury because the finding may reflect the net effect of an inhibition of beneficial as well as detrimental effects of different JNK isoforms. This fact may explain the reported failure of SP600125 to block in vivo liver injury from menadione [Hong et al., 2009], although this finding may reflect that rodent toxicity from menadione is primarily extrahepatic.
Supporting the concept that JNK function varies depending on the nature of the liver injury is that SP600125 has opposing effects on hepatocyte death from oxidant stress and TNF even though both forms of death result from JNK overactivation. SP600125 inhibits death in cells sensitized to TNF toxicity by an inhibition of NF-κB in both primary hepatocytes [Schwabe et al., 2004] and RALA hepatocytes (data not shown). These findings suggest that JNK2 does not function in hepatocyte resistance to TNF. Again the availability of JNK1 and JNK2 specific activity assays might reveal their differential activation by the two types of injury that would explain these findings. Alternatively, JNK2 may exert a protective function relevant to oxidant-but not TNF-induced liver injury.
Chemical or genetic JNK inhibition magnified the ATP depletion and decrease in β-oxidation caused by menadione. The effect of a total JNK knockdown was greater than the loss of JNK2 alone even though the two knockdowns were equivalent in sensitizing to death from menadione. These data suggest that JNK1 also has some protective effect in maintaining these determinants of cellular energy. Although cells died of apoptosis, significant numbers of cells also died of necrosis. Profound ATP depletion favors cell death by necrosis rather than apoptosis as apoptosis is energy dependent [Jaeschke and Lemasters, 2003]. However, the apoptotic machinery fails only with very profound ATP depletion to levels below 15–20% of normal [Jaeschke and Lemasters, 2003]. In the absence of JNK, ATP content after menadione treatment dropped to a mean of 20–30% of normal levels, indicating that some cells likely had sufficient ATP to support apoptosis whereas other cells had lower levels that triggered necrosis. Thatβ-oxidation maintains ATP levels was demonstrated by findings that an inhibition of β-oxidation sensitized RALA hepatocytes to death from menadione in parallel with a decrease in ATP, and theβ-oxidation substrate oleate preserved ATP levels and prevented death from JNK inhibition and menadione. These findings suggest that preservation of ATP levels is the likely mechanism by which β-oxidation mediates cellular resistance to oxidant stress. Oleate blocked necrosis and apoptosis indicating that ATP depletion promoted both forms of cell death. ATP depletion may have promoted mitochondrial dysfunction that triggered apoptosis, and it has recently been reported in neurons and macrophages that ATP depletion triggers the mitochondrial death pathway and apoptosis in these cells [Concannon et al., 2010; Garedew et al., 2010]. That JNK functions to maintain mitochondrial energy homeostasis contrasts with findings that JNK promotes ATP depletion from acetaminophen [Hanawa et al., 2008]. These opposing effects may reflect differences in the two forms of oxidant stress. In contrast to menadione, acetaminophen causes a profound depletion of glutathione that leads to JNK activation [Hanawa et al., 2008].
The result of oxidant-induced JNK activation on cell death therefore reflects the net effect of JNK-mediated resistance and the promotion of cell death by c-Jun. The findings raise the question as to why hepatocytes have components within the same sequential signaling cascade that oppose each other’s function. Presumably the potentially fatal outcome from any instance of prolonged activation of this commonly induced signaling pathway requires safeguards against death. JNK/c-Jun signaling has been implicated in the regulation of a number of forms of experimental liver injury including those mediated by oxidant stress. These models are directly relevant to human diseases such as acetaminophen drug toxicity, nonalcoholic fatty liver disease and cholestatic injury from fatty acids [Gunawan et al., 2006; Malhi et al., 2006; Singh et al., 2009c]. Studies have confirmed that JNK is activated in human livers from patients with obstructive cholestasis, nonalcoholic fatty liver disease and acetaminophen toxicity [Chai et al., 2011; Ferreira et al., 2011; Henderson et al., 2007]. Thus, there is considerable interest in manipulating JNK/c-Jun signaling for the treatment of human diseases [Johnson and Nakamura, 2007].The complex and opposing functions of JNK1 and JNK2 as well as c-Jun in various forms of liver injury render the therapeutic approach of global JNK inhibition impractical in some forms of hepatocellular injury including those from oxidant stress. Function specific inhibitors are required to target this death signal. Alternatively, delineation of downstream effectors of JNK/c-Jun-mediated death may better identify potential therapeutic targets in this critical hepatocyte death signaling pathway.
Acknowledgments
We thank Yongjun Wang for his technical assistance and David Brenner for the adenoviruses.
Grant sponsor: National Institutes of Health; Grant number: DK044234 to MJC and a Training Grant DK007218 for KL
References
- Abdel-aleem S, Li X, Anstadt MP, Perez-Tamayo RA, Lowe JE. Regulation of glucose utilization during the inhibition of fatty acid oxidation in rat myocytes. Horm Metab Res. 1994;26:88–91. doi: 10.1055/s-2007-1000779. [DOI] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradham CA, Hatano E, Brenner DA. Dominant-negative TAK1 induces c-Myc and G0 exit in liver. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1279–G1289. doi: 10.1152/ajpgi.2001.281.5.G1279. [DOI] [PubMed] [Google Scholar]
- Brown PH, Alani R, Preis LH, Szabo E, Birrer MJ. Suppression of oncogene-induced transformation by a deletion mutant of c-jun. Oncogene. 1993;8:877–886. [PubMed] [Google Scholar]
- Chai J, He Y, Cai SY, Jiang Z, Wang H, Li Q, Chen L, Peng Z, He X, Wu X, Xiao T, Wang R, Boyer JL, Chen W. Elevated hepatic MRP3/ABCC3 expression in human obstructive cholestasis is mediated through TNFα and JNK/SAPK signaling pathway. Hepatology. 2011 doi: 10.1002/hep.24801. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, Liu YC, Karin M. The E3 ubiquitin ligase itch couples JNK activation to TNFα-induced cell death by inducing c-FLIPL turnover. Cell. 2006;124:601–613. doi: 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Chou JY. Temperature-sensitive adult liver cell line dependent on glucocorticoid for differentiation. Mol Cell Biol. 1983;3:1013–1020. doi: 10.1128/mcb.3.6.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concannon CG, Tuffy LP, Weisova P, Bonner HP, Davila D, Bonner C, Devocelle MC, Strasser A, Ward MW, Prehn JH. AMP kinase-mediated activation of the BH3-only protein Bim couples energy depletion to stress-induced apoptosis. J Cell Biol. 2010;189:83–94. doi: 10.1083/jcb.200909166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde de la Rosa L, Schoemaker MH, Vrenken TE, Buist-Homan M, Havinga R, Jansen PL, Moshage H. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: involvement of JNK and ERK MAP kinases. J Hepatol. 2006;44:918–929. doi: 10.1016/j.jhep.2005.07.034. [DOI] [PubMed] [Google Scholar]
- Czaja MJ. Cell signaling in oxidative stress-induced liver injury. Semin Liver Dis. 2007;27:378–389. doi: 10.1055/s-2007-991514. [DOI] [PubMed] [Google Scholar]
- Czaja MJ. JNK regulation of hepatic manifestations of the metabolic syndrome. Trends Endocrinol Metab. 2010;21:707–713. doi: 10.1016/j.tem.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaja MJ, Liu H, Wang Y. Oxidant-induced hepatocyte injury from menadione is regulated by ERK and AP-1 signaling. Hepatology. 2003;37:1405–1413. doi: 10.1053/jhep.2003.50233. [DOI] [PubMed] [Google Scholar]
- Devey L, Mohr E, Bellamy C, Simpson K, Henderson N, Harrison EM, Ross JA, Wigmore SJ. c-Jun terminal kinase-2 gene deleted mice overexpress hemeoxygenase-1 and are protected from hepatic ischemia reperfusion injury. Transplantation. 2009;88:308–316. doi: 10.1097/TP.0b013e3181ae3067. [DOI] [PubMed] [Google Scholar]
- Ferreira DM, Castro RE, Machado MV, Evangelista T, Silvestre A, Costa A, Coutinho J, Carepa F, Cortez-Pinto H, Rodrigues CM. Apoptosis and insulin resistance in liver and peripheral tissues of morbidly obese patients is associated with different stages of non-alcoholic fatty liver disease. Diabetologia. 2011;54:1788–1798. doi: 10.1007/s00125-011-2130-8. [DOI] [PubMed] [Google Scholar]
- Garedew A, Henderson SO, Moncada S. Activated macrophages utilize glycolytic ATP to maintain mitochondrial membrane potential and prevent apoptotic cell death. Cell Death Differ. 2010;17:1540–1550. doi: 10.1038/cdd.2010.27. [DOI] [PubMed] [Google Scholar]
- Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, Kaplowitz N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 2006;131:165–178. doi: 10.1053/j.gastro.2006.03.045. [DOI] [PubMed] [Google Scholar]
- Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem. 2008;283:13565–13577. doi: 10.1074/jbc.M708916200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson NC, Pollock KJ, Frew J, Mackinnon AC, Flavell RA, Davis RJ, Sethi T, Simpson KJ. Critical role of c-jun NH2 terminal kinase in paracetamol- induced acute liver failure. Gut. 2007;56:982–990. doi: 10.1136/gut.2006.104372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JY, Lebofsky M, Farhood A, Jaeschke H. Oxidant stress-induced liver injury in vivo: role of apoptosis, oncotic necrosis, and c-Jun NH2-terminal kinase activation. Am J Physiol Gastrointest Liver Physiol. 2009;296:G572–G581. doi: 10.1152/ajpgi.90435.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppel C, DiMarco JP, Tandler B. Riboflavin and rat hepatic cell structure and function. Mitochondrial oxidative metabolism in deficiency states. J Biol Chem. 1979;254:4164–4170. [PubMed] [Google Scholar]
- Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology. 2003;125:1246–1257. doi: 10.1016/s0016-5085(03)01209-5. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Nakamura K. The c-jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim Biophys Acta. 2007;1773:1341–1348. doi: 10.1016/j.bbamcr.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BE, Lo CR, Liu H, Srinivasan A, Streetz K, Valentino KL, Czaja MJ. Hepatocytes sensitized to tumor necrosis factor-α cytotoxicity undergo apoptosis through caspase-dependent and caspase-independent pathways. J Biol Chem. 2000;275:705–712. doi: 10.1074/jbc.275.1.705. [DOI] [PubMed] [Google Scholar]
- Kluwe J, Pradere JP, Gwak GY, Mencin A, De Minicis S, Osterreicher CH, Colmenero J, Bataller R, Schwabe RF. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology. 2010;138:347–359. doi: 10.1053/j.gastro.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama Y, Kisseleva T, Iwaisako K, Miura K, Taura K, De Minicis S, Osterreicher CH, Schnabl B, Seki E, Brenner DA. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology. 2009a;137:1467–1477. doi: 10.1053/j.gastro.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama Y, Taura K, Miura K, Schnabl B, Osawa Y, Brenner DA. Antiapoptotic effect of c-Jun N-terminal Kinase-1 through Mcl-1 stabilization in TNF-induced hepatocyte apoptosis. Gastroenterology. 2009b;136:1423–1434. doi: 10.1053/j.gastro.2008.12.064. [DOI] [PubMed] [Google Scholar]
- Liedtke C, Plumpe J, Kubicka S, Bradham CA, Manns MP, Brenner DA, Trautwein C. Jun kinase modulates tumor necrosis factor-dependent apoptosis in liver cells. Hepatology. 2002;36:315–325. doi: 10.1053/jhep.2002.34615. [DOI] [PubMed] [Google Scholar]
- Liu H, Lo CR, Czaja MJ. NF-κB inhibition sensitizes hepatocytes to TNF-induced apoptosis through a sustained activation of JNK and c-Jun. Hepatology. 2002;35:772–778. doi: 10.1053/jhep.2002.32534. [DOI] [PubMed] [Google Scholar]
- Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- Piva R, Chiarle R, Manazza AD, Taulli R, Simmons W, Ambrogio C, D’Escamard V, Pellegrino E, Ponzetto C, Palestro G, Inghirami G. Ablation of oncogenic ALK is a viable therapeutic approach for anaplastic large-cell lymphomas. Blood. 2006;107:689–697. doi: 10.1182/blood-2005-05-2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuther-Madrid JY, Kashatus D, Chen S, Li X, Westwick J, Davis RJ, Earp HS, Wang CY, Baldwin AS., Jr The p65/RelA subunit of NF-κB suppresses the sustained, antiapoptotic activity of Jun kinase induced by tumor necrosis factor. Mol Cell Biol. 2002;22:8175–8183. doi: 10.1128/MCB.22.23.8175-8183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattenberg JM, Singh R, Wang Y, Lefkowitch JH, Rigoli RM, Scherer PE, Czaja MJ. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology. 2006;43:163–172. doi: 10.1002/hep.20999. [DOI] [PubMed] [Google Scholar]
- Schwabe RF. Cell death in the liver-all roads lead to JNK. Gastroenterology. 2006;131:314–316. doi: 10.1053/j.gastro.2006.05.029. [DOI] [PubMed] [Google Scholar]
- Schwabe RF, Uchinami H, Qian T, Bennett BL, Lemasters JJ, Brenner DA. Differential requirement for c-Jun NH2-terminal kinase in TNFα- and Fas-mediated apoptosis in hepatocytes. FASEB J. 2004;18:720–722. doi: 10.1096/fj.03-0771fje. [DOI] [PubMed] [Google Scholar]
- Shinoura N, Koike H, Furitu T, Hashimoto M, Asai A, Kirino T, Hamada H. Adenovirus-mediated transfer of caspase-8 augments cell death in gliomas: implication for gene therapy. Hum Gene Ther. 2000;11:1123–1137. doi: 10.1089/10430340050015185. [DOI] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009a;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Wang Y, Schattenberg JM, Xiang Y, Czaja MJ. Chronic oxidative stress sensitizes hepatocytes to death from 4-hydroxynonenal by JNK/c-Jun overactivation. Am J Physiol Gastrointest Liver Physiol. 2009b;297:G907–G917. doi: 10.1152/ajpgi.00151.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Wang Y, Xiang Y, Tanaka KE, Gaarde WA, Czaja MJ. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology. 2009c;49:87–96. doi: 10.1002/hep.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theruvath TP, Snoddy MC, Zhong Z, Lemasters JJ. Mitochondrial permeability transition in liver ischemia and reperfusion: role of c-Jun N-terminal kinase 2. Transplantation. 2008;85:1500–1504. doi: 10.1097/TP.0b013e31816fefb5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Schattenberg JM, Rigoli RM, Storz P, Czaja MJ. Hepatocyte resistance to oxidative stress is dependent on protein kinase C-mediated down-regulation of c-Jun/AP-1. J Biol Chem. 2004;279:31089–31097. doi: 10.1074/jbc.M404170200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Singh R, Lefkowitch JH, Rigoli RM, Czaja MJ. Tumor necrosis factor-induced toxic liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. J Biol Chem. 2006;281:15258–15267. doi: 10.1074/jbc.M512953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Singh R, Xiang Y, Czaja MJ. Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology. 2010;52:266–277. doi: 10.1002/hep.23645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Xu Y, Nguyen Q, Novikoff PM, Czaja MJ. Induction of hepatoma cell apoptosis by c-myc requires zinc and occurs in the absence of DNA fragmentation. Am J Physiol. 1996;270:G60–G70. doi: 10.1152/ajpgi.1996.270.1.G60. [DOI] [PubMed] [Google Scholar]
- Xu Y, Bialik S, Jones BE, Iimuro Y, Kitsis RN, Srinivasan A, Brenner DA, Czaja MJ. NF-κB inactivation converts a hepatocyte cell line TNF-α response from proliferation to apoptosis. Am J Physiol. 1998a;275:C1058–C1066. doi: 10.1152/ajpcell.1998.275.4.C1058. [DOI] [PubMed] [Google Scholar]
- Xu Y, Bradham C, Brenner DA, Czaja MJ. Hydrogen peroxide-induced liver cell necrosis is dependent on AP-1 activation. Am J Physiol. 1997;273:G795–G803. doi: 10.1152/ajpgi.1997.273.4.G795. [DOI] [PubMed] [Google Scholar]
- Xu Y, Jones BE, Neufeld DS, Czaja MJ. Glutathione modulates rat and mouse hepatocyte sensitivity to tumor necrosis factor toxicity. Gastroenterology. 1998b;115:1229–1237. doi: 10.1016/s0016-5085(98)70095-2. [DOI] [PubMed] [Google Scholar]