

Abstract
Since the discovery of the Jak2-V617F mutation as the causative agent in a large number of myeloproliferative neoplasms (MPNs), there has been a drive to develop Jak2 specific inhibitors that can be used in therapy for MPN patients and other Jak2-related pathologies. Over the past few years, a number of research groups have sought to develop Jak2 tyrosine kinase inhibitors. These compounds are currently in pre-clinical or clinical trials. Unfortunately, there is still a need for more potent, specific, and orally bioavailable drugs to treat these diseases. Within the past twelve months, a variety of medicinal chemistry techniques have produced several lead compounds that exhibit promising Jak2 inhibitory properties. The majority of these inhibitors target the Jak2 kinase domain in general and the ATP-binding pocket in particular. In this review, we summarize these studies and discuss the structure activity relationship (SAR) properties of several compounds. As we learn more about the key structural components that provide potency and specificity in Jak2 inhibition, we will come closer to finding suitable treatment options for individuals suffering from Jak2-mediated pathologies.
Keywords: JAK2, small molecule inhibitors, medicinal chemistry, structure activity relationships
Introduction
Jak2, a member of the Janus family of cytoplasmic tyrosine kinases, is ubiquitously expressed and plays a key role in signal transduction. Jak2 is activated by a variety of cytokines, growth factors, and GPCR ligands and mediates signaling cascades that regulate cell survival, proliferation, and development. Jak2 activates the Signal Transducers and Activators of Transcription (STAT) proteins, which then translocate to the nucleus to activate transcription of various target genes. Constitutive activation of Jak-STAT signaling promotes aberrant cell proliferation and is linked to hematological malignancies and myeloproliferative neoplasms (MPNs). To date, there are very few treatment options for patients suffering from these diseases. Because of its role in the pathogenesis of human disease, Jak2 serves as a viable therapeutic target.
Interestingly, the history of Jak2 tyrosine kinase inhibitors extends back nearly two decades. The first Jak2 inhibitor to be developed was tyrphostin AG490. It was initially found to inhibit type I Fc epsilon receptor-induced PLC gamma 1 phosphorylation and ensuing inositol phosphate formation [1]. Subsequent work demonstrated that AG490 also inhibited Jak2 tyrosine kinase activity and blocked acute lymphoblastic leukemic cell growth, in vitro and in vivo [2]. While this later work underscored the importance of small molecules in the inhibition of Jak2-mediated disease, it simultaneously called attention to the non-specific nature of AG490. As a consequence, several derivative compounds of AG490 were developed with the hope of making a more potent and/or specific Jak2 inhibitor [3, 4, 5]. While the progress of Jak2 inhibitors moved along at a reasonable pace, the field began growing more rapidly in 2005 when the Jak2-V617F mutation was identified in a large percentage of myeloproliferative neoplasm patients [6, 7, 8, 9, 10].
Using techniques such as library screening, molecular docking, fragment-based drug discovery, scaffold morphing, and derivatization of lead compounds, numerous laboratories have developed Jak2 inhibitors. The majority of these inhibitors target the ATP-binding pocket within the Jak2 kinase domain (JH1). These compounds belong to several different structural classes including pyrazines, pyrimidines, azaindoles, aminoindazoles, deazapurines, stilbenes, benzoxazoles and quinoxalines. Through structure-based optimization, a number of functional groups have been identified that play crucial roles in potent and specific Jak2 inhibition. This review summarizes some of the most recently discovered inhibitors and discusses their structure activity relationships (SARs). The structures and properties of selected compounds are shown in Table 1.
Table 1.
Structures and characteristics of recently developed Jak2 inhibitors. All IC50 values were determined by in vitro kinase assays.
| Name | Structure | Jak2 IC50 (nM) | Jak2 H-bond Interactions | Reference |
|---|---|---|---|---|
| 40 |
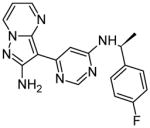
|
0.2 | Leu932, Gly993 | [14] |
| 45 |
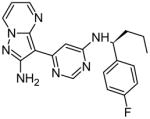
|
0.5 | N/A | [14] |
| CYT387 |
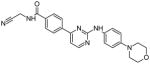
|
18 | Precursor compound: Leu932, Asp994 | [15] |
| 15a |
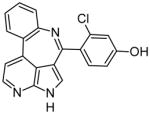
|
0.8 | Leu932, Glu930, Glu898, Phe995 | [17] |
| 9 |
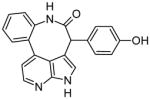
|
1.0 | Leu932, Glu930, Asp994, Phe995, Glu898 | [18] |
| 7c |
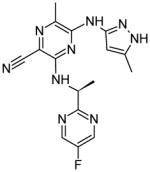
|
<3.0 | N/A | [20] |
| 7h |
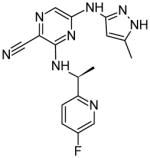
|
<3.0 | N/A | [20] |
| 7b |
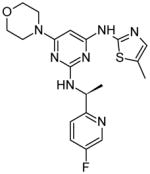
|
5 | N/A | [21] |
| 13 |
|
78 | Glu930, Leu932, Asn981 | [22] |
| G6 |
|
60 | Glu930, Leu932 | [23] |
| 15 |
|
5 | N/A | [24] |
| 3 |
|
12 | Leu932 | [25] |
| 4 |
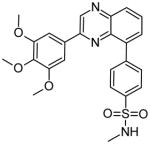
|
42 | Leu932 | [25] |
Derivatization of AG490
AG490 was originally synthesized within a series of tyrosine kinase inhibitors that were shown to block EGF receptor activity and therefore was not designed to selectively inhibit Jak2. However, given its potent anti-tyrosine kinase activity, it was used as a starting point for the possible development of Jak2-specific inhibitors. Several research groups have synthesized derivatives of AG490 and a number of these have shown increased Jak2 potency and/or specificity over the parent compound. In one study, a library of 599 AG490 derivatives was screened and two compounds containing trifluoromethyl groups were shown to have improved activity against IL3- and IL7-dependent cell lines [3]. Specifically, the compound 5H4 showed an approximate twofold increase in potency as determined by cell proliferation assays.
In another study, the AG490 derivative WP1066 was shown to inhibit STAT3 activity and induce apoptosis in malignant glioma cells [4]. This compound also inhibited Jak2-dependent cell growth and led to degradation of the protein in AML cells [5]. WP1066 had highly improved potency and specificity for Jak2 over AG490, but the specific binding of this compound to the protein was never demonstrated, therefore raising the question of whether the inhibitory effects of WP1066 are perhaps independent of Jak2. Interestingly, a compound known as CR4 or LS104, which is structurally similar to AG490, has been shown to be a non-ATP competitive inhibitor of Jak2 [11]. This compound is active against both Philadelphia-positive and -negative ALL cells as well as AML cells [12].
Using in silico molecular modeling, one group docked AG490 into the Jak2 ATP-binding site, but no specific amino acid interactions were reported [13]. Because of its structural similarity to LS104, it is possible that AG490 may also be a non-ATP-competitive inhibitor. Overall, the derivatization of AG490 demonstrates the proof-of-principle use of structure-based optimization of lead compounds for the intent purpose of identifying novel Jak2 inhibitors. The following sections outline the discovery of new generation Jak2 inhibitors through the use of this technique in combination with more advanced methodologies.
Pyrimidines
Vertex Pharmaceuticals recently developed several Jak2 small molecule inhibitors with an aminopyrazolopyrimidine (APP) core. The initial compound of interest contained the APP core attached to a piperidine carboxamide, which was further optimized to produce four potent compounds, named 40, 44, 45, and 46. The APP structure was shown to interact with the hinge region of Jak2 through hydrogen bonds with both the carbonyl and the NH backbone of Leu932. Through X-ray crystallography, compound 40 was also shown to have a unique interaction with Gly993. The unique hydrogen bond was achieved by changing the piperidine carboxamide group to a 4-fluoro-benzylic amine [14]. This work shows that interactions with both the hinge region and amino acids near the DFG motif provide high potency and specificity for Jak2 inhibition. In this case, structure-based optimization techniques led to the development of compounds with novel Jak2 binding properties.
Cytopia identified a group of Jak2 inhibitors containing a phenylaminopyrimidine (PAP) core. Molecular docking of one of the most effective compounds into the Jak2 ATP-binding site revealed potential hydrogen-bonding interactions with Leu932 in the hinge region and with Asp994 in the activation loop. Manipulation of the structure led to a potent compound known as CYT387, which contained a cyanomethylamide in the para position on the 4-phenyl group [15]. These modifications may have enhanced the interaction with Asp994 due to the presence of the nitrile group. Additionally, the pyrimidine ring may have been shifted closer to Leu932, since the added group is larger than the original substituent. While CYT387 had a high inhibitory potential against Jak2, it was also found to be a potent inhibitor of Jak1 using in vitro based assays [16].
Azaindoles and Deazapurines
The previously mentioned Vertex Pharmaceuticals research group also identified a number of potent compounds with azaindole or deazapurine cores. These types of structures bind to the Jak2 hinge region (Glu930 and Leu932) and are present in some additional Jak2 inhibitors such as CP-690550. In the most recent studies, ring-fused 7-azaindoles or deazapurines were synthesized and structurally optimized. The most effective molecules from these experiments contained fluorinated para-hydroxy phenyl substituents. Specifically, compound 15a was shown to form hydrogen bonds with Leu932, Glu930, Glu898, and the backbone NH of Phe995 [17]. In a separate publication, this group described the synthesis of a very similar compound that showed many of the same interactions, with an additional hydrogen bond to the backbone NH of Asp994 [18]. In these studies, the significance of the phenolic hydroxide is highlighted, because it interacts with both Glu898 and Phe995.
Pyrazines
In 2008, a research group at Astra Zeneca developed the Jak2 inhibitor AZ960, a pyrazolo nicotinonitrile with a Jak2 IC50 of < 3 nM [19]. In 2009, the same group developed a series of compounds that contained the hinge binding pyrazol-3-yl amine from AZ960 along with a pyrazine core structure. Optimization led to two compounds, 7c and 7h, which showed Jak2 inhibition with IC50 values similar to that of AZ960 [20]. Subsequent alterations of the hinge binding group led to the development of several compounds containing a thiazol-2-yl amine. These compounds were shown to be as effective as the previous analogs, indicating that the thiazol-2-yl amine could successfully replace the pyrazol-3-yl amine group. Additionally, the pyrazine core was subsequently replaced with a pyrimidine and similar Jak2 IC50 values were achieved [21]. While this work identified a novel structural class of Jak2 inhibitors, it did not identify the sites of physical interaction with Jak2.
Aminoindazoles
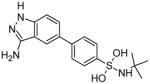
A group at SGX Pharmaceuticals recently developed a series of Jak2 inhibitors through fragment-based drug discovery and structure-based optimization. An initial hit was a 5-bromoaminoindazole that showed hydrogen bonding to Leu932 and Glu930. Replacing bromine with a 5-phenyl substituent led to a 25-fold increase in potency. Further modifications led to compound 13, which contains a 4-tert-butyl sulfonamide attached to the 5-phenyl group. Compound 13 was co-crystallized with Jak2 and the sulfonamide oxygen was found to interact with Asn981 through a water-bridged hydrogen bond [22].
Stilbenes
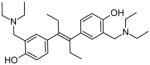
Through structure-based in silico screening, our laboratory identified a novel Jak2 inhibitor with a stilbene core. This compound, known as G6, forms hydrogen bonds with Leu932 and Glu930 and forms a salt bridge with Asp994 [23]. Our group was one of the first to show that Asp994 could be a significant point of interaction for Jak2 inhibition beyond the established hydrophobic interactions with the hinge region of Jak2. We have demonstrated that the stilbene structure is essential for the activity of G6, as non-stilbenoid derivatives show a reduction in Jak2 inhibition in vitro and bind the protein less efficiently in silico (unpublished data).
Benzoxazoles and Quinoxalines
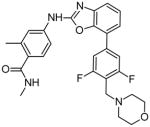
Very recently, Novartis developed a series of benzoxazole Jak2 inhibitors through scaffold morphing and structural optimization. The series was based on a pyrrolopyrimidine derivative, in which substitutions were made at positions 2 and 7 to improve potency and specificity. It was found that increasing Jak2 inhibition was accompanied by increased inhibition of other Jak proteins, but some degree of selectivity for Jak2 still remained. One of the best compounds of the series, compound 15, was shown to have high oral bioavailability in rats, but so far the molecular interactions of this inhibitor with Jak2 have not been characterized [24].
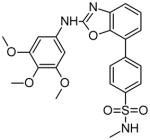
In a separate publication, this group reports the docking of benzoxazole and quinoxaline compounds into the Jak2 ATP-binding pocket [25]. Compound 1 forms two hydrogen bonds with the backbone of Leu932 and forms hydrophobic interactions with Val911, Gly993 and Met929 and with Val863 in the P-loop. Compound 2, a benzoxazole, mimicked these interactions very well. Additionally, scaffold morphing led to the development of the quinoxaline 4, which forms a nonconventional pseudo hydrogen bond with Leu932. Compound 4 was designed to mimic the hydrogen bonding of compound 3, another benzoxazole [25].
Summary, Significance, and Future Directions
Jak2 inhibition can be achieved through a variety of small molecule structures, as shown by the wide range of compounds that have been developed. A common feature of several of these compounds is binding to both the hinge region and the activation loop of Jak2. It is important for an inhibitor to have multiple points of contact within the binding pocket in order to achieve greater potency and/or specificity. The SAR properties of these compounds demonstrate that each has specific advantages.
The pyrimidine compound 40 has a unique hydrogen bond with Gly993 because of the presence of a 4-fluoro-benzylic amine. This unique bond is expected to provide an extra degree of specificity for Jak2, because Jak3 contains an Ala residue at this position [14]. Because many kinase ATP-binding pockets are highly homologous, it can often be difficult to obtain a high degree of specificity. Therefore, developing more compounds that specifically interact with Gly993 could be very useful for targeting Jak2.
Compound 9 has a phenolic hydroxide component that interacts with both Glu898 and Phe995 [18]. This unique interaction highlights the significance of the location of hydroxyl groups within an inhibitor. Another interesting characteristic of the azaindoles and deazapurines is the presence of 7- or 8-membered rings in the core structure. These ring constraints allow for strategic positioning of reactive groups within the binding pocket. This type of structure in combination with phenol groups could be further utilized in Jak2 inhibitory compounds.
Our research group has demonstrated that the stilbene core is a crucial element in G6-mediated inhibition of Jak2 [23]. Through modifications of the stilbene structure, we have found that manipulating the position of the hydroxyls and the size of amine groups has a significant impact on Jak2 inhibitory activity (unpublished data).
The aminoindazole 13 [22] is very interesting because it has a low Jak2 IC50 and was created through a fragment-based design scheme, after relatively few modifications were made to the initial fragment hit. This indicates that fragment-based discovery is indeed a valuable tool for designing Jak2 inhibitors, and could potentially be used to design compounds that interact with the critical amino acids within the binding pocket.
It is well known that the use of fluorinated substituents can often improve the potency of small molecule drugs. This technique does not usually improve the specificity of a drug, but can be used in combination with other modifications. Several of the studies discussed here take advantage of this technique, including those involving pyrazine compounds [20, 21]. It would be interesting to see docking or crystal structures of the pyrazine compounds in complex with Jak2. These would be expected to bind the hinge region because they were designed from a hinge-binding motif, and would likely have additional interactions similar to those formed by many of the other structures.
The benzoxazole and quinoxaline compounds have some interesting features, such as the pseudo hydrogen bond formed by compound 4 [25]. These studies introduce the concept of scaffold morphing as a means of developing Jak2 inhibitors and indicate that taking advantage of pseudo hydrogen bonding may be a practical consideration in future design schemes.
In summary, many structurally distinct Jak2 inhibitors have been developed recently through a wide variety of techniques. It would be difficult to state that any one class of compounds is the most effective, but each type discussed here may have potential for use in future clinical applications. The specificity and potency of Jak2 inhibitors is improving at a considerable rate, but many of the new inhibitors still need to be tested for their in vivo efficacy and toxicity. Future development of novel Jak2 inhibitors could certainly take advantage of the techniques applied in these studies, perhaps by combining multiple strategies. With the variety of structures that are available as starting points, it is possible that minor modifications could have major effects on activity.
Acknowledgments
This work was supported by National Institutes of Health Award R01-HL67277, an American Heart Association Greater Southeast Affiliate Grant in Aid (#0855361E), a University of Florida Opportunity Fund Award, a University of Florida/Moffitt Cancer Center Collaborative Initiative Award, and two Doctoral Fellowships from the University of Florida Alumni Foundation. We would like to thank Sarah Barilovits for proofreading the manuscript.
References
- 1.Schneider H, Cohen-Dayag A, Pecht I. Tyrosine phosphorylation of phospholipase C gamma 1 couples the Fc epsilon receptor mediated signal to mast cells secretion. Int Immunol. 1992;4:447–53. doi: 10.1093/intimm/4.4.447. [DOI] [PubMed] [Google Scholar]
- 2.Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, Lapidot Z, Leeder JS, Freedman M, Cohen A, Gazit A, Levitzki A, Roifman CM. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature. 1996;379:645–8. doi: 10.1038/379645a0. [DOI] [PubMed] [Google Scholar]
- 3.Gu L, Zhuang H, Safina B, Xiao XY, Bradford WW, Rich BE. Combinatorial approach to identification of tyrphostin inhibitors of cytokine signaling. Bioorg Med Chem. 2005;13:4269–78. doi: 10.1016/j.bmc.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 4.Iwamaru A, Szymanski S, Iwado E, Aoki H, Yokoyama T, Fokt I, Hess K, Conrad C, Madden T, Sawaya R, Kondo S, Priebe W, Kondo Y. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene. 2007;26:2435–44. doi: 10.1038/sj.onc.1210031. [DOI] [PubMed] [Google Scholar]
- 5.Ferrajoli A, Faderl S, Van Q, Koch P, Harris D, Liu Z, Hazan-Halevy I, Wang Y, Kantarjian HM, Priebe W, Estrov Z. WP1066 disrupts Janus kinase-2 and induces caspase-dependent apoptosis in acute myelogenous leukemia cells. Cancer Res. 2007;67:11291–9. doi: 10.1158/0008-5472.CAN-07-0593. [DOI] [PubMed] [Google Scholar]
- 6.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D’Andrea A, Frohling S, Dohner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–97. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 7.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 8.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–8. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 9.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 10.Zhao R, Xing S, Li Z, Fu X, Li Q, Krantz SB, Zhao ZJ. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280:22788–92. doi: 10.1074/jbc.C500138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lipka DB, Hoffmann LS, Heidel F, Markova B, Blum MC, Breitenbuecher F, Kasper S, Kindler T, Levine RL, Huber C, Fischer T. LS104, a non-ATP-competitive small-molecule inhibitor of JAK2, is potently inducing apoptosis in JAK2V617F-positive cells. Mol Cancer Ther. 2008;7:1176–84. doi: 10.1158/1535-7163.MCT-07-2215. [DOI] [PubMed] [Google Scholar]
- 12.Grunberger T, Demin P, Rounova O, Sharfe N, Cimpean L, Dadi H, Freywald A, Estrov Z, Roifman CM. Inhibition of acute lymphoblastic and myeloid leukemias by a novel kinase inhibitor. Blood. 2003;102:4153–8. doi: 10.1182/blood-2003-03-0860. [DOI] [PubMed] [Google Scholar]
- 13.Tondel K, Anderssen E, Drablos F. Protein Alpha Shape Similarity Analysis (PASSA): a new method for mapping protein binding sites. Application in the design of a selective inhibitor of tyrosine kinase 2. J Comput Aided Mol Des. 2002;16:831–40. doi: 10.1023/a:1023840914434. [DOI] [PubMed] [Google Scholar]
- 14.Ledeboer MW, Pierce AC, Duffy JP, Gao H, Messersmith D, Salituro FG, Nanthakumar S, Come J, Zuccola HJ, Swenson L, Shlyakter D, Mahajan S, Hoock T, Fan B, Tsai WJ, Kolaczkowski E, Carrier S, Hogan JK, Zessis R, Pazhanisamy S, Bennani YL. 2-Aminopyrazolo [1,5-a]pyrimidines as potent and selective inhibitors of JAK2. Bioorg Med Chem Lett. 2009;19:6529–33. doi: 10.1016/j.bmcl.2009.10.053. [DOI] [PubMed] [Google Scholar]
- 15.Burns CJ, Bourke DG, Andrau L, Bu X, Charman SA, Donohue AC, Fantino E, Farrugia M, Feutrill JT, Joffe M, Kling MR, Kurek M, Nero TL, Nguyen T, Palmer JT, Phillips I, Shackleford DM, Sikanyika H, Styles M, Su S, Treutlein H, Zeng J, Wilks AF. Phenylaminopyrimidines as inhibitors of Janus kinases (JAKs) Bioorg Med Chem Lett. 2009;19:5887–92. doi: 10.1016/j.bmcl.2009.08.071. [DOI] [PubMed] [Google Scholar]
- 16.Pardanani A, Lasho T, Smith G, Burns CJ, Fantino E, Tefferi A. CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia. 2009;23:1441–5. doi: 10.1038/leu.2009.50. [DOI] [PubMed] [Google Scholar]
- 17.Wang T, Ledeboer MW, Duffy JP, Pierce AC, Zuccola HJ, Block E, Shlyakter D, Hogan JK, Bennani YL. A novel chemotype of kinase inhibitors: Discovery of 3,4-ring fused 7-azaindoles and deazapurines as potent JAK2 inhibitors. Bioorg Med Chem Lett. 2010;20:153–6. doi: 10.1016/j.bmcl.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 18.Wang T, Duffy JP, Wang J, Halas S, Salituro FG, Pierce AC, Zuccola HJ, Black JR, Hogan JK, Jepson S, Shlyakter D, Mahajan S, Gu Y, Hoock T, Wood M, Furey BF, Frantz JD, Dauffenbach LM, Germann UA, Fan B, Namchuk M, Bennani YL, Ledeboer MW. Janus kinase 2 inhibitors. Synthesis and characterization of a novel polycyclic azaindole. J Med Chem. 2009;52:7938–41. doi: 10.1021/jm901383u. [DOI] [PubMed] [Google Scholar]
- 19.Gozgit JM, Bebernitz G, Patil P, Ye M, Parmentier J, Wu J, Su N, Wang T, Ioannidis S, Davies A, Huszar D, Zinda M. Effects of the JAK2 inhibitor, AZ960, on Pim/BAD/BCL-xL survival signaling in the human JAK2 V617F cell line SET-2. J Biol Chem. 2008;283:32334–43. doi: 10.1074/jbc.M803813200. [DOI] [PubMed] [Google Scholar]
- 20.Ioannidis S, Lamb ML, Davies AM, Almeida L, Su M, Bebernitz G, Ye M, Bell K, Alimzhanov M, Zinda M. Discovery of pyrazol-3-ylamino pyrazines as novel JAK2 inhibitors. Bioorg Med Chem Lett. 2009;19:6524–8. doi: 10.1016/j.bmcl.2009.10.054. [DOI] [PubMed] [Google Scholar]
- 21.Ioannidis S, Lamb ML, Almeida L, Guan H, Peng B, Bebernitz G, Bell K, Alimzhanov M, Zinda M. Replacement of pyrazol-3-yl amine hinge binder with thiazol-2-yl amine: Discovery of potent and selective JAK2 inhibitors. Bioorg Med Chem Lett. 2010;20:1669–73. doi: 10.1016/j.bmcl.2010.01.091. [DOI] [PubMed] [Google Scholar]
- 22.Antonysamy S, Hirst G, Park F, Sprengeler P, Stappenbeck F, Steensma R, Wilson M, Wong M. Fragment-based discovery of JAK-2 inhibitors. Bioorg Med Chem Lett. 2009;19:279–82. doi: 10.1016/j.bmcl.2008.08.064. [DOI] [PubMed] [Google Scholar]
- 23.Kiss R, Polgar T, Kirabo A, Sayyah J, Figueroa NC, List AF, Sokol L, Zuckerman KS, Gali M, Bisht KS, Sayeski PP, Keseru GM. Identification of a novel inhibitor of JAK2 tyrosine kinase by structure-based virtual screening. Bioorg Med Chem Lett. 2009;19:3598–601. doi: 10.1016/j.bmcl.2009.04.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerspacher M, Furet P, Pissot-Soldermann C, Gaul C, Holzer P, Vangrevelinghe E, Lang M, Erdmann D, Radimerski T, Regnier CH, Chene P, De Pover A, Hofmann F, Baffert F, Buhl T, Aichholz R, Blasco F, Endres R, Trappe J, Drueckes P. 2-Amino-aryl-7-aryl-benzoxazoles as potent, selective and orally available JAK2 inhibitors. Bioorg Med Chem Lett. 2010;20:1724–7. doi: 10.1016/j.bmcl.2010.01.069. [DOI] [PubMed] [Google Scholar]
- 25.Furet P, Gerspacher M, Pissot-Soldermann C. Design of two new chemotypes for inhibiting the Janus kinase 2 by scaffold morphing. Bioorg Med Chem Lett. 2010 doi: 10.1016/j.bmcl.2010.01.151. [DOI] [PubMed] [Google Scholar]