

Abstract
The endocannabinoid (eCB) system plays central roles in the regulation of food intake and energy expenditure. Its alteration in activity contributes to the development and maintenance of obesity. Stimulation of the cannabinoid receptor type 1 (CB1 receptor) increases feeding, enhances reward aspects of eating, and promotes lipogenesis, whereas its blockade decreases appetite, sustains weight loss, increases insulin sensitivity, and alleviates dysregulation of lipid metabolism. The hypothesis has been put forward that the eCB system is overactive in obesity. Hippocampal circuits are not directly involved in the neuronal control of food intake and appetite, but they play important roles in hedonic aspects of eating. We investigated the possibility whether or not diet-induced obesity (DIO) alters the functioning of the hippocampal eCB system. We found that levels of the two eCBs, 2-arachidonoyl glycerol (2-AG) and anandamide, were increased in the hippocampus from DIO mice, with a concomitant increase of the 2-AG synthesizing enzyme diacylglycerol lipase-α and increased CB1 receptor immunoreactivity in CA1 and CA3 regions, whereas CB1 receptor agonist-induced [35S]GTPγS binding was unchanged. eCB-mediated synaptic plasticity was changed in the CA1 region, as depolarization-induced suppression of inhibition and long-term depression of inhibitory synapses were enhanced. Functionality of CB1 receptors in GABAergic neurons was furthermore revealed, as mice specifically lacking CB1 receptors on this neuronal population were partly resistant to DIO. Our results show that DIO-induced changes in the eCB system affect not only tissues directly involved in the metabolic regulation but also brain regions mediating hedonic aspects of eating and influencing cognitive processes.
Introduction
The endocannabinoid (eCB) system is a physiological signaling system that comprises two types of membrane receptors, called CB1 and CB2 receptors, endogenous ligands (eCBs), and the machinery for their biosynthesis and degradation (Pagotto et al., 2006; Lutz, 2002).
This neuromodulatory system is widely expressed in the CNS and modulates synaptic transmission and plasticity. Released from the postsynaptic site, eCBs can act retrogradely on presynaptic CB1 receptors modulating the release of neurotransmitter with both tonic (Neu et al., 2007) and phasic (Mackie, 2006) mechanisms. At the GABAergic inputs of the CA1 hippocampal region, the activation of CB1 receptors leads to different forms of synaptic plasticity, such as depolarization-induced suppression of inhibition (DSI) or long-term depression of inhibitory synapses (I-LTD) (Lovinger, 2008). At glutamatergic synapses, CB1 receptors were also demonstrated to be present and functional (Monory et al., 2006), even though only a few studies (Ohno-Shosaku et al., 2002; Straiker and Mackie, 2005; Hofmann et al., 2008) showed a modulation of synaptic plasticity in the hippocampus, which might be caused by the very low expression of CB1 receptors in this neuronal subpopulation and/or the difficulty in having successful electrophysiological protocols.
Whereas the hippocampal region and related neuronal networks are most commonly associated with memory processing, several studies demonstrated a role of the hippocampus in the control of food intake. Hippocampal damage can result in hyperphagia (Forloni et al., 1986), whereas the hippocampal neuronal network contributes to eating behavior by processing satiety signals, such as cholecystokinin, ghrelin, and motilin (Guan et al., 2003; Davidson and Jarrard, 2004; Diano et al., 2006). Moreover, obese patients tasting a liquid meal showed a decreased activity in the posterior hippocampus compared with lean control subjects (DelParigi et al., 2005), and other imaging studies demonstrated that the desire of food is related to the activation of the hippocampus, reflecting a direct role of this brain area with memories associated with food (Pelchat et al., 2004).
CB1 receptor agonists stimulate feeding and particularly enhance the reward aspects of eating (Cota et al., 2006), whereas CB1 receptor antagonists were shown to consistently reduce body weight in obese patients (Van Gaal et al., 2005; Scheen, 2008). Neuronal expression of CB1 receptors are crucially involved in both motivational and metabolic aspects of these effects (Bellocchio et al., 2010; Quarta et al., 2010). The eCB system in the hippocampus may participate in driving appetite and, thus, determining total energy intake and the severity of obesity.
Here, we tested the hypothesis whether or not the eCB system is altered in the hippocampus of a well established animal model of obesity and its associated conditions (such as insulin resistance, type 2 diabetes, dyslipidemia), the diet-induced obesity (DIO) mice (Buettner et al., 2007), similarly as it was previously found to occur in peripheral organs (particularly in adipose tissue and liver) of obese rodents (Di Marzo, 2008). We also asked whether or not eCB-mediated synaptic processes are altered in the hippocampus of DIO mice compared with standard-diet fed control mice.
Materials and Methods
Animals.
Three-month-old male CB1 receptor-deficient mice (CB1−/−) and littermate (CB1+/+) controls, and male wild-type mice in C57BL/6N background were single-housed in standard laboratory cages. CB1−/−, GABA-CB1−/− mice and corresponding wild-type littermate controls were generated and genotyped as described previously (Marsicano et al., 2002; Monory et al., 2006). Special attention must be given to GABA-CB1−/−. To prevent germ line deletion of the CB1 receptor gene, CB1floxed/floxed;dlx-cre+ male mice must be crossed with CB1floxed/floxed female mice, as the presence of dlx-cre in the female germ line causes the general deletion of the CB1 receptor gene. Genotyping of conditional knock-outs always included primers to monitor whether or not germ line deletion has occurred. By the age of 6 weeks, after 1 week of habituation in single housing, mice were sorted by body weight to obtain homogeneous groups. Mice were fed for 12 weeks with a high-fat diet (HFD) (17.0 kJ/g; Tso's High Fat Diet D03082706; Piccioni Laboratory) or standard mouse chow [standard diet (SD)] (8.0 kJ/g; Altromin). Animals' weights were measured weekly at the same time (9:00 A.M.; light phase for 12 h, light on at 7:00 A.M.). C57BL/6N mice (under SD and HFD) were used for determination of eCB levels, Western blot analyses, [35S]GTPγS binding assays, immunohistochemistry, and electrophysiological experiments, whereas CB1−/− and GABA-CB1−/− and corresponding wild-type littermate controls were used for body weight curves. In all experiments, in which animals were killed for anatomical or electrophysiological experiments, mice were fed ad libitum and killed at 9:00 A.M. Brain and visceral fat depots (all the fat tissue present in the abdominal cavity) were collected and analyzed. All experimental procedures were approved by the local Ethics Committee on Animal Health and Care of the State Rhineland-Palatinate.
Endocannabinoid measurement.
Hippocampi of SD and HFD ad libitum fed mice were punched from the frozen brains using a cryostat and cylindrical brain punchers (Fine Science Tools; internal diameter, 1.0 mm). Location and length of the punches were chosen on the basis of a stereotaxic atlas (Paxinos and Franklin, 2001) as follows: 1.2 mm for dorsal hippocampus (starting at 1.6 mm posterior to bregma), including dentate gyrus, CA1, and CA3 hippocampal regions; 1.2 mm for ventral hippocampus (starting at 2.5 mm posterior to bregma), including DG, CA1, and CA3 hippocampal regions; 1 mm for prefrontal cortex (starting at 2.8 mm anterior to bregma); 1.2 mm for basolateral amygdala (BLA) (starting at 0.58 mm posterior to bregma); 1 mm for cerebellum (starting at 5.8 mm posterior to bregma). After punching, the actual site of dissection was checked by histological analysis of toluidine blue-stained sequential sections. Punches not corresponding to the expected anatomical region were excluded from additional analysis. Brain punches were kept frozen at −80°C until processing for eCB analysis. Pieces of tissue were weighed, homogenized, and extracted by liquid–liquid extraction with ethyl acetate/n-hexane (9:1, v/v). The extracts were evaporated under nitrogen, and the reconstituted samples were analyzed for arachidonoyl ethanolamide (anandamide; AEA), palmitoyl ethanolamide (PEA), 2-arachidonoyl glycerol (2-AG), 1-arachidonoyl glycerol (1-AG), and oleoyl ethanolamide (OEA). The respective deuterated analytes AEA-d8, PEA-d4, 2-AG-d5, 1-AG-d5, and OEA-d2 were used as internal standards. All analytes were from Cayman Chemical. HPLC analysis was done under gradient conditions using a Luna HST C18(2) column (100 mm length × 2 mm inner diameter, 2.5 μm particle size; Phenomenex). Mass spectrometry (MS) and MS/MS analyses were performed on an API5000 triple-quadrupole tandem mass spectrometer with a Turbo V source (Applied Biosystems) in the negative-ion mode. Precursor-to-product ion transitions of m/z 346→259 for AEA, m/z 354→86 for AEA-d8, m/z 298→268 for PEA, m/z 302→272 for PEA-d4, m/z 377→303 for 2-AG and 1-AG, m/z 382→303 for 2-AG-d5 and 1-AG-d5, m/z 324→86 for OEA, and m/z 326→86 for OEA-d2 were used for the multiple-reaction monitoring with a dwell time of 75 ms. Concentrations of the calibration standards, quality controls, and unknowns were evaluated by Analyst software (version 1.4; Applied Biosystems). Variations in accuracy and intraday and interday precision (n = 6 for each concentration, respectively) were <15% over the range of the whole calibration range.
Immunohistochemistry.
Mice were anesthetized with pentobarbital and transcardially perfused with 4% paraformaldehyde (PFA) in PBS. Brains were removed and postfixed for 24 h in 4% PFA solution, and then treated with 30% sucrose in PBS for 48 h and stored at −80°C until use. Thirty-micrometer-thick brain slices were prepared and stored at −20°C in cryoprotection solution (25% glycerol, 25% ethylene glycol, and 50% PBS) until use. CB1 receptor was detected by an affinity-purified polyclonal antibody against the last 15 residues of the rat CB1 receptor (Bodor et al., 2005). All incubation steps were performed in six-well plate (4 ml of solution per well) on a wave shaker (Heidolph). Free-floating sections were washed with Tris-buffered saline (TBS) (25 mm Tris/HCl, 150 mm NaCl, pH 7.6; for 10 min). Sections were preincubated in blocking solution (5% normal donkey serum, 2.5% bovine serum albumin, 0.3% Triton X-100 in TBS) for 1 h. After blocking, sections were incubated with the primary antibody diluted in blocking solution for overnight at 4°C. Sections were washed in 1× TBS five times for 5 min at room temperature (RT) and then incubated for 2 h with a FITC-labeled anti-rabbit IgG from goat (dilution, 1:100; Jackson ImmunoResearch). Unbound secondary antibody was removed by washing in 1× TBS-T (1× TBS/0.1% Triton X-100). Sections were counterstained with Hoechst 33258 (2 μg/ml). Sections were washed twice for 2 min in distilled water and then mounted onto glass slides to dry for 2–4 h at 37°C. To clear off the remaining salt, slides were dipped for 2 s into distilled water and dried overnight in a dust-free area at RT, and then covered with Moviol mounting medium. As negative controls, brain sections from CB1−/− mice and staining without primary antiserum, respectively, were used.
In situ hybridization.
Radioactive in situ hybridization (ISH) experiments were performed as previously described (Marsicano and Lutz, 1999). Mice in SD and HFD were anesthetized with isoflurane and decapitated. Brains were isolated, immediately frozen on dry ice, and stored at −80°C. Brains were mounted on Tissue Freezing Medium (Jung) and 18-μm-thick coronal sections were cut on a Microm HM560 cryostat (Microm). Sections were mounted on frozen SuperFrost Plus slides (Menzel) and stored at −20°C until use. CB1 receptor riboprobes were prepared as previously described (Marsicano and Lutz, 1999). After linearization with BamHI, the antisense riboprobe was synthesized with T3 RNA polymerase (Roche). For the generation of the sense riboprobe, XhoI was used for linearization and T7 RNA polymerase (Roche) for RNA synthesis. The incubation with sense riboprobes did not show any signal.
Agonist-stimulated [35S]GTPγS binding.
Hippocampi from SD and HFD mice were isolated and homogenized in 1 ml of ice-cold membrane buffer (50 mm Tris-HCl, pH 7.4, 3 mm MgCl2, 0.2 mm EGTA, Complete protease inhibitor; Roche), appropriate aliquots were stored at −80°C until use. Homogenates were thawed, diluted in assay buffer, and homogenized, and protein content was determined by Bradford assay. To remove endogenous adenosine (Breivogel and Childers, 2000), samples were preincubated for 10 min at 30°C in 0.004 U/ml adenosine deaminase (240 U/mg of protein; Sigma-Aldrich). Hippocampus homogenates (5–8 μg of protein) were incubated with 0.05 nm [35S]GTPγS and appropriate concentrations of 3-(1,1-dimethylheptyl)-(−)-11-hydroxy-Δ8-tetrahydrocannabinol (HU-210) in assay buffer (50 mm Tris-HCl, pH 7.4, 3 mm MgCl2, 0.2 mm EGTA, 100 mm NaCl) containing 30 μm GDP and 0.1% BSA in a final volume of 0.5 ml for 60 min at 30°C. Nonspecific binding was determined in presence of 30 μm GTPγS and subtracted to yield specific binding values. Bound [35S]GTPγS was harvested by vacuum filtration through Whatman GF/B filters with a Brandel Cell Harvester and three times washed with 3 ml of ice-cold 50 mm Tris-HCl buffer, pH 7.4. Radioactivity was determined by liquid scintillation counting after overnight incubation of filters in 2.5 ml of scintillation mixture Aquasafe 300 plus from Zinsser Analytic.
Western blot analysis.
Hippocampi from SD and HFD mice were isolated and homogenized. Twenty micrograms of total protein were resolved by 15% SDS-PAGE and electroblotted onto nitrocellulose membrane (HyBond-C Extra; GE Healthcare). Primary antibodies used were CB1 receptor (1:1000; Cayman), fatty acid amide hydrolase (FAAH) [1:500; molecular weight (MW), 70 kDa], monoacylglycerol lipase (MAGL) (1:500; MW, 35/37 kDa) (Straiker et al., 2009), and diacyglycerol lipase isoforms α and β (DAGL-α and DAGL-β) (MW, 100 and 70 kDa, respectively; 1:500) (Berghuis et al., 2007); α-tubulin (MW, 55 kDa) (Sigma-Aldrich; 1:400,000) was used as loading control. Secondary antibodies were horseradish peroxidase-conjugated anti-rabbit IgG (Dianova). Western blot analysis was performed using a chemiluminescence system (Luminol). Detection was made by autoradiography. Relative protein levels were calculated after normalization to the loading control (α-tubulin).
Electrophysiology.
For DSI experiments in CA1 region (Wilson et al., 2001), sagittal hippocampal slices (300 μm thick) were obtained from mice and maintained in artificial CSF (ACSF) for at least 40 min before recording. ACSF contained the following (in mm): 125 NaCl, 1.25 NaH2PO4, 25 glucose, 2.5 KCl, 2.5 CaCl2, 2 MgCl2, 25 NaHCO3, 0.005 NBQX [2,3-dihyro-6-nitro-7-sulfamoyl-benzo(f)quinoxaline], 0.05 APV [(2R)-amino-5-phosphonovaleric acid], gassed with carbogen (95% O2/5% CO2) at 25°C. 4-Nitrophenyl-4-[bis(1,3-benzodioxol-5-yl)(hydroxy)methyl] piperidine-1-carboxylate (JZL184) (1 mm) (from Cayman) was initially dissolved in 100% DMSO. Sixty to 120 min before recordings were performed, slices were incubated with JZL184 at the final concentration of 100 nm (containing 0.01% DMSO). Neurons were recorded in the whole-cell voltage-clamp mode (holding potential, −60 mV), using electrodes (open tip resistance, 2–5 MΩ) filled with a solution containing the following (in mm): 100 CsCH3SO3, 60 CsCl, 10 HEPES, 0.2 EGTA, 1 MgCl2, 1 MgATP, 0.3 Na3GTP, 5 QX-314 (lidocaine N-ethyl bromide), pH 7.3, 275 mOsm. Recordings were accepted only if the holding current was <100 pA. Current was low-pass filtered at 1 kHz and digitized at 3 kHz. IPSCs in CA1 pyramidal cells were evoked using a bipolar tungsten electrode placed within the CA1 stratum radiatum. DSI tests consisted of 30 stimuli at 0.33 Hz and a depolarization step to 0 mV for 3 s after the 13th stimulus. DSI amplitude was calculated using the mean of the three evoked IPSCs (eIPSCs) just before and after depolarization. I-LTD was commonly induced after 20 min of stable baseline by high-frequency stimulation (HFS), which consisted of two trains (20 s apart), each containing 100 pulses at 100 Hz. The magnitude of I-LTD was calculated by comparing averaged responses 35–40 min after HFS with baseline-averaged responses.
Data analysis.
Signals from in situ hybridization, immunohistochemistry and Western blots were quantified using NIH ImageJ (Image Processing and Analysis in Java) and expressed as integrated density (pixel number/area). For immunohistochemistry experiments, hippocampal regions CA1, CA3, and dentate gyrus were chosen and defined on the basis of a stereotaxic atlas (Paxinos and Franklin, 2001) (n = 3 per group; three sections per animal). Data were analyzed using t test and a two-way ANOVA followed by t test. For electrophysiology, data are presented as mean ± SEM. For DSI strength, data were analyzed using two-way ANOVA for repeated measures, whereas failures in I-LTD induction were measured using Fisher's exact test. For JZL184, two-way ANOVA and Bonferroni's post hoc test were used. For agonist-stimulated [35S]GTPγS binding, data were analyzed with GraphPad Prism 4.0 software (GraphPad Prism Software) using nonlinear regression and sigmoidal curve fitting to obtain potency (EC50) and efficacy (Emax) values. Basal activities were measured in the absence of receptor agonists and defined as 0% in each experiment. All data are expressed as percentage stimulation above basal [35S]GTPγS binding and are the means ± SEM of two experiments, each performed in quadruplicate. The unpaired, two-tailed t test was used to evaluate statistical significance with p < 0.05 as level of statistical significance.
Results
Involvement of CB1 receptors on GABAergic neurons in diet-induced obesity
First, in replication of experiments performed previously (Cota et al., 2003; Ravinet Trillou et al., 2004), male CB1+/+ and CB1−/− were fed ad libitum on a SD and HFD from week 6 to week 18 of age, and body weight was recorded weekly. Starting from week 6, CB1−/− mice had a lower body weight than CB1+/+ mice on either diet, and CB1−/− mice maintained a significantly reduced body weight compared with CB1+/+ mice on either diet throughout the whole period of observation (p < 0.05) (Fig. 1A). Thus, CB1−/− mice were lean under SD, and under HFD, they were resistant to DIO under HFD. We also observed, as previously reported (Cota et al., 2003; Ravinet Trillou et al., 2004), a reduced caloric intake in the CB1−/− mice in both SD and HFD compared with respective CB1+/+ controls (data not shown).
Figure 1.
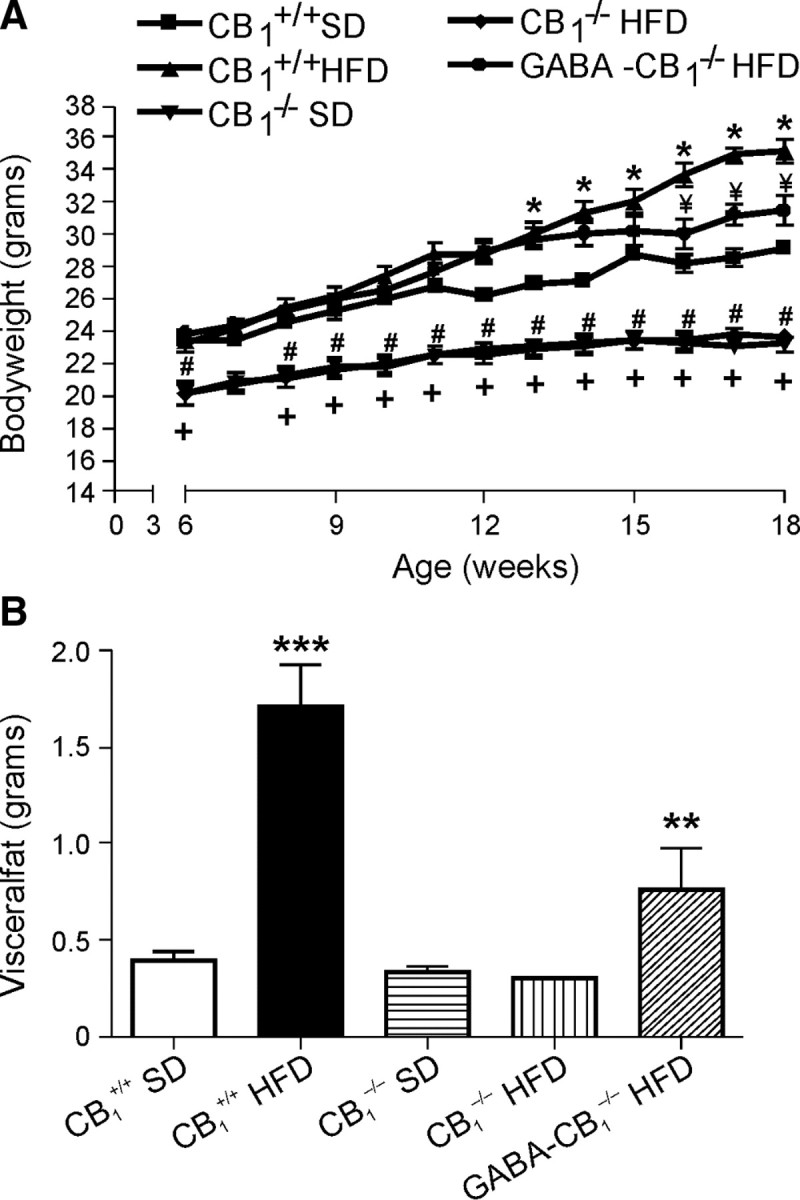
A, Growth curve of wild-type (CB1+/+) and CB1 knock-out (CB1−/−) mice during 12 weeks of SD and HFD treatment. Data are expressed as mean ± SEM. CB1−/−; CB1+/+, n = 10 mice per group; GABA-CB1−/−, n = 7 mice. *p < 0.05, CB1+/+ SD vs CB1+/+ HFD; #p < 0.05, CB1+/+ SD vs CB1−/− SD; +p < 0.05, CB1+/+ SD vs CB1−/− HFD; ¥p < 0.001, CB1+/+ HFD vs GABA-CB1−/− HFD; two-way ANOVA analysis, Bonferroni's post hoc test. B, Total amount of visceral fat (in grams) in CB1+/+, CB1−/− mice, and GABA-CB1−/− under SD and HFD, respectively, measured at the end of the treatment. ***p < 0.001, CB1+/+ SD vs CB1+/+ HFD; **p < 0.01, CB1+/+ HFD vs GABA-CB1−/− HFD; t test analysis.
As CB1 receptors are abundantly expressed in forebrain GABAergic neurons, in particular in the hippocampus, we asked whether or not specific loss of CB1 receptor on GABAergic neurons (GABA-CB1−/−) (Monory et al., 2006) affects DIO. Under SD, GABA-CB1−/− mice and wild-type controls had the same body weight curve (data not shown). Interestingly, however, under HFD diet, GABA-CB1−/− mice and CB1+/+ controls showed the same body weight only during the first 10 weeks of HFD treatment (Fig. 1A). After this time point, the body weight of the GABA-CB1−/− mice was significantly lower than the body weight of the CB1+/+ control mice. It is known that a prominent effect of HFD is to significantly increase the amount of visceral fat in the CB1+/+ mice, whereas no change of visceral fat is present in CB1−/− mice (Ravinet Trillou et al., 2004) (Fig. 1B). At the end of the HFD treatment, GABA-CB1−/− mice also showed a reduced amount of visceral fat compared with CB1+/+ mice (Fig. 1B) (t test analysis, **p < 0.01, ***p < 0.001). We did not find any significant difference in caloric intake under HFD in GABA-CB1−/− compared with wild-type controls (data not shown), suggesting that CB1 receptors on GABAergic terminals influence peripheral metabolic homeostatic mechanisms, such as energy uptake and expenditure. In summary, our HFD feeding schedule is able to induce obesity, a process that is absent in CB1−/− mice. In addition, we found that the expression of CB1 receptor in GABAergic neurons plays a central role in body weight regulation under HFD, preventing in part the development of DIO.
Effects of diet-induced obesity on endocannabinoid levels in the hippocampus
As CB1 receptors are critically involved in the development of DIO, we asked the complementary question of whether or not DIO may lead to an enhancement of the activity of the eCB system. Such an alteration under DIO was previously reported in the hypothalamus and peripheral organs (Di Marzo, 2008). For the determination of eCB levels, wild-type C57BL/6N mice were fed for 12 weeks with SD and HFD. At the end of the treatment, hippocampal regions were isolated by punching of cryosections, and eCB levels were determined by liquid chromatography–MS/MS. Levels of 2-AG and anandamide (AEA) were significantly increased in the HFD group compared with the SD group (Fig. 2A,B) (SD group, n = 4; HFD group, n = 8; t test analysis, *p < 0.05, **p < 0.01). To test whether the increase in eCB levels is a general phenomenon, we analyzed several other brain regions. No significant differences were observed in the BLA and cerebellum (Fig. 2C–F) (for BLA: SD group, n = 7; HFD group, n = 4; for cerebellum: SD group, n = 5; HFD group, n = 3). We observed a slight reduction of AEA levels in the prefrontal cortex (PFC) of HFD-fed mice (Fig. 2H) (*p < 0.05; SD group, n = 4; HFD group, n = 4). No significant differences were found in the levels of PEA, 1-AG, and OEA in any of the regions analyzed. Thus, increased levels of eCBs in the hippocampus might enhance eCB signaling in neurons.
Figure 2.

2-AG and anandamide levels in different brain regions. A, B, Increase of 2-AG and anandamide in hippocampus of HFD-fed mice compared with SD-fed mice (n = 4, SD group; n = 8 mice, HFD group; t test analysis, *p < 0.05, **p < 0.01). C–F, No differences were observed in BLA and cerebellum. G, H, In the prefrontal cortex of mice fed with HFD, the levels of anandamide were slightly reduced (t test analysis, p < 0.01). Data are expressed in picomoles/milligram wet tissue ± SEM.
Diet-induced obesity influences CB1 receptor expression in hippocampus
Our finding of increased levels of the CB1 receptor agonists, 2-AG and AEA, in the hippocampus prompted us to evaluate mRNA and protein levels of CB1 in hippocampi from mice fed with SD or HFD. In the CA1 and CA3 neurons of HFD-fed mice, the amount of CB1 receptor mRNA was only slightly, but significantly increased (212.43 ± 1.50 vs 217.00 ± 0.80 arbitrary units for SD and HFD, respectively; t test, p < 0.01). No differences were observed in the dentate gyrus region (data not shown). To test whether CB1 receptor protein levels are altered, CB1 receptor immunoreactivity was quantified in the CA1, CA3, and dentate gyrus of hippocampi of SD- and HFD-fed mice, respectively. CB1 receptor protein levels were found to be slightly, but significantly increased in hippocampi from mice in HFD, both in CA1 and CA3 area, whereas no differences in CB1 receptor expression levels were found in the dentate gyrus (Fig. 3) (n = 3 per group; two-way ANOVA analysis, **p < 0.01, ***p < 0.001). Interestingly, this difference was only observed in the area of the stratum radiatum (Fig. 3B), but not in stratum pyramidale (Fig. 3C). The specificity of the CB1 receptor immunoreactivity was evidenced by performing immunostaining with the CB1 receptor antibody on coronal brain sections from CB1−/− mice. In these tissue samples, no CB1 receptor immunoreactivity was detected (data not shown). Thus, increased levels of CB1 receptor protein was detected specifically in hippocampal regions CA1 and CA3, but not in the dentate gyrus of DIO mice, which predicts, together with the increased levels of eCBs, an enhanced signaling via CB1 receptors.
Figure 3.

A, CB1 receptor immunoreactivity in hippocampi of SD-fed and HFD-fed animals (green signal: CB1 receptor). B, Quantification of CB1 receptor immunoreactivity in the area of the stratum radiatum, as indicated in A. C, CB1 receptor quantification in the layer of pyramidal cell bodies as indicated in A. Signal was quantified in CA1, CA3, and dentate gyrus (DG). Scale bar, 250 μm. Data are expressed in integrated density ± SEM. n = 3 mice per group. Two-way ANOVA analysis, Bonferroni's post hoc test: **p < 0.01, ***p < 0.001.
Stimulation of [35S]GTPγS binding by HU-210
Although increased eCB and CB1 receptor protein levels may suggest enhanced eCB signaling capacity, intracellular coupling mechanisms will also determine CB1 signaling efficiency. Therefore, we investigated whether CB1 receptor agonist stimulated G-protein activation is changed in DIO. We performed HU-210-stimulated [35S]-GTPγS binding in whole hippocampal homogenates of SD-fed and HFD-fed mice (Fig. 4). However, G-protein activation via CB1 receptors in hippocampal homogenates of HFD-fed mice did not show any significant differences compared with SD-fed mice.
Figure 4.

Stimulation of [35S]GTPγS binding in hippocampal homogenates of HFD-fed (filled circles) and SD-fed (open circles) mice by various concentrations of the cannabinoid receptor agonist HU-210. Assays were performed in the presence of GDP (30 μm) and [35S]GTPγS (0.05 nm) and were incubated for 60 min at 30°C as described in Materials and Methods. Nonspecific binding was determined in the presence of unlabeled GTPγS (30 μm). Basal binding was measured in the absence of receptor agonist and defined as 0% in each experiment. Data are expressed as percentage stimulation above basal [35S]GTPγS binding and are the means ± SEM, n = 2, all performed in quadruplicate. EC50 = 2.29 ± 0.04 nm (SD), 2.45 ± 0.06 nm (HFD); Emax = 68.83 ± 0.90 (SD), 68.74 ± 1.40 (HFD). Unpaired t test analysis, two-tailed: p = 0.9802.
Diet-induced obesity induces increased endocannabinoid biosynthesis pathway in hippocampus
As we found enhanced eCB levels in the hippocampus of DIO mice, we used Western blot analysis to quantify the levels of enzymes important in eCB synthesis and degradation: DAGL-α, DAGL-β, MAGL, and FAAH (Fig. 5A–D). No differences were observed in the levels of MAGL and FAAH protein, the enzymes involved in degradation of 2-AG and anandamide, respectively (Fig. 5C,D). However, the levels of DAGL-α protein were significantly increased (Fig. 5A) (N = 3; *p < 0.05, t test analysis), but DAGL-β protein levels were unchanged (Fig. 5B).
Figure 5.

Western blot analysis of hippocampus homogenates of SD-fed and HFD-fed mice. A, DAGL-α protein levels were significantly increased in HFD-fed mice. B, DAGL-β protein levels were unchanged. C, D, Protein levels of MAGL and FAAH, two major eCB degradation enzymes, were unchanged. N = 3; data are normalized for α-tubulin. *p < 0.05, t test analysis. Error bars indicate SEM.
Depolarization-induced suppression of inhibition and CB1 agonist application
DIO increases eCBs, eCB synthetic capacity, and CB1 receptor protein levels, and consequently may enhance CB1 receptor signaling at synapses. Given the involvement of eCBs as retrograde neurotransmitters at GABAergic synapses, we evaluated hippocampal DSI, a CB1 receptor-mediated suppression of GABAergic transmission (Wilson et al., 2001). As shown in Figure 6, A and B, 3 s of depolarization produced a significant reduction of evoked GABAergic currents in both groups (HFD: p < 0.001, n = 12; SD: p < 0.01, n = 11), but the HFD group showed a significantly higher inhibition compared with the SD group (p < 0.05). To confirm the CB1 receptor dependency of this process, we performed these recordings with brain slices of CB1−/− mice fed a SD (Marsicano et al., 2002). As previously described in the literature (Wilson et al., 2001), a 3 s depolarization did not evoke any DSI (p > 0.05; n = 4) (Fig. 6B). As 2-AG, rather than AEA, is thought to be the eCB responsible for DSI (Pan et al., 2009; Straiker et al., 2009; Gao et al., 2010; Tanimura et al., 2010), we next evaluated the effect of the blockade of 2-AG degradation on DSI using the MAGL inhibitor, JZL184. As expected, JZL184 at 100 nm strongly increased DSI magnitude in SD-fed mice (Fig. 6C) (SD, n = 16, vs SD plus JZL184, n = 10; p < 0.01). Interestingly, JZL184 was unable to increase DSI magnitude in HFD-fed mice (Fig. 6C) (HFD, n = 7, vs HFD plus JZL184, n = 14; p > 0.05), suggesting a saturation of endogenous CB1 receptor signaling in HFD mice.
Figure 6.

A, DSI in CA1 hippocampal pyramidal neurons of HFD- and age-matched SD-fed mice. Bottom panel, Normalized eIPSCs amplitude with 3 s depolarization steps (HFD-fed mice: n = 12, filled circles; SD-fed mice: n = 11, open circles). Top panel, Representative traces of eIPSCs before (1) and after (2) DSI induction. B, Summary of DSI magnitude of SD- and HFD-fed wild-type mice, and CB1−/− SD-fed mice. Data are mean ± SEM. **p < 0.001, ***p < 0.01 versus respective baseline before depolarization (100%, dotted line). C, Effect of the inhibitor of 2-AG degradation JZL184, 100 nm on DSI. Bottom panel, Summary of DSI magnitude in SD-fed and HFD-fed mice before and after application of JZL184. Data are mean ± SEM. *p < 0.05, **p < 0.01 versus SD-fed mice without JZL184 treatment. Top panel, Representative traces of eIPSCs, 3 s after depolarization in untreated (1) and JZL184-treated (2) slices. D, Application of the CB1 agonist WIN-55,212-2 (WIN) (5 μm) strongly reduced GABAergic currents with no significant difference between two diet groups.
We also addressed the question whether or not HFD enhances CB1 receptor signaling per se. To do this, we applied the CB1 receptor agonist (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo(1,2,3-de)-1,4-benzoxazin-6-yl]-1-napthalenylmethanone (WIN-55,212-2) at a concentration of 5 μm. This treatment strongly reduced GABAergic currents in both SD- and HFD-fed mice to the same extent (p > 0.05) (Fig. 6D) (SD, −54.15 ± 9.64%, p < 0.05; HFD, −52.25 ± 3.48%, p < 0.01).
Long-term depression of inhibitory synapses
Endocannabinoids and CB1 receptors are also involved in the long-term depression of certain inhibitory synapses in the hippocampus (I-LTD) (Chevaleyre and Castillo, 2003). HFS of Schaffer collaterals activates group I metabotropic glutamate receptors in the CA1 area, triggering a persistent reduction of GABA release at CA1 pyramidal cells, which is mediated by eCBs and CB1 receptors (Chevaleyre and Castillo, 2003). In our experiments, HFS produced a long-term reduction of GABAergic transmission compared with baseline in both SD and HFD group (Fig. 7A,B) (HFD: p < 0.001, n = 11; SD: p < 0.001, n = 5). Like DSI, this suppression was more pronounced in HFD-fed mice compared with the SD-fed group (Fig. 7B) (p < 0.01). Because of the long duration that the mice were on their respective diets, electrophysiological recordings were done in slices prepared from 18- to 22-week-old mice. SD-fed mice showed a high failure ratio in the generation of I-LTD (58.33%). Interestingly, this ratio was significantly lower in obese mice (8.33%; p < 0.01), as shown in Figure 7C. These results suggest HFD mice have an increased susceptibility to I-LTD, and likely other forms of eCB-mediated synaptic plasticity.
Figure 7.

HFS induces a stronger I-LTD at CA1 pyramidal cells of HFD-fed mice compared with SD-fed mice. A, Summary of I-LTD induced in HFD-fed mice (n = 12; filled circles) compared with SD-fed mice (n = 5; open circles). B, Top panel, Representative traces of eIPSCs before (1) and after (2) I-LTD induction. Bottom panel, Summary of I-LTD amplitudes obtained by calculating the averaged responses 25–30 min after HFS with the last 5 min of baseline-averaged and normalized responses (**p < 0.01; ***p < 0.001). Data are mean ± SEM. C, Failure ratio in eliciting I-LTD between HFD-fed (filled bars) and SD-fed mice (open bars; n = 12 per group).
Discussion
Hippocampal circuits play an important role in learning and memory, but also in the hedonic aspects of eating. They also provide one of the main external inputs to hypothalamic circuits, the key brain region for the control of food intake and energy balance. Since eCBs have been demonstrated to regulate synaptic transmission and plasticity in several brain areas including the hippocampus and hypothalamus (Lovinger, 2008; Kano et al., 2009), they are strongly involved in central and peripheral neuronal modulation of food intake pathways (Cota et al., 2006; Di Marzo, 2008).
For many years, it has been assumed that the effects of DIO primarily result in cardiovascular and metabolic dysfunctions (Kalmijn, 2000), overlooking an effect of diet on brain areas that are not directly related to processes controlling food intake. However, an elegant study (Molteni et al., 2002) showed that DIO can influence hippocampal structures via the regulation of neurotrophins, such as BDNF (brain-derived neurotrophic factor), or transcription factors such as CREB (cAMP response element-binding protein). Moreover, DIO was demonstrated to alter the activity of eCBs in extrahypothalamic brain areas. However, these analyses were restricted to the determination of CB1 receptor expression and ligand binding characteristics (Harrold et al., 2002; South and Huang, 2008), without examining changes of synaptic transmission or plasticity.
DIO could potentially affect several components of the eCB system, including receptor density and functionality, eCB levels, and their synthetic or degradation machinery. Here, we provide strong evidence that DIO causes profound hippocampal alterations: eCB levels are increased, with a concomitant increase in the 2-AG synthesizing enzyme DAGL-α. This ultimately leads to an enhanced CB1 receptor-dependent suppression of GABA transmission.
As expected, 12 weeks of high-fat diet treatment made mice obese, typically characterized by a marked increase in body weight and visceral fat. As previously described (Ravinet Trillou et al., 2004), CB1−/− mice showed a resistance to develop DIO. Here, we report for the first time the involvement of CB1 receptor expression in GABAergic neurons in controlling body homeostasis. In the late period of HFD treatment, despite similar caloric intake, GABA-CB1−/− mice maintained a body weight significantly lower than in HFD-fed CB1+/+ mice, although their weight was still more than in HFD-fed CB1−/− mice. Thus, GABA-CB1−/− mice are partly resistant to DIO, evidenced by their reduced adiposity. In conclusion, in DIO, we found a correlation between changes in GABAergic transmission (i.e., enhanced DSI, I-LTD) and the functional importance of CB1 receptors expressed on GABAergic neurons regarding the resistance to DIO.
A pertinent question for the role of the eCB system in the development of obesity is whether or not the increased activity of the eCB system is the cause or the consequence of obesity. The finding that GABA-CB1−/− mice under HFD are partly resistant only in the later phase in the emergence of obesity might argue for a scenario in which CB1 receptors on GABAergic neurons are required for a second phase of the obese state development (e.g., maintenance, “metabolic consolidation”), but not for the initiation of an obese state. However, additional investigations are needed to gain additional insights into this process. GABA-CB1−/− mice fed a HFD might be an interesting model system for the investigation of this intriguing question.
Previous studies described hypothalamic alterations of eCB levels caused by obesity (Di Marzo et al., 2001). Nevertheless, the possibility that DIO might affect extrahypothalamic areas has not been yet investigated in detail. Remarkably, we found that hippocampal levels of both major endocannabinoids (2-AG and anandamide) were significantly increased in DIO mice compared with lean control mice. To see whether the increase of eCB levels is a generalized phenomenon, eCB levels in BLA, cerebellum, and prefrontal cortex were measured. Of these regions, significant changes were seen only in PFC, where anandamide was decreased. Thus, hippocampus appears to be particularly prone to eCB dysregulation.
A possible explanation for the enhancement of hippocampal eCB levels might be an unbalanced ratio between eCB synthesis and degradation. No differences were found in the expression levels of FAAH and MAGL, two degrading enzymes for anandamide and 2-AG, respectively, whereas a significant increase of DAGL-α was detected, suggesting that DIO leads to increased production of 2-AG, which is not counterbalanced by the eCB degrading machinery. Our findings may represent an important step in understanding the role or the eCB system in the development of obesity and related pathologies.
To detail our study further, we investigated the possibility whether DIO also alters the expression and functionality of CB1 receptors. CB1 receptor mRNA and protein levels were found to be significantly increased in the hippocampus of DIO mice. CB1 immunoreactivity was particularly enhanced around pyramidal cell dendrites in the stratum radiatum. The increased mRNA levels found in this study are not in concordance with the study of Harrold et al. (2002). This discrepancy may be explained by the fact that, in the latter study, ligand binding capacity to CB1 receptor on brain sections was measured, whereas in our work, mRNA and protein levels were determined by ISH and immunohistochemistry, respectively. Thus, it seems that these different characteristics do not necessarily overlap. In our study, [35S]GTPγS binding did not reveal any difference in the CB1 receptor signaling/expression levels between the experimental groups. This might be attributable to the fact that, for these experiments, whole hippocampi were used, leading to a loss in spatial resolution. There are other possible explanations for this discrepancy: two different species were used (mouse vs rat), and it might be that the food composition and duration of HFD might influence the levels of protein and mRNA. At any rate, under our experimental conditions, we were able to demonstrate an overactivation of the eCB signaling system in the hippocampus in DIO.
We finally focused our studies on eCB-mediated synaptic plasticity. We found that DIO mice showed a stronger DSI compared with SD-fed mice, confirming an altered plasticity at GABAergic synapses in these animals. Such a change in short-term plasticity might be attributable to increased eCB levels, turnover rate of eCBs, and/or changes in receptor functionality. To address this crucial point, we first treated hippocampal slices with JZL184, the inhibitor of 2-AG degradation. 2-AG has been demonstrated to be the sole mediator in DSI (Szabo et al., 2006, Pan et al., 2009; Straiker et al., 2009; Gao et al., 2010; Tanimura et al., 2010). JZL184 was able to strongly increase DSI magnitude in slices from SD-fed mice, reaching the levels of DSI comparable with those seen in slices from DIO mice. Surprisingly, JZL184 failed to increase DSI magnitude in slices from DIO mice, suggesting a tonic saturation of 2-AG levels in these mice. However, application of the CB1 agonist WIN-55,212-2 reduced GABAergic currents to the same extent in both experimental groups, excluding a possible desensitization of CB1 receptors in slices from HFD-fed mice. Together, these results demonstrate that the increased short-term plasticity in DIO mice is evidently attributable to the elevated eCB synthesis, presumably 2-AG, considering that at this time no other eCB has been described that is capable to evoke DSI, and not to changes in receptor properties.
We also examined a long-term form of synaptic plasticity mediated by CB1 receptors at GABAergic synapses. DIO mice showed a stronger I-LTD compared with SD-fed mice, confirming our initial hypothesis that the eCB system is overactive in DIO mice. Interestingly, 60% of SD-fed mice failed to develop I-LTD with our induction protocol. A possible explanation for such a high rate of failures might be the fact that 6- to 7-month-old mice were used for the experiments because of the 12 weeks of diet treatment. Such old mice showed a reduced synaptic plasticity with a 40% reduction in I-LTD magnitude (F. Massa, unpublished data) and increased failure rate when compared with younger animals that are usually used in in vitro electrophysiological experiments. By contrast, in DIO mice, we found a normal elicitation and magnitude of I-LTD, comparable with younger mice. A plausible reason for this unexpected result might be found in the overactivation of the eCB system in obese mice, which leads to an elevated eCB tone, being able to counterbalance aging processes at GABAergic synapses. Our observations showed that the development/maintenance of the obesity state is strictly related to a dysregulation of the eCB system in its main components.
Overall, our findings add to previous reports and outline a direct and prominent role of the eCB system in the obesity-related pathophysiology of extrahypothalamic neuronal circuits. Future studies will have to identify the specific cell types producing eCBs in DIO mice, and whether it is a restricted neuronal mechanism or whether it involves glia and immune cells. Moreover, it will be interesting to address how these alterations in DSI and I-LTD may be related to changes in behaviors in DIO mice. To date, an enhanced hippocampal DSI in a pathophysiological context was reported in a rat model for febrile seizures, in which increases in CB1 receptor expression in GABAergic interneurons occurred concomitantly with an enhanced DSI and lowered seizure threshold (Chen et al., 2003, 2007). The role of DSI and I-LTD regarding the control and modulation of memory processing and emotional behaviors, however, is unclear and requires additional investigations (Lutz, 2009).
Footnotes
This work was supported in part by Deutsche Forschungsgemeinschaft Grants LU 775/3-1 and LU 775/4-1 (in the context of FOR926) (B.L.) and GE 695/3-1 (G.G.), European Union Grant REPROBESITY FPVII-223713 (B.L.), the LOEWE Lipid Signaling Forschungszentrum Frankfurt (G.G., H.S.), National Institutes of Health Grants DA11322 and DA21696 (K.M.), and a grant from the European Foundation for the Studies of Diabetes (G.M.). We thank Andrea Conrad and Martin Purrio for the help in the animal facility, Claudia Schwitter and Anne Rohrbacher for tissue preparation, Ruth Jelinek for Western blot experiments, Dr. Krisztina Monory for critically reading this manuscript, Dr. Sodikdjon Kodirov for the experimental help at the electrophysiology setup, and Dr. Giovanni Marsicano for providing CB1−/− mice.
The authors declare no competing financial interests.
References
- Bellocchio L, Lafenêtre P, Cannich A, Cota D, Puente N, Grandes P, Chaouloff F, Piazza PV, Marsicano G. Bimodal control of stimulated food intake by the endocannabinoid system. Nat Neurosci. 2010;13:281–283. doi: 10.1038/nn.2494. [DOI] [PubMed] [Google Scholar]
- Berghuis P, Rajnicek AM, Morozov YM, Ross RA, Mulder J, Urbán GM, Monory K, Marsicano G, Matteoli M, Canty A, Irving AJ, Katona I, Yanagawa Y, Rakic P, Lutz B, Mackie K, Harkany T. Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science. 2007;316:1212–1216. doi: 10.1126/science.1137406. [DOI] [PubMed] [Google Scholar]
- Bodor AL, Katona I, Nyíri G, Mackie K, Ledent C, Hájos N, Freund TF. Endocannabinoid signaling in rat somatosensory cortex: laminar differences and involvement of specific interneuron types. J Neurosci. 2005;25:6845–6856. doi: 10.1523/JNEUROSCI.0442-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breivogel CS, Childers SR. Cannabinoid agonist signal transduction in rat brain: comparison of cannabinoid agonists in receptor binding, G-protein activation, and adenylyl cyclase inhibition. J Pharmacol Exp Ther. 2000;295:328–336. [PubMed] [Google Scholar]
- Buettner R, Schölmerich J, Bollheimer LC. High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity. 2007;15:798–808. doi: 10.1038/oby.2007.608. [DOI] [PubMed] [Google Scholar]
- Chen K, Ratzliff A, Hilgenberg L, Gulyás A, Freund TF, Smith M, Dinh TP, Piomelli D, Mackie K, Soltesz I. Long-term plasticity of endocannabinoid signaling induced by developmental febrile seizures. Neuron. 2003;39:599–611. doi: 10.1016/s0896-6273(03)00499-9. [DOI] [PubMed] [Google Scholar]
- Chen K, Neu A, Howard AL, Földy C, Echegoyen J, Hilgenberg L, Smith M, Mackie K, Soltesz I. Prevention of plasticity of endocannabinoid signaling inhibits persistent limbic hyperexcitability caused by developmental seizures. J Neurosci. 2007;27:46–58. doi: 10.1523/JNEUROSCI.3966-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Cota D, Marsicano G, Tschöp M, Grübler Y, Flachskamm C, Schubert M, Auer D, Thöne-Reinecke C, Ortmann S, Cervino C, Linthorst A, Pasquali R, Lutz B, Stalla GK, Pagotto U. Decreased fat mass in mice deficient for cannabinoid receptor 1 is due to decreased orexigenic drive and impaired adipocyte differentiation. J Clin Invest. 2003;112:423–431. doi: 10.1172/JCI17725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota D, Tschop MH, Horvath TL, Levine AS. Cannabinoids, opioids and eating behavior: the molecular face of hedonism? Brain Res Brain Res Rev. 2006;51:85–107. doi: 10.1016/j.brainresrev.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Davidson TL, Jarrard LE. The hippocampus and inhibitory learning: a “Gray” area? Neurosci Biobehav Rev. 2004;28:261–271. doi: 10.1016/j.neubiorev.2004.02.001. [DOI] [PubMed] [Google Scholar]
- DelParigi A, Chen K, Salbe AD, Reiman EM, Tataranni PA. Sensory experience of food and obesity: a positron emission tomography study of the brain regions affected by tasting a liquid meal after a prolonged fast. Neuroimage. 2005;24:436–443. doi: 10.1016/j.neuroimage.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Diano S, Farr SA, Benoit SC, McNay EC, da Silva I, Horvath B, Gaskin FS, Nonaka N, Jaeger LB, Banks WA, Morley JE, Pinto S, Sherwin RS, Xu L, Yamada KA, Sleeman MW, Tschöp MH, Horvath TL. Ghrelin controls hippocampal spine synapse density and memory performance. Nat Neurosci. 2006;9:381–388. doi: 10.1038/nn1656. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. Targeting the endocannabinoid system: to enhance or reduce? Nat Rev Drug Discov. 2008;7:438–455. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Goparaju SK, Wang L, Liu J, Bátkai S, Járai Z, Fezza F, Miura GI, Palmiter RD, Sugiura T, Kunos G. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature. 2001;410:822–825. doi: 10.1038/35071088. [DOI] [PubMed] [Google Scholar]
- Forloni G, Fisone G, Guaitani A, Ladinsky H, Consolo S. Role of the hippocampus in the sex-dependent regulation of eating behavior: studies with kainic acid. Physiol Behav. 1986;38:321–326. doi: 10.1016/0031-9384(86)90101-0. [DOI] [PubMed] [Google Scholar]
- Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang MY, Strassle BW, Lu P, Mark L, Piesla MJ, Deng K, Kouranova EV, Ring RH, Whiteside GT, Bates B, Walsh FS, Williams G, Pangalos MN, et al. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J Neurosci. 2010;30:2017–2024. doi: 10.1523/JNEUROSCI.5693-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Tang M, Jiang Z, Peeters TL. Excitatory effects of motilin in the hippocampus on gastric motility in rats. Brain Res. 2003;984:33–41. doi: 10.1016/s0006-8993(03)03016-6. [DOI] [PubMed] [Google Scholar]
- Harrold JA, Elliott JC, King PJ, Widdowson PS, Williams G. Down-regulation of cannabinoid-1 (CB-1) receptors in specific extrahypothalamic regions of rats with dietary obesity: a role for endogenous cannabinoids in driving appetite for palatable food? Brain Res. 2002;952:232–238. doi: 10.1016/s0006-8993(02)03245-6. [DOI] [PubMed] [Google Scholar]
- Hofmann ME, Nahir B, Frazier CJ. Excitatory afferents to CA3 pyramidal cells display differential sensitivity to CB1 dependent inhibition of synaptic transmission. Neuropharmacology. 2008;55:1140–1146. doi: 10.1016/j.neuropharm.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmijn S. Fatty acid intake and the risk of dementia and cognitive decline: a review of clinical and epidemiological studies. J Nutr Health Aging. 2000;4:202–207. [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Lovinger DM. Presynaptic modulation by endocannabinoids. Handb Exp Pharmacol. 2008;184:435–477. doi: 10.1007/978-3-540-74805-2_14. [DOI] [PubMed] [Google Scholar]
- Lutz B. Molecular biology of cannabinoid receptors. Prostaglandins Leukot Essent Fatty Acids. 2002;66:123–142. doi: 10.1054/plef.2001.0342. [DOI] [PubMed] [Google Scholar]
- Lutz B. Endocannabinoid signals in the control of emotion. Curr Opin Pharmacol. 2009;9:46–52. doi: 10.1016/j.coph.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Mackie K. Mechanisms of CB1 receptor signaling: endocannabinoid modulation of synaptic strength. Int J Obes (Lond) 2006;30(Suppl 1):S19–S23. doi: 10.1038/sj.ijo.0803273. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci. 1999;11:4213–4225. doi: 10.1046/j.1460-9568.1999.00847.x. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgänsberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Molteni R, Barnard RJ, Ying Z, Roberts CK, Gómez-Pinilla F. A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience. 2002;112:803–814. doi: 10.1016/s0306-4522(02)00123-9. [DOI] [PubMed] [Google Scholar]
- Monory K, Massa F, Egertová M, Eder M, Blaudzun H, Westenbroek R, Kelsch W, Jacob W, Marsch R, Ekker M, Long J, Rubenstein JL, Goebbels S, Nave KA, During M, Klugmann M, Wölfel B, Dodt HU, Zieglgänsberger W, Wotjak CT, et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51:455–466. doi: 10.1016/j.neuron.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu A, Földy C, Soltesz I. Postsynaptic origin of CB1-dependent tonic inhibition of GABA release at cholecystokinin-positive basket cell to pyramidal cell synapses in the CA1 region of the rat hippocampus. J Physiol. 2007;578:233–247. doi: 10.1113/jphysiol.2006.115691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Tsubokawa H, Mizushima I, Yoneda N, Zimmer A, Kano M. Presynaptic cannabinoid sensitivity is a major determinant of depolarization-induced retrograde suppression at hippocampal synapses. J Neurosci. 2002;22:3864–3872. doi: 10.1523/JNEUROSCI.22-10-03864.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagotto U, Marsicano G, Cota D, Lutz B, Pasquali R. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr Rev. 2006;27:73–100. doi: 10.1210/er.2005-0009. [DOI] [PubMed] [Google Scholar]
- Pan B, Wang W, Long JZ, Sun D, Hillard CJ, Cravatt BF, Liu QS. Blockade of 2-arachidonoylglycerol hydrolysis by selective monoacylglycerol lipase inhibitor 4-nitrophenyl 4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate (JZL184) enhances retrograde endocannabinoid signaling. J Pharmacol Exp Ther. 2009;331:591–597. doi: 10.1124/jpet.109.158162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. San Diego: Academic; 2001. [Google Scholar]
- Pelchat ML, Johnson A, Chan R, Valdez J, Ragland JD. Images of desire: food-craving activation during fMRI. Neuroimage. 2004;23:1486–1493. doi: 10.1016/j.neuroimage.2004.08.023. [DOI] [PubMed] [Google Scholar]
- Quarta C, Belloccio L, Mancini G, Mazza R, Cervino C, Braulke LJ, Fekete C, Latorre R, Nanni C, Bucci M, Clemens LE, Heldmaier G, Watanabe M, Leste-Lassere T, Maitre M, Tedesco L, Fanelli F, Reuss S, Klaus S, Srivastava RK, et al. CB1 signalling in forebrain and sympathetic neurons is a key determinant of endocannabinoid actions on energy balance. Cell Metab. 2010;11:273–285. doi: 10.1016/j.cmet.2010.02.015. [DOI] [PubMed] [Google Scholar]
- Ravinet-Trillou C, Delgorge C, Menet C, Arnone M, Soubrié P. CB1 cannabinoid receptor knockout in mice leads to leanness, resistance to diet-induced obesity and enhanced leptin sensitivity. Int J Obes Relat Metab Disord. 2004;28:640–648. doi: 10.1038/sj.ijo.0802583. [DOI] [PubMed] [Google Scholar]
- Scheen AJ. CB1 receptor blockade and its impact on cardiometabolic risk factors: overview of the RIO programme with rimonabant. J Neuroendocrinol. 2008;20(Suppl 1):S139–S146. doi: 10.1111/j.1365-2826.2008.01681.x. [DOI] [PubMed] [Google Scholar]
- South T, Huang XF. Temporal and site-specific brain alterations in CB1 receptor binding in high fat diet-induced obesity in C57BL/6 mice. J Neuroendocrinol. 2008;20:1288–1294. doi: 10.1111/j.1365-2826.2008.01785.x. [DOI] [PubMed] [Google Scholar]
- Straiker A, Mackie K. Depolarization-induced suppression of excitation in murine autaptic hippocampal neurones. J Physiol. 2005;569:501–517. doi: 10.1113/jphysiol.2005.091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Hu SS, Long JZ, Arnold A, Wager-Miller J, Cravatt BF, Mackie K. Monoacylglycerol lipase limits the duration of endocannabinoid-mediated depolarization-induced suppression of excitation in autaptic hippocampal neurons. Mol Pharmacol. 2009;76:1220–1227. doi: 10.1124/mol.109.059030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo B, Urbanski MJ, Bisogno T, Di Marzo V, Mendiguren A, Baer WU, Freiman I. Depolarization-induced retrograde synaptic inhibition in the mouse cerebellar cortex is mediated by 2-arachidonoylglycerol. J Physiol. 2006;577:263–280. doi: 10.1113/jphysiol.2006.119362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, Watanabe M, Sakimura K, Kano M. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron. 2010;65:320–327. doi: 10.1016/j.neuron.2010.01.021. [DOI] [PubMed] [Google Scholar]
- Van Gaal LF, Rissanen AM, Scheen AJ, Ziegler O, Rössner S. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet. 2005;365:1389–1397. doi: 10.1016/S0140-6736(05)66374-X. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA. Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron. 2001;31:453–462. doi: 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]