

Abstract
Under stress, chlamydiae can enter a non-infectious but viable state termed persistence. In the absence of a tractable genetic system, persistence induction provides an important experimental tool with which to study these fascinating organisms. This review will discuss examples of: i) persistence studies that have illuminated critical chlamydiae/host interactions; and ii) novel persistence models that will do so in the future.
Keywords: Chlamydial persistence, Aberrant body, Tissue tropism, Bacterial/viral co-infection, Sexually-transmitted infection, Chlamydiaceae
1. Introduction
The chlamydiae are Gram-negative, obligate intracellular pathogens that infect a wide variety of hosts, ranging from humans to amoebae. This review will focus on the species within the Chlamydiaceae [1]. The genus Chlamydia currently contains 9 species, which differ in their host species and disease spectrum. Those discussed in this review are summarized in Table 1 . The Chlamydiaceae share a unique biphasic developmental cycle (Fig. 1 ) during which they exist in one of two developmental forms: the EB (or elementary body) and RB (reticulate body). The EB is the smaller (0.2 μm), metabolically inert, infectious, extracellular form of the organism. After host cell entry, EB-containing endosomes resist acidification and may, or may not, fuse to form a membrane-bound, protective intracellular replicative niche termed an inclusion. The inclusion develops in close proximity to the host nucleus, ER and Golgi compartments, from which raw materials, such as sphingomyelin, needed for inclusion expansion are obtained. Once the inclusion is formed, the EB within develop into larger (0.8 μm), metabolically active, non-infectious RB. The RB then use ATP and host cell metabolites to grow and divide. RB generally undergo 8-12 rounds of cell division, after which they asynchronously differentiate back into EB. The trigger that initiates RB to EB conversion is unknown, although disruption of type III secretion by RB/inclusion membrane detachment [2] may be involved. At 30–80 h post-infection (hpi), depending upon the chlamydial species examined, RB mature into infectious EB and are released by either host cell lysis or extrusion of the inclusion [3]. Though chlamydiae can successfully infect many cell types in culture, many of the species that infect humans and non-human animals target mucosal epithelial cells in vivo.
Table 1.
Chlamydia species discussed in this review.
| Species | Biovar (Serovar) | Host species | Some associated diseases |
|---|---|---|---|
| C. trachomatis | Trachoma biovar (serovars A, B, Ba and C) | Humans | Trachoma – leading infectious cause of blindness worldwide |
| Non-invasive genital biovars (serovars D-K) | Humans | Inflammatory urogenital infections leading to urethritis and epididimitis in men; pelvic inflammatory disease, atopic pregnancy and sterility in women, may be associated with cervical cancer - most common bacterial STD agents worldwide | |
| LGV biovar (serovars L1–L3) | Humans | Lymphogranuloma venereum | |
| C. pneumoniae | Humans, also reptiles, amphibians and some marsupials | “Walking pneumonia” in humans, may be associated with atherosclerosis, progressive neurologic disorders and lung cancer | |
| C. muridarum | Mice | Respiratory or urogenital inflammation depending upon inoculation route – commonly used experimental animal model system | |
| C. psittaci | Birds, also humans, swine, ruminants | Infected humans may develop the respiratory disease psittacosis, may be associated with ocular adnexal lymphoma | |
| C. suis | Swine | Enteritis and urogenital inflammation | |
| C. pecorum | Ruminants, swine | Enteritis, urogenital inflammation, abortion | |
| C. caviae | Guinea pigs | Ocular and urogenital inflammation depending upon inoculation route – commonly used experimental animal model system | |
| C. abortus | Ruminants, swine | Abortion | |
Fig. 1.
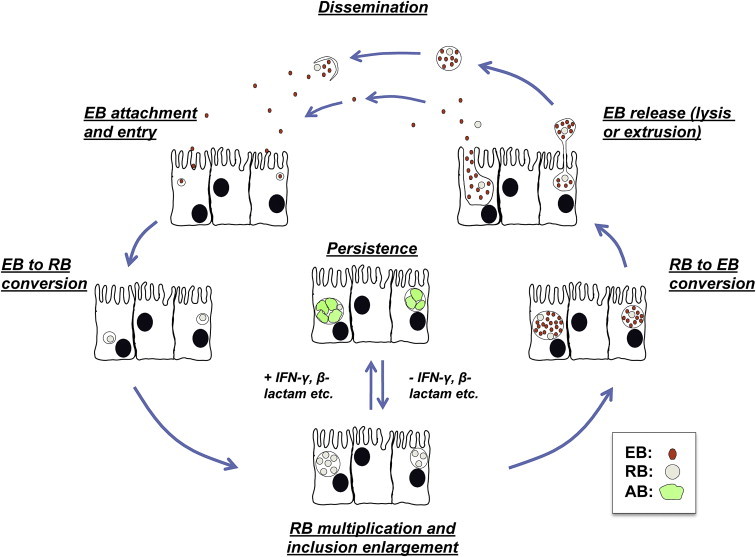
General overview of the chlamydial developmental cycle.
Over the past 50 years, ever more sophisticated analyses suggest that viewing the chlamydial developmental cycle as a simple “biphasic” process may significantly underestimate it’s complexity. In 1961, Galasso and Manire demonstrated that penicillin- and antiserum-exposed Chlamydia psittaci could be maintained in culture for up to 9 months in a non-infectious, but viable, state; when the stressor was removed, developmental cycle progression and production of infectious EB ensued [4]. Subsequent transmission electron microscopy (TEM) analyses of penicillin G-exposed chlamydiae revealed abnormal developmental forms that did not correspond to “normal” EB or RB (variously termed aberrant RB, persistent bodies or aberrant bodies (AB)). Penicillin-exposure inhibited C. psittaci RB to EB conversion, forming greatly enlarged, irregular, less electron-dense RB (i.e. AB) that shed membrane blebs [5]. Amoxicillin-exposed, Chlamydia muridarum-infected murine epithelial cells show the characteristic AB morphology (Fig. 2C). While AB are occasionally observed within normally-developing inclusions (compare Fig. 2A to B), they are more abundant when the chlamydiae are exposed to physiologic stressors, such as β-lactam antibiotics (Fig. 2C).
Fig. 2.
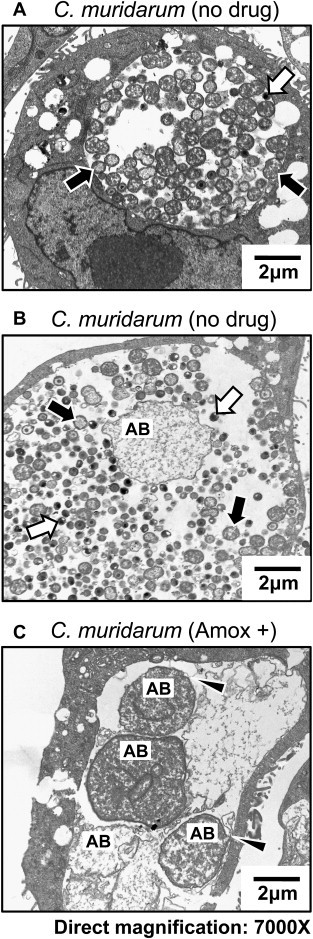
Transmission electron micrographs of Chlamydia muridarum-infected BM1.11 murine oviduct epithelial cells. Panels A and B. BM1.11 cells were infected at 1MOI, refed with culture medium + ddH2O at 8 hpi and harvested at 30 hpi. Panel C. C. muridarum-infected cultures were re-fed at 8 hpi with medium + 0.605 ug/ml amoxicillin and incubated for an additional 22 h. Infected cells were subjected to TEM as described [10]. RB are indicated by black arrows, EB by white arrows, and AB by “AB”. Arrows in panel C indicate AB membrane blebbing.
Numerous subsequent studies (recently reviewed in [6], [7] have made it abundantly clear that when they encounter specific stimuli, developing chlamydiae can divert from the productive developmental cycle into a state termed persistence. In the chlamydial field, persistence is defined as a developmental stage in which the chlamydiae are viable but non-infectious. This usage can be somewhat confusing, as most microbiologists use the term “persistence” to refer to a long-term infection within a host. During persistence, chlamydial metabolism is slowed and RB division, as well as differentiation into EB, halts, which reduces infectious particle production and stimulates AB formation. Persistent chlamydiae are viable, as indicated by continued synthesis of unprocessed 16S rRNA and genomic DNA replication. This state is also reversible; persistent chlamydiae can be maintained for weeks or months in culture, after which they continue productive replication if the inducer is removed. Persistence, therefore, appears to be a mechanism that allows the chlamydiae to “ride out” hostile conditions and maintain long-term infection within a host cell. It is important to note that persistent (i.e. viable but not infectious) chlamydiae have been primarily studied in culture. Although AB have been demonstrated in vivo [6], [7], whether or not chlamydiae enter this developmental state in order to establish chronic host infections has not yet been determined.
Several different stimuli induce persistence in culture; the best characterized are: i) IFN-γ- and penicillin-exposure; ii) chlamydiaphage infection; iii) heat shock; and iv) amino acid, glucose and iron deprivation. These stimuli do not alter the developmental cycle if they are delivered after RB to EB maturation initiates. Chlamydiae can also establish persistent infections in monocytes, as well as during continuous culture in other cells [6]. Since 2004, several new culture models of chlamydial persistence have been described: i) exposure to cigarette smoke components [8] and extracellular adenosine [9]; and ii) host cell co-infection with Herpes Simplex Virus (HSV) [10] or Porcine Epidemic Diarrhea Virus (PEDV) [11]. As many of the “older” persistence models have been exhaustively reviewed, this discussion will focus upon the relationship between IFN-γ-induced persistence and Chlamydia trachomatis tissue tropism, in order to illustrate how culture models of persistence have been used as tools to explore chlamydia/host interactions. In addition, several recently-developed, novel persistence models will be described. Note that given length and reference limitations for this review, in some cases it was necessary to reference recent reviews rather than multiple original observations.
2. Definitions matter: phenotypic characteristics of persistent chlamydiae
When studying chlamydial persistence, one must first address how to experimentally define this developmental stage. The accepted definition requires simultaneous demonstration of: i) reduced or absent production of infectious progeny EB, usually determined by sub-passage infectious titer assay, and ii) continued presence of viable organisms, demonstrated by continued accumulation of genomic DNA, pre-rRNA and/or other chlamydial mRNAs. Continuation of productive replication post-inducer removal also demonstrates continued viability. TEM demonstration of AB formation is strongly supportive, although there is significant variation in the reported morphological characteristics of “persistent” chlamydiae. For example, penicillin-exposed C. psittaci-infected cells contain greatly enlarged AB. In contrast, iron-restriction does not induce significant RB enlargement in C. trachomatis serovar E-infected HEC-1B cells. Inclusions within iron-starved cells do, however, contain aberrantly-shaped RB with dense, wavy membranes (reviewed in [6]). Thus, though helpful, TEM analyses must be combined with other experimental measures to confirm persistent infection.
Numerous investigators have examined differential chlamydial gene expression during persistence, in the hope of identifying chlamydial proteins and/or transcripts that are consistently up- or down-regulated in different persistence models. Such “persistence markers” could prove useful for confirming that persistent chlamydiae exist in vivo [7]. Initial studies suggested that reduced major outer membrane protein (MOMP) expression, increased chlamydial heat shock protein 60 (cHSP60) accumulation as well as decreased expression of gene products predicted to function in RB division (such as ftsK and ftsW) were most associated with persistence. However, none of these markers are consistently modulated in all characterized models of chlamydial persistence (reviewed in [6]).
More recently, several investigators have compared the transcriptional response for a single chlamydial species between at least two different persistence inducers within the same culture model system [12], [13], [14], [15], [16]. Timms et al. suggest that the chlamydial genes hctB, omcB, ompA and crpA are most consistently down-regulated when different persistence models and chlamydial species are compared. The htrA gene is the most consistently up-regulated [16]. However, there are several considerations that complicate interpretation of published gene expression data. First, protein and transcript expression are not always coordinately regulated during persistence. For example, Mukhopadhyay et al., demonstrated that Chlamydia pneumoniae OmpA protein was increased 1.6 to 2.3-fold in IFN-γ-exposed and iron-restricted HEp-2 cultures at 48 hpi [13]. The same group subsequently reported that the C. pneumoniae omp A transcript accumulation decreased up to 5-fold under similar conditions [16]. Indeed, Ouellette et al. reported that IFN-γ-exposure reduces C. pneumoniae-specific 35S-incorporation by >95% at 96 hpi, at which time bacterial transcript accumulation is globally increased [14]. Secondly, in most of the studies where accumulation of chlamydial transcripts have been compared across multiple persistence inducers [12], [15], [16], the quantity of each chlamydial mRNA of interest was normalized to 16S rRNA levels to control for differences in host cell chlamydial load and RNA recovery between samples. If, however, there are temporal differences in rRNA accumulation under the experimental conditions used, this approach becomes problematic. Goellner et al. demonstrated that several chlamydial mRNAs identified as significantly different compared to controls when normalized to 16S rRNA were not so when normalized to the gyrA transcript [12]. Thus, discrimination of significant differences can clearly be influenced by choice of normalization strategy. In a subsequent study, Ouellette et al. noted that: i) C. pneumoniae rRNA accumulation rate was not constant throughout the developmental cycle; and ii) rRNA accumulation profiles differed in productive versus persistent infection. When chlamydial DNA copy number was used to normalize expression data, a global increase in C. pneumoniae late gene mRNA accumulation was observed by both Q-PCR and microarray in IFN-exposed cultures. Normalization of these same data to 16S rRNA levels obscured the IFN-induced, global late gene increase [14]. Interestingly, this group previously reported that IFN-γ-exposure actually suppressed C. trachomatis late gene accumulation, however, the data were normalized to rRNA [17]. When the same transcript accumulation data from IFN-γ-exposed, C. trachomatis cultures was normalized to genome copy number, the observation that late genes are globally up-regulated was reproduced [14], again illustrating that the choice of normalization method greatly influences the resulting data.
3. An elegant story unfolds: interferon-γ-induced persistence and C. trachomatis tissue and host tropism
In 1963, Sueltenfuss and Pollard demonstrated that interferon-containing culture supernatants inhibited C. psittaci development [18]. Subsequently, IFN-γ-exposure of human epithelial cell lines was shown to reduce C. psittaci [19] infectious particle yield in a dose-dependent fashion. Byrne et al. also showed that addition of exogenous tryptophan to IFN-γ-exposed, infected T24 cells rescued C. psittaci inclusion development. IFN-γ-exposure also increased accumulation of labeled kynurenine within T24 cells fed with L- [3H]tryptophan, suggesting that IFN-γ-induced indoleamine-2,3-dioxygenase (IDO) activity reduced host intracellular tryptophan concentration and restricted chlamydial development (Fig. 3A) [19]. This model was supported by subsequent observations that IFN-γ-exposure simultaneously restricted chlamydial development and increased tryptophan catalysis in C. psittaci-infected human macrophages [20] and C. trachomatis-infected human conjunctival epithelial cells [21]. The IDO-dependence of this effect was confirmed by the observation that C. trachomatis serovar A replication in a human IDO-deficient cell line was unaltered by IFN-γ-exposure [22].
Fig. 3.
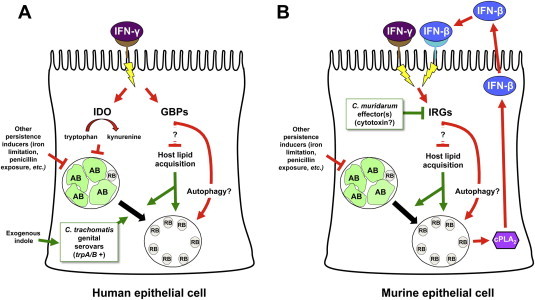
IFN-γ elicits different anti-chlamydial responses in human and murine cells. Panel A. IFN-γ/receptor interaction activates at least 2 distinct pathways in human cells that limit chlamydial development. The first increases indoleamine-2,3-dioxygenase (IDO) activity, activating tryptophan to kynurenine catalysis and reducing host intracellular tryptophan concentration, which induces chlamydial persistence [19]. Genital C. trachomatis serovars can use exogenous indole to synthesize tryptophan, thus evading this response. In contrast, ocular serovars, which lack functional trpA/B genes, cannot utilize indole [29], [30]. The IFN-γ-inducible 65 kD guanylate binding proteins (GPBs) may also restrict C. trachomatis growth in human epithelial cells by activating autophagosomal destruction of the chlamydiae and/or by blocking chlamydial lipid acquisition [39]. As host sphingomyelin synthesis is required for efficient reactivation of C. trachomatis persistence [40], the GBPs may also function to keep persistent chlamydiae (induced by tryptophan limitation or other stimuli) from re-entering the productive developmental cycle until they can be disposed of by the host autophagic machinery. Panel B. IFN-γ/receptor interaction on murine cells inhibits chlamydial development primarily by inducing immunity-related GTPases (IRGs). C. trachomatis-infection can also increase IRG accumulation via a cPLA2 and IFN-β-dependent pathway [38]. IRGs do not induce persistence but may restrict both normal chlamydial development and recovery from persistence by inhibiting sphingomyelin transport [33] and/or by increasing host autophagic activity [34]. C. muridarum is more resistant to the anti-chlamydial effects of murine IRGs than are human C. trachomatis serovars due to the production of secreted inhibitory effectors [36], [41], one of which may be the cytotoxin [33]. Red lines represent those pathways that restrict chlamydial development; green lines those that promote chlamydial productive replication; arrowheads represent activation events and “T”s indicate inhibition.
Several TEM studies demonstrated that IFN-γ-exposure of chlamydiae-infected cells resulted in accumulation of abnormal RB, which do not differentiate into EB while IFN-restriction is maintained. However, the chlamydial ultrastructural alterations observed varied depending upon the IFN concentration, host cell and chlamydial species used. For example, exposure to 0.2 ng/ml IFN-γ completely abrogated recovery of infectious EB from C. trachomatis serovar A-infected HeLa cells; smaller inclusions containing AB were observed under these conditions. Chlamydial growth in tryptophan-deficient medium reproduced these morphologic alterations [23]. In contrast, though C. trachomatis L2 inclusions were both smaller and contained fewer RB in IFN-γ-exposed murine L cells, no AB were observed [24]. As it is now apparent that the IFN-γ-restriction is mediated by different mechanisms in murine and human cells (Fig. 3), it is not surprising that IFN-γ-exposed chlamydiae would appear morphogically different in these cell types. Notably, Beatty et al. maintained C. trachomatis serovar A within IFN-γ-exposed HeLa cultures for more than 30 days post-infection (pi). During the entire IFN-exposure period: i) AB expressing increased cHSP-60 and reduced MOMP were observed; and ii) the persistent chlamydiae recovered infectivity upon IFN removal [25]. These data, and others, strongly suggest that fluctuations in IFN-γ-exposure in vivo could allow periodic productive replication and release of infectious chlamydiae from persistently-infected epithelial cells.
More recent data suggest that evasion of IFN-γ-induced developmental cycle restriction is an important determinant of C. trachomatis tissue tropism (Fig. 3; reviewed in [26]). All sequenced C. trachomatis serovars lack some of the eubacterial tryptophan biosynthetic genes (reviewed in [26]) and are, thus, tryptophan auxotrophs, which accounts for their IFN-γ sensitivity in HeLa cells [27]. C. trachomatis ocular serovars retain genes encoding the tryptophan synthase α (trpA) and β (trpB) subunits, yet they are generally interrupted by frame-shift or null mutations (reviewed in [26]). The C. trachomatis genital serovars (D-K and L1-L3), in contrast, encode uninterrupted copies of these genes [26]. Indeed, Shaw et al. observed that C. trachomatis serovar A and L2 express tryptophan synthase α-subunits of different sizes under IFN-γ-restriction, suggesting that trpA polymorphisms might influence pathogenesis [28]. Subsequent studies demonstrated that, in contrast to ocular serovars, genital C. trachomatis serovars encode a functional tryptophan synthase enzyme, which can utilize indole as a substrate for tryptophan synthesis. As a result, indole supplementation allows genital, but not ocular, serovars to escape IFN-γ-mediated growth suppression in human genital epithelial cells [29], [30]. Genital tract normal flora can secrete indole, suggesting that genital serovars have retained the capacity to use exogenous indole present in their specific environmental niche to synthesize tryptophan. This would, in turn, allow these serovars to evade host IFN-γ-mediated growth restriction, thus promoting long term establishment of genital tract infection in humans [26], [29], [30].
Evasion of the IFN-γ-induced anti-chlamydial response is also a determinant of host tropism (Fig. 3), as suggested by the observation that C. trachomatis serovar D is shed longer than C. muridarum from genitally-infected IFN-γ knock out mice [31]. C. muridarum is also less sensitive to IFN-γ-exposure in murine cell lines than is C. trachomatis [31]. Comparison of growth restriction in a large number of human and murine cell lines demonstrated that IFN-γ-mediated inhibition of both C. muridarum and C. trachomatis L2 growth was primarily mediated by IDO in human cells. In contrast, IFN-γ-exposure inhibited C. trachomatis L2 replication in murine cell lines much more strongly than that of C. muridarum and this block was not reversed by tryptophan or indole supplementation [32]. In another report, IFN-γ-exposure strongly inhibited C. trachomatis L2, but not C. muridarum, development in primary mouse oviduct epithelial (MEC) cells. This inhibition was not due to either iNOS or IDO induction. However, siRNA-mediated knock down of the IFN-inducible p47 GTPase, Iigp1/Irga6, partially reversed the IFN-γ-induced block to C. trachomatis L2 replication in murine cells. IFN-γ-exposure also decreased C. trachomatis inclusion size and blocked transport of fluorescent sphingomyelin to the inclusion in MECs. These data suggest that IFN-induced Iigp1/Irga6 expression disrupts lipid transport to the developing inclusion in C. trachomatis, but not C. muridarum-infected, murine cells [33]. Other observations also indicate that Irga6 is a murine IFN-induced anti-chlamydial effector [34]. IFN-γ-exposure simultaneously decreased C. trachomatis L2 infectivity and increased inclusion/LAMP1 co-localization in mouse embryo fibroblasts (MEFs), indicating increased inclusion:lysosomal fusion. IFN-γ-exposure had little effect on C. trachomatis L2 infectivity or LAMP1 co-localization in Atg5 knockout MEFs, suggesting that autophagosome formation is also required. Irga6 also co-localized with C. trachomatis L2 inclusions, but not to those of C. muridarum, in an IFN-γ-dependent manner. Finally, LAMP-1/inclusion co-localization was reduced in Irga6 −/− MEF, which also supported C. trachomatis L2 development equally well in the presence and absence of IFN-γ. The authors concluded that IFN-γ-exposure inhibits C. trachomatis, but not C. muridarum, development in murine cells by targeting the chlamydiae to autophagosomes via an Irga6-dependent mechanism [34]. Though both studies indicate that IFN-γ-induced murine cell resistance to C. trachomatis is Irga6-dependent, they proposed different mechanisms: sphingomyelin transport inhibition [33] and increased autophagy [34]. It seems likely, therefore, that either IFN-induced murine effectors inhibit C. trachomatis development at more than one point or that there are several different IFN-activated inhibitory pathways that are cell-type specific (Fig. 3B).
Other IFN-γ-responsive immunity-related GTPases (IRGs) may also restrict chlamydial development in mouse cells. Irgp10 inhibits C. trachomatis L2 but not C. muridarum infection in both IFN-γ-exposed murine fibroblasts and systemically-infected knockout mice [35], [36]. The IRGs Iigp2 and Irgp10 also determine susceptibility to C. psittaci infection in mice. Furthermore, Iigp2 mediates part of the IFN-γ-induced inhibition of C. psittaci development in MEF cells [37]. Vignola et al., also recently demonstrated that MEFs from cPLA2 (calcium-dependent phospholipase A2) knockout mice were 5-fold more permissive to C. trachomatis L2 than wildtype MEFs. In contrast, C. muridarum infectivity in MEFs was unaltered by cPLA2 ablation. Notably, C. trachomatis-infected MEFs spontaneously produced IFN-β by a cPLA2-dependent mechanism. IFN-β exposure increased accumulation of Irgb6 protein and reduced yield of infectious C. trachomatis in both cPLA2 −/− and wildtype MEFs [38]. These observations demonstrate that multiple stimuli can both up-regulate IRG expression and diminish C. trachomatis development in murine cells (Fig. 3B), which greatly complicates interpretation of existing studies exploring the in vivo relevance of the IRGs. Finally, Tietzel et al. demonstrated that the IFN-γ-inducible 65 kD guanylate binding proteins (GPBs) hGBP1 and 2 may also restrict C. trachomatis growth in human genital epithelial cells (Fig. 3A). Co-localization studies indicated that tagged hGBP1 and 2 associate with the inclusion membrane in serovar B-infected cells. Over-expression of hBP1 in infected HeLa cells reduced inclusion size for all three C. trachomatis serovars tested. C. trachomatis serovar B inclusions in hGBP1-siRNA transfected, IFN-γ-exposed cells were significantly larger than those in IFN-exposed controls, suggesting that hBP1 cooperates with IDO to restrict C. trachomatis replication in human cells. The authors hypothesize that hBP1, which localizes to LC3-positive autophagosomes, may potentiate autophagosomal destruction of the chlamydiae or, alternatively, that the GBPs may alter chlamydial nutrient acquisition [39]. However, IFN-γ-exposure blocks sphingomyelin transport to the inclusion [33] and host sphingomyelin synthesis is required for efficient reactivation of C. trachomatis persistence [40]. These data suggest that IFN-induced IRGs may function to keep persistent chlamydiae from re-entering the productive developmental cycle if IDO-induction or activity is temporarily reduced by fluctuations in local IFN-γ-concentrations or increased exogenous tryptophan availability. Other IRGs might simultaneously function to increase trafficking of persistent chlamydiae to the autophagic pathway, so that they could ultimately be disposed of (Fig. 3). Thus, IFN-induced GPBs may serve a vital role in limiting chlamydial dissemination in vivo by inhibiting recovery from persistence.
The C. muridarum genome encodes three full-length copies of a cytotoxin that has homology to the clostridial cytotoxin (TcdB) and to the type III secreted cystiene protease YopT, both of which inactivate host cellular GTPases. Because C. trachomatis serovars contain only a single cytotoxin copy with a disrupted YopT-homology domain (reviewed in [26]), Nelson et al. proposed that the C. muridarum cytotoxin relieves the murine-specific block imposed by IFN-induced p47 GTPases (Fig. 3B) [33]. The chlamydial cytotoxin may also inhibit hBP1-mediated inhibition of inclusion enlargement, as C. caviae and C. muridarum, both of which carry full-length cytotoxin genes, are essentially insensitive to hBP1 over-expression [39]. Coers et al., however, maintain that cytotoxin-mediated Iigp1/Irga6 inactivation is unlikely to mediate C. trachomatis restriction in mouse cells because: i) IFN-γ restriction of C. trachomatis L2 DNA accumulation is not relieved in Irga6 (Iigp1) knockout fibroblasts; and ii) there is no difference in spleen bacterial DNA load when Irga6 knockout mice are C. trachomatis L2-infected via the IV route [36]. However, it is possible that functional redundancy between murine p47 GTPases obscures the effect of the Irga6 knockout in vivo. Alternatively, the contribution of the various murine p47 GTPases to IFN-γ-induced C. trachomatis developmental blockade may differ in different host cell types.
Regardless of whether or not the cytotoxin is involved, co-infection with UV-irradiated C. muridarum EB, but not with inactivated C. caviae or C. trachomatis, rescues C. trachomatis L2 productive replication in IFN-γ-exposed murine cells, suggesting that C. muridarum secretes pre-formed effectors into the host cytoplasm, where they block p47 GTPase-mediated inhibition of chlamydial development [41]. Microscopic analyses of co- and singly-infected, IFN-γ-exposed, MEF cells indicated that localization of Irgb10 to C. trachomatis L2 inclusions is essentially eliminated when C. muridarum EB are added at a 5:1 ratio, which is consistent with the prediction that a C. muridarum secreted effector inactivates murine p47 GTPases [36]. Collectively, these data indicate that: i) the IFN-γ-induced anti-chlamydial response differs in a host cell species-specific manner; and ii) that chlamydiae have evolved mechanisms to evade IFN-γ-elicited host responses that are specific to their natural host/tissue type (Fig. 3). Although the IFN-induced GTPases appear to inhibit inclusion development, rather than inducing persistence, they may also play a role in limiting the transition from persistent to productive infection. Moreover, these data provide an elegant demonstration of how culture studies of IFN-γ-induced chlamydial persistence have ultimately led to a deeper understanding of chlamydial host and tissue tropism.
4. Persistence or death: regulation of the developmental cycle by purinergic receptor activation
Extracellular ATP (ATPe) and adenosine (Ado) are two of the many cellular components released into the extracellular milieu when cells are damaged or destroyed. Activation of purinergic receptors by ATPe- or Ado-binding is a critical component of the mammalian defense against intracellular pathogens (reviewed in [42]). Not surprisingly, purinergic receptor activation also limits intracellular development of chlamydiae. Thirty-minute exposure to 5 mM ATPe strongly induced apoptosis in the murine J774 macrophage line. Exposure of C. caviae-infected J774 cells to 5 mM ATPe for 30 min at 24 hpi reduced infectious titer by >50%. However, chlamydial infection reduced apoptosis nearly to background levels, suggesting that C. caviae killing was not merely an indirect effect of apoptosis induction [43]. In C. muridarum-infected J774 cells, ATPe-exposure increased co-localization of vesicles staining with anti-MOMP and anti-LAMP-1, indicating that purinergic receptor activation kills incoming chlamydiae by promoting inclusion-lysosomal fusion. In contrast, ATPe-exposure did not kill chlamydiae in peritoneal macrophages from P2X7 receptor knock-out mice, directly demonstrating a requirement for this receptor [44]. Exposure to 5 mM ATPe also reduced C. muridarum infectivity in HeLa cultures through a mechanism that required P2X7 receptor function. Interestingly, 0.5 mM ATPe reduced yield of infectious C. muridarum from J774 cells by 50% [44], while eliciting no significant titer reduction in HeLa cultures [45], demonstrating that epithelial cells respond differently to ATPe than do macrophages. ATPe-stimulation of P2X7 and P2X4 receptors also activates cPLA2 activity in murine macrophages [46] and rat epithelial cells [47]. Additionally, activation of cPLA2 is required for autocrine IFN-β production and subsequent IRG up-regulation in C. trachomatis-infected mouse cells [38]. Finally, C. psittaci-infection alone elicits IFN-β-dependent, IDO up-regulation in human monocyte-derived macrophages [48]. Thus, purinergic receptor stimulation could inhibit chlamydial development via IFN-β-induced up-regulation of IRG or IDO activity (or both). ATPe-exposure increases inclusion/LAMP1 co-localization in C. muridarum-infected J774 cells [44], as do IFN-γ-induced GTPases [34], which is consistent with the hypothesis that purinergic signaling inhibits chlamydial development by activating IRGs. It would, therefore, be interesting to determine whether either IDO or IRGs mediates P2X7 receptor-based restriction of chlamydial replication and, if so, do the mechanisms differ in humans and mice.
Previous studies suggested that ATPe/P2X7 stimulation killed developing chlamydiae, rather than inducing persistent infection [43], [44], [45]. Pettengill et al. also recently demonstrated that extracellular Ado-exposure reduces infectious EB production by both C. trachomatis L2 and serovar D in HeLa cells. Notably, though recovery of serovar D was not tested, L2 recovered infectivity after Ado-exposure was terminated, suggesting persistence induction. Knockdown experiments demonstrated that the A2b receptor was primarily responsible for the observed inhibition. Fluorescent microscopic and TEM analyses confirmed that inclusions within Ado-exposed, infected cells were sparsely populated with normally-sized RB and few EB, which contrasts significantly with morphologic findings in many persistence models. Though morphologically different from other models, the authors concluded that Ado-exposure induces C. trachomatis persistence [9].
Purinergic receptor activation appears to kill C. caviae in a macrophage line [43] as well as C. muridarum in macrophages and HeLa cells [44], [45]. In contrast, C. trachomatis L2 remains viable but not infectious when infected HeLa cells are exposed to a similar insult [9]. This may result from differences in the inducer used (ATPe versus Ado) or the receptor activated (P2X7 versus A2b). Alternatively, C. caviae and C. muridarum may be more sensitive to purinergic receptor-mediated growth restriction than C. trachomatis L2. The C. muridarum and C. caviae genomes both differ significantly from that of C. trachomatis L2 in the plasticity zone (PZ), which encodes the cytotoxin and tryptophan biosynthetic genes. Thus, one or more of these genes may determine sensitivity to purinergic receptor-dependent inhibition. However, exploration of this possibility awaits further mechanistic characterization of Ado- and ATPe-mediated inhibition as well as direct comparison of the effects of purinergic receptor activation upon different chlamydial species within the same experimental system.
5. Tobacco use may be hazardous to developing chlamydiae: induction of C. pneumoniae persistence by cigarette smoke exposure
Smokers may have a higher rate of C. pneumoniae DNA carriage in blood and atheromatous lesions [49] than do individuals who do not smoke. Wiedeman et al. hypothesized that that such increased carriage might be due, at least in part, to increased establishment of persistent infection in the lung due to cigarette smoke exposure. Thus, they examined the developmental cycle of C. pneumoniae VR1360 in HEp-2 monolayers exposed to cigarette smoke-conditioned medium (CSM). Exposure to CSM immediately post-infection reduced production of infectious EB by 56% and 64% at 72 and 96 hpi, respectively. CSM-exposure did not reduce HEp-2 viability or human cytomegalovirus (hCMV) production in control cultures, suggesting that the effect was not due to host cell toxicity but was, rather, chlamydia-specific. Accumulation of chlamydial chromosomes increased from 24 to 96 hpi during CSM-exposure, indicating continued viability. Developmental forms present after 72 hour CSM-exposure were nearly 100% enlarged AB with an average size of 4 μm, similar to those observed during penicillin or IFN-γ-exposure [8]. These studies were later extended to C. pneumoniae-infected primary human aortic endothelial cells (HAEC), where CSM and vapor-phase smoke-exposure similarly reduced chlamydial infectivity; continued DNA accumulation and AB formation were also observed. CSM-exposed chlamydiae did not spontaneously recover after CSM removal; however, infectious titer more than doubled after CSM removal if cultures were supplemented with l-tryptophan. Because l-tryptophan addition produced a greater increase in infectivity of IFN-γ-exposed controls, the authors concluded that CSM-exposure induces persistence via several mechanisms, one of which is tryptophan deprivation (Fig. 4A) [50]. Cigarette smoke contains approximately 1015 free radicals per puff and nitric oxide, which is present at 500–1000 ppm, can react with superoxide anions to produce other reactive nitrogen species [51]. Nitrogen radicals can damage l-tryptophan [52], which could produce a state of tryptophan starvation within CSM-exposed cells (Fig. 4A) [50]. IDO expression can also be induced by either IFN-α or γ in human lung cells [53], suggesting that CSM exposure could stimulate IFN secretion, followed by IDO-induced tryptophan starvation and persistence. However, this possibility seems less likely given the observations that cigarette smoke actually inhibits IFN-α production from lung epithelial cells [54] and that alveolar macrophages isolated from smokers have reduced capacity to respond to IFN-γ [55].
Fig. 4.
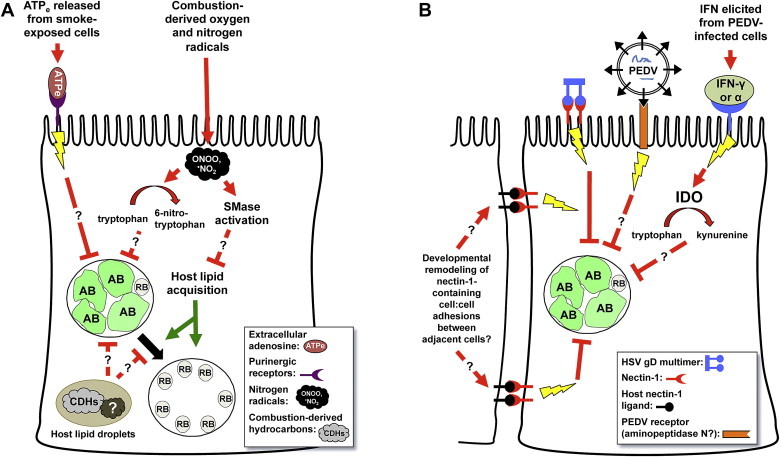
Induction of chlamydial persistence by cigarette smoke and host cellular viral co-infection. Panel A. There are several likely mechanism(s) by which components of cigarette smoke induce C. pneumoniae persistence [8]. ATPe or Ado released from smoke-exposed cells could induce chlamydial persistence by activating purinergic receptor-linked signaling pathways [9]. Nitrogen radicals present in cigarette smoke could also react with intracellular tryptophan [52], inducing persistence via tryptophan depravation [50]. As nitrogen radicals also activate intracellular sphingomyelinase (SMase) activity [60], [61], subsequent reduction in intracellular sphingomyelin would be predicted to restrict recovery from persistence [40] and/or productive chlamydial development. Finally, because smoke components are concentrated in host lipid droplets [66], these compounds may directly alter chlamydial development when lipid droplets are brought within close proximity to the developing inclusion [65]. Panel B. Herpes Simplex Virus (HSV) co-infection [10] and HSV glycoprotein D (gD)/host nectin-1 interaction [76], [78] appears to activate a currently unidentified host cellular signaling pathway that induces chlamydial persistence. Since nectin-1 also interacts with other host ligands on adjacent epithelial cells, remodeling of nectin-1 containing adherens junctions may also regulate chlamydial development in the absence of HSV infection. Porcine Epidemic Diarrhea Virus (PEDV) co-infection also induces persistence [11] and may do so directly by interacting with a host cell receptor (aminopeptidase N), in a manner analogous to that stimulated by HSVgD/nectin-1 interaction. Alternatively, PEDV infection may activate IDO expression and tryptophan limitation through an IFN-γ- or -α-dependent mechanism. Red lines represent those pathways that restrict chlamydial development; green lines those that promote chlamydial productive replication; arrowheads represent activation events and “T”s indicate inhibition.
Since tryptophan-depletion alone does not appear to account for CSM-induced persistence induction, other stimuli that influence chlamydial development may also play a role. As previously discussed, ATPe- and Ado-exposure reduces C. caviae, C. muridarum and C. trachomatis L2 infectivity through the P2X7 [43], [45] and A2b receptors [9]. Furthermore, A2b stimulation induces chlamydial persistence [9]. Notably, ATPe concentrations are increased in the lungs of cigarette smoke-exposed mice [56] as well as in bronchioalveolar lavage fluid isolated from smokers [57]. Up-regulation of purinergic receptors, including P2X7, has also been observed in the lungs of smoke-exposed mice [56]. Cigarette smoke may, therefore, activate C. pneumoniae persistence by increasing host cell purinergic receptor expression and ATPe- or Ado-release (Fig. 4A).
Depletion of host cellular sphingomyelin is another mechanism by which CSM could interfere with C. pneumoniae development (Fig. 4A). Like C. trachomatis and C. psittaci, C. pneumoniae [58] acquires host sphingomyelin from the Golgi apparatus. Inhibition of host sphingolipid biosynthesis reduces C. trachomatis L2 EB production and inclusion number [59] as well as disrupting C. trachomatis serovar E inclusion stability [40], indicating that host-derived sphingolipids are critical for developmental cycle progression. Cigarette smoke contains reactive oxygen and nitrogen species, both of which increase neutral (nSMase) and acidic (aSMase) sphingomyelinase activity in primary primate tracheobronchial epithelial and human bronchial epithelial (HAE) cells, respectively. This simultaneously decreases intracellular sphingomyelin concentration and increases that of ceramide, ultimately inducing caspase-3-dependent apoptosis [60], [61]. Cigarette smoke similarly activates nSMase-dependent apoptosis in human bronchial epithelial cells, which can be abrogated by addition of the antioxidant, glutathione [62]. Although SMase induction might be expected to induce apoptosis, rather than persistence, in chlamydiae-infected cells, persistent C. pneumoniae block caspase-3-induced apoptosis in human epithelial cell lines [63]. Although extracellular SMase-exposure does not alter C. trachomatis growth [64], free radicals activate intracellular SMase activity, which could access both internal host membranes and the developing inclusion and might, thus, be more likely to alter chlamydial development. Finally, C. trachomatis serovar B requires host sphingomyelin to transition efficiently from IFN-γ- or penicillin-induced persistence back to an actively replicating state [40]. Because low intracellular sphingomyelin levels presumably impede recovery from persistence, the observation that tryptophan supplementation rescues IFN-γ-exposed chlamydiae more efficiently than those that are CSM-exposed [50] is entirely consistent with the hypothesis that cigarette smoke induces persistence by simultaneously reducing intracellular tryptophan concentration and increasing SMase activity (Fig. 4A). It is also possible that cigarette smoke components affect chlamydiae directly. C. trachomatis directs translocation of host cytoplasmic lipid droplets into the developing inclusion [65]. As combustion-derived hydrocarbons from cigarette smoke are concentrated in cytoplasmic lipid droplets [66], the chlamydiae may be directly exposed to significant concentrations of these compounds, which could block developmental cycle progression (Fig. 4A).
Because C. pneumoniae establishes respiratory tract infection, it is easy to envision that the organisms would be exposed to high levels of smoke components in smokers or those exposed to second-hand smoke. However, do these observations have any applicability to persistence of non-respiratory chlamydial species? Nicotine [67] and other cigarette smoke components [68] have been detected in cervical fluids of smokers at significantly higher concentrations than observed in serum. The frequency of benzo[a]pyrene DNA adducts observed in ovarian tissue [69] isolated from women who smoke is also increased compared to that in non-smokers, suggesting that benzo[a]pyrene from cigarette smoke reaches the genital tract in quantities sufficient to inflict host DNA damage. These data indicate that cigarette smoke components reach the genital tract, as well as other sites, in concentrations high enough to alter host, and possibly chlamydial, physiological function. Finally, it should be pointed out that these data are not just applicable to chlamydiae-infected smokers: many compounds in tobacco smoke, such as polycyclic aromatic hydrocarbons, are also found in other combustion products, including coal, wood and diesel smoke [70]. Given that both humans and non-human animals are more-or-less constantly exposed to these environmental pollutants, smoke component-induced persistence may have considerably more impact upon chlamydial development in vivo than one might at first predict.
6. When pathogens don’t share well: alteration of chlamydial development by viral co-infection
Because experimental systems exploring the interaction between a single pathogen and cell type do not always accurately reflect host–pathogen interplay in vivo, some investigators are testing interactions between multiple pathogenic microorganisms. Several groups have studied Herpes Simplex Virus type 2 (HSV-2)/chlamydial co-infection in culture. TEM analyses demonstrated that Vero cells infected with C. trachomatis serovar L2 and super-infected with HSV-2 contained swollen inclusions with few RB or EB [71]. Chiarini et al. reported that in HSV-2/C. trachomatis serovar D co-infected HeLa cells, the number of cells positive for chlamydiae by anti-MOMP immunofluorescence was reduced [72]. Finally, HSV-2 pre-infection of HT-1376 human bladder cells reduced production of infectious C. trachomatis EB by about 15-fold [73]. Though these data suggest that HSV co- or super-infection influences chlamydial development, neither the effect nor the mechanism was defined. Deka et al., infected HeLa cells with C. trachomatis serovar E followed 24 h later with either HSV-1 or HSV-2. TEM analyses indicated that co-infected cells contained numerous AB and, few, or no EB. HSV co-infection also reduced C. trachomatis infectious titer, while accumulation of pre-16S rRNA was unaffected. Finally, HSV super-infection reduced MOMP accumulation by 50% while simultaneously increasing that of cHSP60-1 [10]. These data demonstrate that co-infection with either HSV-2 or HSV-1, both of which commonly cause genital herpes infections, induces chlamydial persistence in human genital epithelial cells. Neither cyclohexamide addition nor co-infection with UV-inactivated virions abrogates this effect, demonstrating that neither productive HSV replication nor de novo host protein synthesis is required. Furthermore, these data suggest that viral attachment and/or entry, rather than subsequent replication steps, stimulate C. trachomatis persistence [74].
There are a number of known persistence stimuli that could account for the observed effect of HSV super-infection upon chlamydial development. For example, HSV infection could induce IFN-γ secretion and activate IDO expression. Conversely, viral replication could deplete nutrients essential for continued chlamydial development. However, Luminex and RT-PCR studies revealed that co-infection did not induce host IFN-γ, IFN-α, TNF-α, IDO, lymphotoxin-α or iNOS expression. Nutrient supplementation experiments indicated that HSV-induced persistence was not mediated by glucose, amino acid or iron starvation. Finally, HSV co-infection did not alter inclusion expansion or transport of C6-NBD-ceramide to the inclusion [75]. These data indicate that HSV co-infection alters chlamydial development via a mechanism distinct from previously characterized models of chlamydial persistence (Fig. 4B). Lastly, persistent chlamydiae recover infectivity after a single inoculation of UV-inactivated HSV-2 indicating that viral-induced persistence is reversible [76], as observed in other models. However, the recent report by Pettengill et al. suggests that HSV-induced Ado release followed by A2b receptor engagement is also a possible mechanism. Varicella-Zoster Virus, which is closely related to HSV, strongly down-regulates A2b-receptor transcript expression in infected human cells; the human herpes virus hCMV does so as well [77]. Also, Ado-exposed chlamydiae are morphologically very different [9] compared to those observed within co-infected cells [10], [74], suggesting that these persistence models differ mechanistically.
If HSV does induce persistence by a novel mechanism, the question remains - how does it work? Since viral attachment or entry appear sufficient for persistence induction [74], Vanover et al. examined the role of HSV envelope glycoprotein gD in this process. Glycoprotein D facilitates viral entry by interacting with one of several host cell surface co-receptors, including herpes virus entry mediator (HVEM), nectin-1 (nec-1) and nectin-2 (nec-2). Pre-incubation of virions with anti-gD, but not with control antibodies, prevented HSV-1-induced persistence. Addition of purified recombinant HSV-2 gD protein to C. trachomatis pre-infected cells also reduced chlamydial titer to a degree indistinguishable from that observed during HSV co-infection, demonstrating that gD/host cell contact, in the absence of co-infection, alters chlamydial development [76]. Finally, co-infection with a nec-1-specific mutant herpes virus also strongly inhibited infectious EB production and induced AB formation, suggesting that persistence induction specifically requires HSV gD/nec-1 interaction (Fig. 4B) [78].
The nectins are immunoglobulin super-family members required for establishment of apical-basal polarity and formation of adherens (AJ) and tight junctions between polarized epithelial cells. The nec-1 extracellular domain interacts not only with HSV gD but also with four different endogenous cellular surface ligands: nec-1, nec-3, nec-4 and αvβ3 integrin. Trans-interactions between nec-1 and it’s ligands on adjacent cells activate the small G proteins Rap1 and Cdc42 through a Src-dependent pathway (reviewed in [79]). Not surprisingly, HSV/host receptor interactions similarly activate intracellular signaling. HSV-1 attachment to human fibroblasts activates several different intracellular signaling pathways, increasing transcription of down-stream genes [80]. Also, HSV-1 binding-induced intracellular calcium release in human CaSki cervical epithelial cells is nec-1 dependent [81], indicating that nec-1 participates in HSV-binding-induced epithelial cell signaling events. Finally, both HSV infection and soluble gD can disrupt nec-1 trans-interactions, suggesting that the nec-1 extracellular domain binding sites for gD overlap with those for nec-1 endogenous ligands [82]. Taken together, these data suggest that HSV gD/nec-1 interaction initiates an epithelial cell signaling cascade that ultimately restricts C. trachomatis development. The observation that HSV-binding and nec-1 trans-interactions with endogenous host ligands activate over-lapping host signaling cascades also suggest that nec-1-associated signaling may induce persistence in the absence of HSV co-infection (Fig. 4B). If so, these data have significant implications for host cell-mediated regulation of chlamydial development.
In another recently established co-infection model, Vero monkey kidney cells were infected with either C. suis, C. abortus or C. pecorum and the porcine Coronavirus, PEDV, all of which cause intestinal infections in swine. Initial IFA analyses of C. pecorum/PEDV co-infected Vero cells revealed inclusions containing enlarged AB similar to those described in other persistence models [83]. Subsequently, Borel et al. carried out a more detailed investigation to specifically determine if PEDV co-infection induces chlamydial persistence. In this study, Vero monolayers were infected with either C. pecorum 1710S or C. abortus S26/3. After 14 h, cultures were super-infected with PEDV and incubated for an additional 24 h. Chlamydial titer analyses indicate that, while PEDV co-infection greatly reduced infectious EB production from both chlamydial species tested, C. pecorum was more profoundly affected than C. abortus. Likewise, TEM experiments demonstrated that C. pecorum inclusions in co-infected cells contained almost entirely AB up to 2 μm in diameter. In contrast, C. abortus/PEDV co-infected cultures, contained a mix of chlamydial morphotypes: some inclusions were indistinguishable from those in chlamydiae-singly infected cells, while others contained a mix of AB, normal RB and a few EB. Finally, C. pecorum pre-infection reduces PEDV-driven syncytium formation by >85%, suggesting that chlamydial co-infection reduces either viral receptor expression/availability or decreases expression, localization or function of the PEDV envelope glycoprotein. The authors conclude that: i) PEDV super-infection induces chlamydial persistence in a species-specific manner; and ii) C. abortus is more resistant to co-infection than is C. pecorum. Borel et al. also propose that the observed difference in chlamydial strain sensitivity may be due to an increased sensitivity of C. pecorum 1710S to nutrient depletion [11].
The mechanism by which PEDV super-infection induces chlamydial persistence is currently unknown. However, some known features of coronavirus biology allow one to make predictions. First, coronavirus infection strongly induces IFN production in certain cell types but not in others [84]. Borel et al. assert that PEDV persistence is unlikely to be due to IFN production because Vero cells are unable to produce IFN [11]. It is important to note, however, that while Vero cells lack a functional IFN-β gene [85], they do respond to IFN-α and -γ, albeit about 10 times more weakly than do human epithelial cells [86]. IFN-α and -γ inhibition of measles virus replication in Vero cells is directly proportional to the quantity of IDO activity induced by each IFN type and is abrogated by addition of exogenous L-tryptophan [86]. Thus, PEDV-induced chlamydial persistence could be mediated by either IFN-α or, more likely, by IFN-γ (Fig. 4B). Indeed, if C. pecorum is more sensitive to tryptophan deprivation than is C. abortus, low-level IFN-γ-stimulated IDO induction could easily account for the observed species-specific differences in response PEDV co-infection [11]. Additional indirect evidence that IFN-γ may be mediating PEDV-induced persistence is provided by the observation that IFN-γ-exposure reduces expression of aminopeptidase N, the putative Vero cell PEDV receptor [87], in HL-60 cells [88]. As syncytium formation requires interaction between PEDV virion glycoproteins and host receptors on adjacent cells, production of IFN-γ in co-infected cultures may account for both PEDV-induced chlamydial persistence and the reduction of PEDV-directed syncytium formation observed in C. pecorum co-infected cells [11]. If chlamydiae- or co-infection induced-IFN-γ release down-regulates PEDV receptor expression on neighboring gut epithelial cells in vivo, viral replication, pathogenesis and subsequent transmission could also be significantly altered. Of course, PEDV super-infection could also alter chlamydial development directly by consuming host amino acids or other metabolites required for productive chlamydial replication or indirectly by activating host cellular anti-microbial defenses (Fig. 4B). Clearly, additional studies are needed to clarify the mechanism(s) and in vivo consequences of this intriguing example of viral co-infection induced chlamydial persistence.
7. Conclusions
There are many unanswered queries regarding the role of the “viable but non-infectious” developmental state in chlamydial biology; however, the two most critical unanswered questions are: “Do persistent chlamydiae, similar to those observed in culture models, actually exist in vivo?” and “If so, what are the consequences of persistence for the chlamydiae and the host?” Various data suggest that persistent chlamydial forms exist in vivo. For example, Pospischil et al. recently used TEM analyses to convincingly demonstrate C. suis AB in intestinal tissues from infected swine [89]. However, as TEM studies cannot demonstrate viability, we should continue to carefully examine clinical samples from infected humans or non-human animals for evidence of persistent infection using the full panoply of available molecular techniques [7]. Answering the second question is actually much more difficult than the first because it requires a tractable animal model system in which: i) infection with non-replicative, viable chlamydiae can be convincingly demonstrated; and ii) persistent infection can be turned “on” or “off” by the investigator, so that the contributions to pathogenesis can be assessed. If an exogenous inducer is used to create such a system, it must have few or no toxic or immunomodulatory effects on the host. IFN-γ-exposure, though well-studied, is unlikely to prove suitable. While IFN-γ production can be knocked out in vivo, it is difficult to envision a method by which IFN-γ could be consistently “added back” to an animal without causing additional immunomodulatory effects. Moreover, development of such a model in mice would require a “humanized” strain, since the IFN-γ-dependent anti-chlamydial response in mice is mediated by IRG proteins, which restrict inclusion development, rather than IDO, which induces the persistent state. Although construction of such mouse strains have been proposed [26], none are yet available. In vivo chlamydial persistence models based on heat shock, nutrient-starvation or phage infection are likely even less feasible. However, penicillin, the first characterized persistence inducer, may be an ideal candidate for use in such an in vivo system. Penicillins are: i) relatively non-toxic to the host; ii) easily administered to an animal over a wide concentration range; iii) well understood pharmokinetically; iv) not produced naturally in the mammalian host; and v) targeted directly to the developing chlamydiae, rather than functioning through host-dependent pathways. Transgenic mouse strains that inducibly-express HSV gD protein might also be used to develop such a model. Though it may prove very difficult, development of an experimental in vivo persistence model is absolutely necessary if we are to directly explore the contribution of persistent chlamydiae to pathogenesis.
In conclusion, chlamydial persistence, as a tissue culture phenomenon, has been the subject of increasingly intense experimental scrutiny over the past 30 years. In particular, the cumulative data obtained from studies of IFN-γ-induced persistence have revealed the molecular basis of C. trachomatis tissue and host tropism. Given the absence of a tractable system for genetic manipulation of chlamydiae, both well-established and newly-characterized culture persistence models remain valuable experimental tools for dissecting chlamydia/host interactions. Thus, continued exploration of these systems is expected to expose intimate details of the biology of these remarkable intracellular pathogens.
Acknowledgements
The author would like to thank Drs. Priscilla B. Wyrick, Jennifer Vanover-Hall and Michael Kruppa for critical review of the manuscript. Funding was provided by NIH/NIAID grants AI078373-01A1 and AI082322-01 to RVS. This review is dedicated to the memory of Dr. Jane Raulston; colleague, mentor and friend.
References
- 1.Stephens R.S., Myers G., Eppinger M., Bavoil P.M. Divergence without difference: phylogenetics and taxonomy of Chlamydia resolved. FEMS Immunol. Med. Microbiol. 2009;55:115–119. doi: 10.1111/j.1574-695X.2008.00516.x. [DOI] [PubMed] [Google Scholar]
- 2.Wilson D.P., Timms P., McElwain D.L., Bavoil P.M. Type III secretion, contact-dependent model for the intracellular development of chlamydia. Bull. Math. Biol. 2006;68:161–178. doi: 10.1007/s11538-005-9024-1. [DOI] [PubMed] [Google Scholar]
- 3.Hybiske K., Stephens R.S. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc. Natl. Acad. Sci. U.S.A. 2007;104:11430–11435. doi: 10.1073/pnas.0703218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galasso G.J., Manire G.P. Effect of antiserum and antibiotics on persistent infection of HeLa cells with meningopneumonitis virus. J. Immunol. 1961;86:382–385. [PubMed] [Google Scholar]
- 5.Matsumoto A., Manire G.P. Electron microscopic observations on the effects of penicillin on the morphology of Chlamydia psittaci. J. Bacteriol. 1970;101:278–285. doi: 10.1128/jb.101.1.278-285.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hogan R.J., Mathews S.A., Mukhopadhyay S., Summersgill J.T., Timms P. Chlamydial persistence: beyond the biphasic paradigm. Infect. Immun. 2004;72:1843–1855. doi: 10.1128/IAI.72.4.1843-1855.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wyrick P.B. Chlamydia trachomatis persistence in vitro: an overview. J. Infect. Dis. 2010;201(Suppl. 2):S88–S95. doi: 10.1086/652394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiedeman J.A., Kaul R., Heuer L.S., Thao N.N., Pinkerton K.E., Wenman W.M. Tobacco smoke induces persistent infection of Chlamydophila pneumoniae in HEp-2 cells. Microb. Pathog. 2004;37:141–148. doi: 10.1016/j.micpath.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Pettengill M.A., Lam V.W., Ojcius D.M. The danger signal adenosine induces persistence of chlamydial infection through stimulation of A2b receptors. PLoS One. 2009;4:e8299. doi: 10.1371/journal.pone.0008299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deka S., Vanover J., Dessus-Babus S., Whittimore J., Howett M.K., Wyrick P.B., Schoborg R.V. Chlamydia trachomatis enters a viable but non-cultivable (persistent) state within herpes simplex virus type 2 (HSV-2) co-infected host cells. Cell Microbiol. 2006;8:149–162. doi: 10.1111/j.1462-5822.2005.00608.x. [DOI] [PubMed] [Google Scholar]
- 11.Borel N., Dumrese C., Ziegler U., Schifferli A., Kaiser C., Pospischil A. Mixed infections with Chlamydia and porcine epidemic diarrhea virus - a new in vitro model of chlamydial persistence. BMC Microbiol. 2010;10:201. doi: 10.1186/1471-2180-10-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goellner S., Schubert E., Liebler-Tenorio E., Hotzel H., Saluz H.P., Sachse K. Transcriptional response patterns of Chlamydophila psittaci in different in vitro models of persistent infection. Infect. Immun. 2006;74:4801–4808. doi: 10.1128/IAI.01487-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mukhopadhyay S., Miller R.D., Sullivan E.D., Theodoropoulos C., Mathews S.A., Timms P., Summersgill J.T. Protein expression profiles of Chlamydia pneumoniae in models of persistence versus those of heat shock stress response. Infect Immun. 2006;74:3853–3863. doi: 10.1128/IAI.02104-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ouellette S.P., Hatch T.P., AbdelRahman Y.M., Rose L.A., Belland R.J., Byrne G.I. Global transcriptional upregulation in the absence of increased translation in Chlamydia during IFN gamma-mediated host cell tryptophan starvation. Mol. Microbiol. 2006;62:1387–1401. doi: 10.1111/j.1365-2958.2006.05465.x. [DOI] [PubMed] [Google Scholar]
- 15.Klos A., Thalmann J., Peters J., Gerard H.C., Hudson A.P. The transcript profile of persistent Chlamydophila (Chlamydia) pneumoniae in vitro depends on the means by which persistence is induced. FEMS Microbiol. Lett. 2009;291:120–126. doi: 10.1111/j.1574-6968.2008.01446.x. [DOI] [PubMed] [Google Scholar]
- 16.Timms P., Good D., Wan C., Theodoropoulos C., Mukhopadhyay S., Summersgill J., Mathews S. Differential transcriptional responses between the interferon-gamma-induction and iron-limitation models of persistence for Chlamydia pneumoniae. J. Microbiol. Immunol. Infect. 2009;42:27–37. [PubMed] [Google Scholar]
- 17.Belland R.J., Nelson D.E., Virok D., Crane D.D., Hogan D., Sturdevant D., Beatty W.L., Caldwell H.D. Transcriptome analysis of chlamydial growth during IFN-gamma-mediated persistence and reactivation. Proc. Natl. Acad. Sci. U.S.A. 2003;100:15971–15976. doi: 10.1073/pnas.2535394100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sueltenfuss E.A., Pollard M. Cytochemical assay of interferon produced by duck hepatitis virus. Science. 1963;139:595–596. doi: 10.1126/science.139.3555.595. [DOI] [PubMed] [Google Scholar]
- 19.Byrne G.I., Lehmann L.K., Landry G.J. Induction of tryptophan catabolism is the mechanism for gamma-interferon-mediated inhibition of intracellular Chlamydia psittaci replication in T24 cells. Infect. Immun. 1986;53:347–351. doi: 10.1128/iai.53.2.347-351.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carlin J.M., Borden E.C., Byrne G.I. Interferon-induced indoleamine 2,3-dioxygenase activity inhibits Chlamydia psittaci replication in human macrophages. J. Interferon. Res. 1989;9:329–337. doi: 10.1089/jir.1989.9.329. [DOI] [PubMed] [Google Scholar]
- 21.Rapoza P.A., Tahija S.G., Carlin J.P., Miller S.L., Padilla M.L., Byrne G.I. Effect of interferon on a primary conjunctival epithelial cell model of trachoma. Invest. Ophthal. Vis. Sci. 1991;32:2919–2923. [PubMed] [Google Scholar]
- 22.Beatty W.L., Belanger T.A., Desai A.A., Morrison R.P., Byrne G.I. Tryptophan depletion as a mechanism of gamma interferon-mediated chlamydial persistence. Infect. Immun. 1994;62:3705–3711. doi: 10.1128/iai.62.9.3705-3711.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beatty W.L., Byrne G.I., Morrison R.P. Morphologic and antigenic characterization of interferon gamma-mediated persistent Chlamydia trachomatis infection in vitro. Proc. Natl. Acad. Sci. U.S.A. 1993;90:3998–4002. doi: 10.1073/pnas.90.9.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rothermel C.D., Byrne G.I., Havell E.A. Effect of interferon on the growth of Chlamydia trachomatis in mouse fibroblasts (L cells) Infect. Immun. 1983;39:362–370. doi: 10.1128/iai.39.1.362-370.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beatty W.L., Morrison R.P., Byrne G.I. Reactivation of persistent Chlamydia trachomatis infection in cell culture. Infect. Immun. 1995;63:199–205. doi: 10.1128/iai.63.1.199-205.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McClarty G., Caldwell H.D., Nelson D.E. Chlamydial interferon gamma immune evasion influences infection tropism. Curr. Opin. Microbiol. 2007;10:47–51. doi: 10.1016/j.mib.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 27.Morrison R.P. Differential sensitivities of Chlamydia trachomatis strains to inhibitory effects of gamma interferon. Infect. Immun. 2000;68:6038–6040. doi: 10.1128/iai.68.10.6038-6040.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shaw A.C., Christiansen G., Roepstorff P., Birkelund S. Genetic differences in the Chlamydia trachomatis tryptophan synthase alpha-subunit can explain variations in serovar pathogenesis. Microbes Infect. 2000;2:581–592. doi: 10.1016/s1286-4579(00)00368-3. [DOI] [PubMed] [Google Scholar]
- 29.Fehlner-Gardiner C., Roshick C., Carlson J.H., Hughes S., Belland R.J., Caldwell H.D., McClarty G. Molecular basis defining human Chlamydia trachomatis tissue tropism. A possible role for tryptophan synthase. J. Biol. Chem. 2002;277:26893–26903. doi: 10.1074/jbc.M203937200. [DOI] [PubMed] [Google Scholar]
- 30.Caldwell H.D., Wood H., Crane D., Bailey R., Jones R.B., Mabey D., Maclean I., Mohammed Z., Peeling R., Roshick C., Schachter J., Solomon A.W., Stamm W.E., Suchland R.J., Taylor L., West S.K., Quinn T.C., Belland R.J., McClarty G. Polymorphisms in Chlamydia trachomatis tryptophan synthase genes differentiate between genital and ocular isolates. J. Clin. Invest. 2003;111:1757–1769. doi: 10.1172/JCI17993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perry L.L., Su H., Feilzer K., Messer R., Hughes S., Whitmire W., Caldwell H.D. Differential sensitivity of distinct Chlamydia trachomatis isolates to IFN-gamma-mediated inhibition. J. Immunol. 1999;162:3541–3548. [PubMed] [Google Scholar]
- 32.Roshick C., Wood H., Caldwell H.D., McClarty G. Comparison of gamma interferon-mediated antichlamydial defense mechanisms in human and mouse cells. Infect. Immun. 2006;74:225–238. doi: 10.1128/IAI.74.1.225-238.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nelson D.E., Virok D.P., Wood H., Roshick C., Johnson R.M., Whitmire W.M., Crane D.D., Steele-Mortimer O., Kari L., McClarty G., Caldwell H.D. Chlamydial IFN-gamma immune evasion is linked to host infection tropism. Proc. Natl. Acad. Sci. U.S.A. 2005;102:10658–10663. doi: 10.1073/pnas.0504198102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Al-Zeer M.A., Al-Younes H.M., Braun P.R., Zerrahn J., Meyer T.F. IFN-gamma-inducible Irga6 mediates host resistance against Chlamydia trachomatis via autophagy. PLoS One. 2009;4:e4588. doi: 10.1371/journal.pone.0004588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bernstein-Hanley I., Coers J., Balsara Z.R., Taylor G.A., Starnbach M.N., Dietrich W.F. The p47 GTPases Igtp and Irgb10 map to the Chlamydia trachomatis susceptibility locus Ctrq-3 and mediate cellular resistance in mice. Proc. Natl. Acad. Sci. U.S.A. 2006;103:14092–14097. doi: 10.1073/pnas.0603338103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coers J., Bernstein-Hanley I., Grotsky D., Parvanova I., Howard J.C., Taylor G.A., Dietrich W.F., Starnbach M.N. Chlamydia muridarum evades growth restriction by the IFN-gamma-inducible host resistance factor Irgb10. J. Immunol. 2008;180:6237–6245. doi: 10.4049/jimmunol.180.9.6237. [DOI] [PubMed] [Google Scholar]
- 37.Miyairi I., Tatireddigari V.R., Mahdi O.S., Rose L.A., Belland R.J., Lu L., Williams R.W., Byrne G.I. The p47 GTPases Iigp2 and Irgb10 regulate innate immunity and inflammation to murine Chlamydia psittaci infection. J. Immunol. 2007;179:1814–1824. doi: 10.4049/jimmunol.179.3.1814. [DOI] [PubMed] [Google Scholar]
- 38.Vignola M.J., Kashatus D.F., Taylor G.A., Counter C.M., Valdivia R.H. cPLA2 regulates the expression of type I interferons and intracellular immunity to Chlamydia trachomatis. J. Biol. Chem. 2010;285:21625–21635. doi: 10.1074/jbc.M110.103010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tietzel I., El-Haibi C., Carabeo R.A. Human guanylate binding proteins potentiate the anti-chlamydia effects of interferon-gamma. PLoS One. 2009;4:e6499. doi: 10.1371/journal.pone.0006499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robertson D.K., Gu L., Rowe R.K., Beatty W.L. Inclusion biogenesis and reactivation of persistent Chlamydia trachomatis requires host cell sphingolipid biosynthesis. PLoS Pathog. 2009;5:e1000664. doi: 10.1371/journal.ppat.1000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nelson D.E., Taylor L.D., Shannon J.G., Whitmire W.M., Crane D.D., McClarty G., Su H., Kari L., Caldwell H.D. Phenotypic rescue of Chlamydia trachomatis growth in IFN-gamma treated mouse cells by irradiated Chlamydia muridarum. Cell Microbiol. 2007;9:2289–2298. doi: 10.1111/j.1462-5822.2007.00959.x. [DOI] [PubMed] [Google Scholar]
- 42.Coutinho-Silva R., Correa G., Sater A.A., Ojcius D.M. The P2X(7) receptor and intracellular pathogens: a continuing struggle. Purinergic Signal. 2009;5:197–204. doi: 10.1007/s11302-009-9130-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coutinho-Silva R., Perfettini J.L., Persechini P.M., Dautry-Varsat A., Ojcius D.M. Modulation of P2Z/P2X(7) receptor activity in macrophages infected with Chlamydia psittaci. Am. J. Physiol. Cell Physiol. 2001;280:C81–C89. doi: 10.1152/ajpcell.2001.280.1.C81. [DOI] [PubMed] [Google Scholar]
- 44.Coutinho-Silva R., Stahl L., Raymond M.N., Jungas T., Verbeke P., Burnstock G., Darville T., Ojcius D.M. Inhibition of chlamydial infectious activity due to P2X7R-dependent phospholipase D activation. Immunity. 2003;19:403–412. doi: 10.1016/s1074-7613(03)00235-8. [DOI] [PubMed] [Google Scholar]
- 45.Darville T., Welter-Stahl L., Cruz C., Sater A.A., Andrews C.W., Jr., Ojcius D.M. Effect of the purinergic receptor P2X7 on Chlamydia infection in cervical epithelial cells and vaginally infected mice. J. Immunol. 2007;179:3707–3714. doi: 10.4049/jimmunol.179.6.3707. [DOI] [PubMed] [Google Scholar]
- 46.Ulmann L., Hirbec H., Rassendren F. P2X4 receptors mediate PGE2 release by tissue-resident macrophages and initiate inflammatory pain. EMBO J. 2010;29:2290–2300. doi: 10.1038/emboj.2010.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alzola E., Perez-Etxebarria A., Kabre E., Fogarty D.J., Metioui M., Chaib N., Macarulla J.M., Matute C., Dehaye J.P., Marino A. Activation by P2X7 agonists of two phospholipases A2 (PLA2) in ductal cells of rat submandibular gland. Coupling of the calcium-independent PLA2 with kallikrein secretion. J. Biol. Chem. 1998;273:30208–30217. doi: 10.1074/jbc.273.46.30208. [DOI] [PubMed] [Google Scholar]
- 48.Paguirigan A.M., Byrne G.I., Becht S., Carlin J.M. Cytokine-mediated indoleamine 2,3-dioxygenase induction in response to Chlamydia infection in human macrophage cultures. Infect. Immun. 1994;62:1131–1136. doi: 10.1128/iai.62.4.1131-1136.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berger M., Schroder B., Daeschlein G., Schneider W., Busjahn A., Buchwalow I., Luft F.C., Haller H. Chlamydia pneumoniae DNA in non-coronary atherosclerotic plaques and circulating leukocytes. J. Lab. Clin. Med. 2000;136:194–200. doi: 10.1067/mlc.2000.108941. [DOI] [PubMed] [Google Scholar]
- 50.Wiedeman J.A., Kaul R., Heuer L.S., Thao N.N., Pinkerton K.E., Wenman W.M. Tobacco smoke induces a persistent, but recoverable state in Chlamydia pneumoniae infection of human endothelial cells. Microb. Pathog. 2005;39:197–204. doi: 10.1016/j.micpath.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 51.Pryor W.A., Stone K. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann. N.Y. Acad. Sci. 1993;686:12–27. doi: 10.1111/j.1749-6632.1993.tb39148.x. (discussion 27–18) [DOI] [PubMed] [Google Scholar]
- 52.Suzuki T., Mower H.F., Friesen M.D., Gilibert I., Sawa T., Ohshima H. Nitration and nitrosation of N-acetyl-l-tryptophan and tryptophan residues in proteins by various reactive nitrogen species. Free Radic. Biol. Med. 2004;37:671–681. doi: 10.1016/j.freeradbiomed.2004.05.030. [DOI] [PubMed] [Google Scholar]
- 53.Yasui H., Takai K., Yoshida R., Hayaishi O. Interferon enhances tryptophan metabolism by inducing pulmonary indoleamine 2,3-dioxygenase: its possible occurrence in cancer patients. Proc. Natl. Acad. Sci. U.S.A. 1986;83:6622–6626. doi: 10.1073/pnas.83.17.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bauer C.M., Dewitte-Orr S.J., Hornby K.R., Zavitz C.C., Lichty B.D., Stampfli M.R., Mossman K.L. Cigarette smoke suppresses type I interferon-mediated antiviral immunity in lung fibroblast and epithelial cells. J. Interfer. Cytokine Res. 2008;28:167–179. doi: 10.1089/jir.2007.0054. [DOI] [PubMed] [Google Scholar]
- 55.Dhillon N.K., Murphy W.J., Filla M.B., Crespo A.J., Latham H.A., O’Brien-Ladner A. Down modulation of IFN-gamma signaling in alveolar macrophages isolated from smokers. Toxicol. Appl. Pharmacol. 2009;237:22–28. doi: 10.1016/j.taap.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cicko S., Lucattelli M., Muller T., Lommatzsch M., De Cunto G., Cardini S., Sundas W., Grimm M., Zeiser R., Durk T., Zissel G., Boeynaems J.M., Sorichter S., Ferrari D., Di Virgilio F., Virchow J.C., Lungarella G., Idzko M. Purinergic receptor inhibition prevents the development of smoke-induced lung injury and emphysema. J. Immunol. 2010;185:688–697. doi: 10.4049/jimmunol.0904042. [DOI] [PubMed] [Google Scholar]
- 57.Lommatzsch M., Cicko S., Muller T., Lucattelli M., Bratke K., Stoll P., Grimm M., Durk T., Zissel G., Ferrari D., Di Virgilio F., Sorichter S., Lungarella G., Virchow J.C., Idzko M. Extracellular adenosine triphosphate and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010;181:928–934. doi: 10.1164/rccm.200910-1506OC. [DOI] [PubMed] [Google Scholar]
- 58.Wolf K., Hackstadt T. Sphingomyelin trafficking in Chlamydia pneumoniae-infected cells. Cell Microbiol. 2001;3:145–152. doi: 10.1046/j.1462-5822.2001.00098.x. [DOI] [PubMed] [Google Scholar]
- 59.van Ooij C., Kalman L., van I., Nishijima M., Hanada K., Mostov K., Engel J.N. Host cell-derived sphingolipids are required for the intracellular growth of Chlamydia trachomatis. Cell Microbiol. 2000;2:627–637. doi: 10.1046/j.1462-5822.2000.00077.x. [DOI] [PubMed] [Google Scholar]
- 60.Goldkorn T., Balaban N., Shannon M., Chea V., Matsukuma K., Gilchrist D., Wang H., Chan C. H2O2 acts on cellular membranes to generate ceramide signaling and initiate apoptosis in tracheobronchial epithelial cells. J. Cell Sci. 1998;111:3209–3220. doi: 10.1242/jcs.111.21.3209. [DOI] [PubMed] [Google Scholar]
- 61.Castillo S.S., Levy M., Thaikoottathil J.V., Goldkorn T. Reactive nitrogen and oxygen species activate different sphingomyelinases to induce apoptosis in airway epithelial cells. Exp. Cell Res. 2007;313:2680–2686. doi: 10.1016/j.yexcr.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 62.Levy M., Khan E., Careaga M., Goldkorn T. Neutral sphingomyelinase 2 is activated by cigarette smoke to augment ceramide-induced apoptosis in lung cell death. Am. J. Physiol. Lung Cell Mol. Physiol. 2009;297:L125–L133. doi: 10.1152/ajplung.00031.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Airenne S., Surcel H.M., Alakarppa H., Laitinen K., Paavonen J., Saikku P., Laurila A. Chlamydia pneumoniae infection in human monocytes. Infect. Immun. 1999;67:1445–1449. doi: 10.1128/iai.67.3.1445-1449.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hatch G.M., McClarty G. Phospholipid composition of purified Chlamydia trachomatis mimics that of the eucaryotic host cell. Infect. Immun. 1998;66:3727–3735. doi: 10.1128/iai.66.8.3727-3735.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cocchiaro J.L., Kumar Y., Fischer E.R., Hackstadt T., Valdivia R.H. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc. Natl. Acad. Sci. U.S.A. 2008;105:9379–9384. doi: 10.1073/pnas.0712241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Murphy G., Jr., Rouse R.L., Polk W.W., Henk W.G., Barker S.A., Boudreaux M.J., Floyd Z.E., Penn A.L. Combustion-derived hydrocarbons localize to lipid droplets in respiratory cells. Am. J. Respir. Cell Mol. Biol. 2008;38:532–540. doi: 10.1165/rcmb.2007-0204OC. [DOI] [PubMed] [Google Scholar]
- 67.McCann M.F., Irwin D.E., Walton L.A., Hulka B.S., Morton J.L., Axelrad C.M. Nicotine and cotinine in the cervical mucus of smokers, passive smokers, and nonsmokers. Cancer Epidemiol. Biomarkers Prev. 1992;1:125–129. [PubMed] [Google Scholar]
- 68.Sasson I.M., Haley N.J., Hoffmann D., Wynder E.L., Hellberg D., Nilsson S. Cigarette smoking and neoplasia of the uterine cervix: smoke constituents in cervical mucus. N. Engl. J. Med. 1985;312:315–316. doi: 10.1056/nejm198501313120516. [DOI] [PubMed] [Google Scholar]
- 69.Zenzes M.T., Puy L.A., Bielecki R. Immunodetection of benzo[a]pyrene adducts in ovarian cells of women exposed to cigarette smoke. Mol. Hum. Reprod. 1998;4:159–165. doi: 10.1093/molehr/4.2.159. [DOI] [PubMed] [Google Scholar]
- 70.Boffetta P., Jourenkova N., Gustavsson P. Cancer risk from occupational and environmental exposure to polycyclic aromatic hydrocarbons. Cancer Causes Control. 1997;8:444–472. doi: 10.1023/a:1018465507029. [DOI] [PubMed] [Google Scholar]
- 71.Pontefract R.D., Ng C.W., Bergeron G. Vero cells co-infected with Chlamydia trachomatis and herpes simplex virus type 2: a scanning and transmission electron microscope study. Sex Transm. Dis. 1989;16:74–78. doi: 10.1097/00007435-198904000-00006. [DOI] [PubMed] [Google Scholar]
- 72.Chiarini F., Mansi A., Pisani S., Seganti L., Brunori S., Gentile V., Di Silverio F. In vitro study of a double infection by herpes simplex virus type 2 and Chlamydia trachomatis. New Microbiol. 1996;19:263–266. [PubMed] [Google Scholar]
- 73.Superti F., Longhi C., Di Biase A.M., Tinari A., Marchetti M., Pisani S., Gallinelli C., Chiarini F., Seganti L. Herpes simplex virus type 2 modulates the susceptibility of human bladder cells to uropathogenic bacteria. Med. Microbiol. Immunol. 2001;189:201–208. doi: 10.1007/s004300100067. [DOI] [PubMed] [Google Scholar]
- 74.Deka S., Vanover J., Sun J., Kintner J., Whittimore J., Schoborg R.V. An early event in the herpes simplex virus type-2 replication cycle is sufficient to induce Chlamydia trachomatis persistence. Cell Microbiol. 2007;9:725–737. doi: 10.1111/j.1462-5822.2006.00823.x. [DOI] [PubMed] [Google Scholar]
- 75.Vanover J., Sun J., Deka S., Kintner J., Duffourc M.M., Schoborg R.V. Herpes simplex virus co-infection-induced Chlamydia trachomatis persistence is not mediated by any known persistence inducer or anti-chlamydial pathway. Microbiology. 2008;154:971–978. doi: 10.1099/mic.0.2007/012161-0. [DOI] [PubMed] [Google Scholar]
- 76.Vanover J., Kintner J., Whittimore J., Schoborg R.V. Interaction of herpes simplex virus type 2 (HSV-2) glycoprotein D with the host cell surface is sufficient to induce Chlamydia trachomatis persistence. Microbiology. 2010;156:1294–1302. doi: 10.1099/mic.0.036566-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Browne E.P., Wing B., Coleman D., Shenk T. Altered cellular mRNA levels in human cytomegalovirus-infected fibroblasts: viral block to the accumulation of antiviral mRNAs. J. Virol. 2001;75:12319–12330. doi: 10.1128/JVI.75.24.12319-12330.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schoborg R.V., Sun J., Daniels C., Whittimore J., Kintner J. The host nectin-1 protein regulates chlamydial development. In: Schachter J., Byrne G., Caldwell H., Chernesky M., Clarke I., Mabey D., Paavonen J., Saikku P., Starnbach M., Stary A., Stephens R., Timms P., Wyrick P., editors. Proceedings of the 12th International Symposium on Human Chlamydial Infections. Hof bei Salzburg; Austria: 2010. pp. 95–98. [Google Scholar]
- 79.Takai Y., Miyoshi J., Ikeda W., Ogita H. Nectins and nectin-like molecules: roles in contact inhibition of cell movement and proliferation. Nat. Rev. Mol. Cell Biol. 2008;9:603–615. doi: 10.1038/nrm2457. [DOI] [PubMed] [Google Scholar]
- 80.MacLeod I.J., Minson T. Binding of herpes simplex virus type-1 virions leads to the induction of intracellular signalling in the absence of virus entry. PLoS One. 2010;5:e9560. doi: 10.1371/journal.pone.0009560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cheshenko N., Liu W., Satlin L.M., Herold B.C. Multiple receptor interactions trigger release of membrane and intracellular calcium stores critical for herpes simplex virus entry. Mol. Biol. Cell. 2007;18:3119–3130. doi: 10.1091/mbc.E07-01-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Krummenacher C., Baribaud I., Sanzo J.F., Cohen G.H., Eisenberg R.J. Effects of herpes simplex virus on structure and function of nectin-1/HveC. J. Virol. 2002;76:2424–2433. doi: 10.1128/jvi.76.5.2424-2433.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stuedli A., Grest P., Schiller I., Pospischil A. Mixed infections in vitro with different Chlamydiaceae strains and a cell culture adapted porcine epidemic diarrhea virus. Vet. Microbiol. 2005;106:209–223. doi: 10.1016/j.vetmic.2004.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yoshikawa T., Hill T.E., Yoshikawa N., Popov V.L., Galindo C.L., Garner H.R., Peters C.J., Tseng C.T. Dynamic innate immune responses of human bronchial epithelial cells to severe acute respiratory syndrome-associated coronavirus infection. PLoS One. 2010;5:e8729. doi: 10.1371/journal.pone.0008729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mosca J.D., Pitha P.M. Transcriptional and posttranscriptional regulation of exogenous human beta interferon gene in simian cells defective in interferon synthesis. Mol. Cell Biol. 1986;6:2279–2283. doi: 10.1128/mcb.6.6.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Obojes K., Andres O., Kim K.S., Daubener W., Schneider-Schaulies J. Indoleamine 2,3-dioxygenase mediates cell type-specific anti-measles virus activity of gamma interferon. J. Virol. 2005;79:7768–7776. doi: 10.1128/JVI.79.12.7768-7776.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li B.X., Ge J.W., Li Y.J. Porcine aminopeptidase N is a functional receptor for the PEDV coronavirus. Virology. 2007;365:166–172. doi: 10.1016/j.virol.2007.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gabrilovac J., Breljak D., Cupic B., Ambriovic-Ristov A. Regulation of aminopeptidase N (EC 3.4.11.2; APN; CD13) by interferon-gamma on the HL-60 cell line. Life Sci. 2005;76:2681–2697. doi: 10.1016/j.lfs.2004.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pospischil A., Borel N., Chowdhury E.H., Guscetti F. Aberrant chlamydial developmental forms in the gastrointestinal tract of pigs spontaneously and experimentally infected with Chlamydia suis. Vet. Microbiol. 2009;135:147–156. doi: 10.1016/j.vetmic.2008.09.035. [DOI] [PubMed] [Google Scholar]